

CASE REPORT
Carcinogens in tobacco smoke are the greatest contributor to the worldwide epidemic of lung cancers. Yet 15% to 20% of patients with lung cancers have never smoked. The etiology of lung cancers in never smokers remains poorly understood. Of interest, previous reports have identified germline mutations in driver oncogenes that are associated with lung cancers, such as epidermal growth factor receptor (EGFR),1-6 receptor tyrosine-protein kinase erbB-2 (ERBB2),7 and possibly others,8 which suggests that heritable predisposition to lung cancer is a contributor in some cases. Although rare, these cases provide important insight into the etiology and biology of lung cancers and highlight the increasing role of germline genetics in this area. In this work, we describe a novel germline EGFR V769M variant in a patient with multiple lung cancers, each of which harbors co-occurring somatic mutations in EGFR. To our knowledge, we also describe for the first time the activating and sensitizing characteristics of EGFR V769M in preclinical and in silico structural models.
A 57-year-old man was found to have five distinct lung nodules and no mediastinal or hilar lymphadenopathy, which raised the possibility of multiple synchronous lung cancers. The patient was a remote former smoker (34 pack-years), quitting 21 years before presentation. He underwent several procedures to resect the five distinct lung nodules, each of which was determined to be lung adenocarcinoma. Molecular analysis was performed on the five tumors and each revealed an EGFR V769M alteration. In addition, the presence of distinct G719 mutations in EGFR in several tumors suggested that these tumors represented independent primary lung cancers (Table 1 and Appendix Fig A1, online only).
Table 1.
Summary of Histology, Genetics, and Stage of Individual Tumors

As V769M is predicted to have a significant functional impact (PolyPhen9), the possibility of a germline variant was considered. The patient was referred for genetic counseling and informed consent was obtained for germline testing. Sequencing of peripheral blood leukocyte DNA confirmed the presence of a germline EGFR V769M variant (Fig 1). The co-occurring somatic variant in exon 20 (EGFR S768I) was found on the same allele as the germline EGFR V769M. It was not possible to determine conclusively whether somatic mutations in exon 18 were similarly in cis with the germline variant given the genomic distance between the variants, which were captured separately and sequenced on distinct short reads in the genomic DNA-based assay that was performed. Although the patient had a maternal family history of various cancers, there was no history of lung cancers in the pedigree (Fig 2). Individualized genetic counseling with consideration of EGFR genetic testing and presymptomatic low-dose computed tomography scans were recommended for the patient’s first- and second-degree relatives. Postoperatively, the patient has been treated with erlotinib as a result of several small lung nodules that were found. He has remained without evidence of progressive disease or new lesions for more than 3 years.
Fig 1.

Spectrum of EGFR variants across each tumor and normal tissue. Integrative genomics viewer (IGV) screen shots of variants identified in exons 18 and 20 of EGFR. Single nucleotide variant highlighted in colored letters for each tumor with the corresponding amino acid change denoted at the top of each screen shot.
Fig 2.
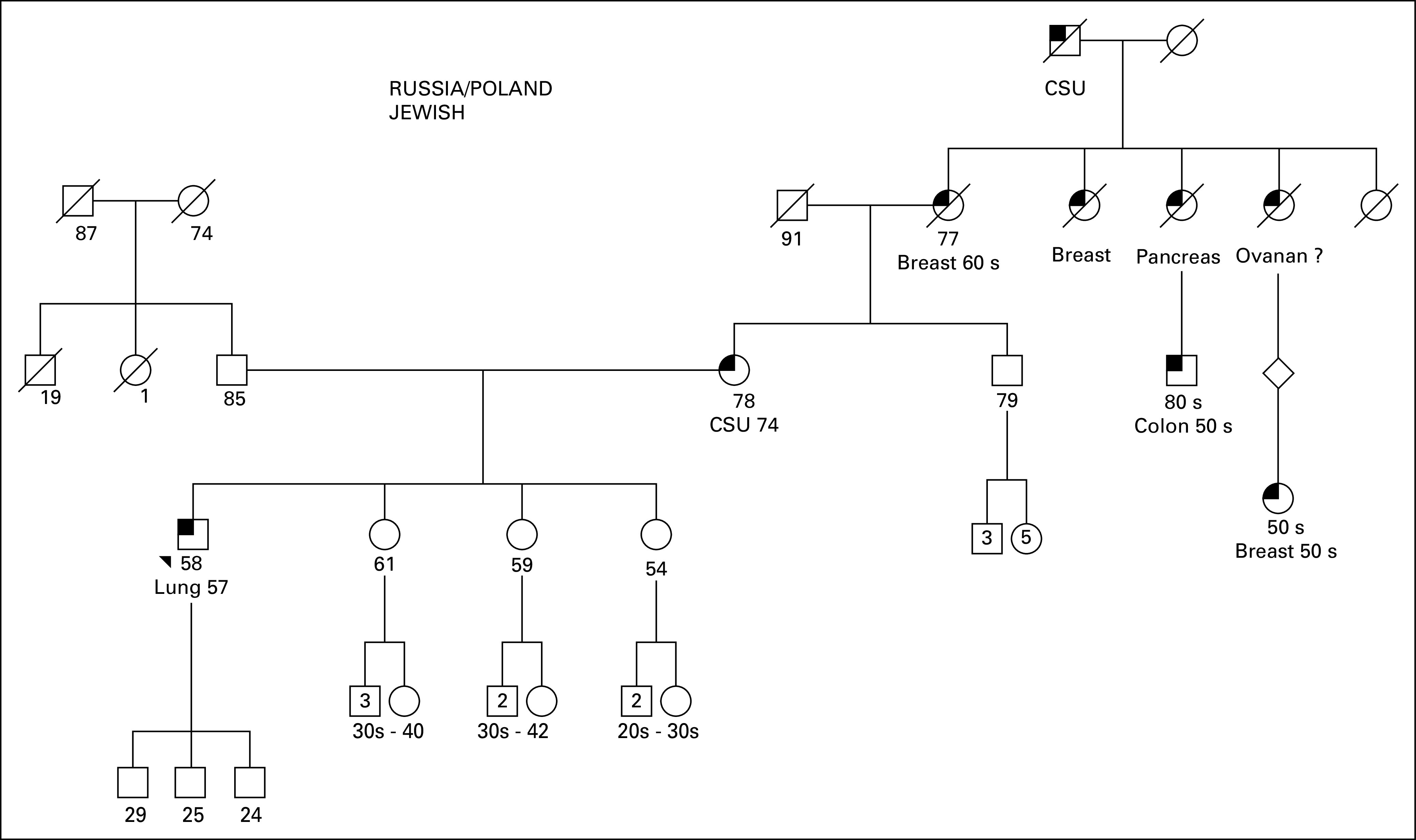
Family pedigree. Male family members are represented by squares and female members by circles. The patient is noted with the black triangle. Known current age (years) for alive patients is noted with the numbers below the square or circles. Age at death, if known, for deceased family members, are represented by diagonal strikes, is noted with the numbers below the square or circles. Known diagnoses of malignancy are annotated, with age at diagnosis. CSU, cancer, site unknown.
The EGFR V769 codon is in exon 20 and corresponds to the region just after the C-helix of the EGFR kinase domain.10 This variant is uncommonly identified in public databases (2 of 120,770 alleles in Exome Aggregation Consortium11; 1 of 13,005 alleles in Exome Variant Server12). Cases of (presumably) somatic EGFR V769M mutations have been reported13-16; one patient received treatment with EGFR tyrosine kinase inhibitors with no clinical response.14 As the functional and sensitizing impact of EGFR V769M has not been assessed experimentally, we examined EGFR V769M in vitro. EGFR V769M was introduced into full-length wild-type EGFR cDNAs (QuikChange II XL Site-Directed Mutagenesis Kit; Agilent Technologies, Santa Clara, CA) and transfected into 293T human cells (FuGENE6; Roche, Indianapolis, IN), a human cell line with a low level of endogenous EGFR expression (Appendix, online only). Consistent with an activated phenotype, p-AKT (S473)/AKT and p-ERK (T202/Y204)/ERK were increased in EGFR V769M mutant compared with wild-type cells in an EGF ligand-dependent setting (Fig 3). To examine the sensitivity of EGFR V769M mutant cells to tyrosine kinase inhibition, transfected 293T cells were treated with increasing concentrations of erlotinib. Compared with cells with the sensitizing EGFR L858R mutation and those with the EGFR T790M gatekeeper mutation, EGFR V769M–bearing cells had intermediate sensitivity to erlotinib. The percent activation, quantified by the ratio of pEGFR (Y1068) to total EGFR, was inhibited by 50% in EGFR V769M cells at an erlotinib concentration of 0.4 μm compared with 0.01 μm in EGFR L858R cells; 50% inhibition was not reached in EGFR T790M cells at the doses tested (up to 2.5 μm; Figs 4A and 4B). To explore the functional impact of comutations in EGFR on EGFR V769M, we also examined 293T cells that were transfected with EGFR V769M + G719C. Activation of downstream pathways and sensitivity to erlotinib were both enhanced in the setting of EGFR V769M plus G719C compared with EGFR V769M alone (Appendix Figs A2 and A3, online only).
Fig 3.

EGFR V769M mutation activates mitogen-activated protein kinase and AKT signaling. 293T cells were transiently transfected with plasmids that encode wild type (WT) EGFR or EGFR mutants with the following changes: V769M, L858R, and T790M. After 36 hours, cells were serum starved for 24 hours, then harvested for immunoblot analyses (Appendix). Quantification of the protein expression of phospho-epidermal growth factor receptor (pEGFR), total EGFR, phospho-ERK (pERK), total-ERK, phospho-AKT (pAKT), and total AKT was analyzed by using ImageJ. Protein levels relative to cells transfected with WT are expressed as fold changes (mean ± standard deviation; n = 3). (*)P < .05, Student’s t test, each condition compared to WT.
Fig 4.

EGFR V769M mutation is sensitive to erlotinib. (A and B) 293T cells were transiently transfected with plasmids that encode wild-type EGFR or EGFR mutants with the following changes: V769M, L858R, T790M. After 36 hours, cells were serum starved for 24 hours, treated with a different concentration of erlotinib for 3 hours, then harvested for immunoblot analyses. (A) Quantification of the protein expression was analyzed by using ImageJ, and (B) data were fitted using GraphPad Prism 6.0f (Appendix). Compared with sensitizing EGFR L858R and resistant T790M mutation, EGFR V769M mutation shows intermediate inhibition of phospho (p)-EGFR/total-EGFR by erlotinib. EGFR, epidermal growth factor receptor; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
We also used qualitative in silico structural modeling to assess the predicted functional impact of V769M on EGFR kinase activity. We examined the location and interactions of the original valine residue in the three-dimensional structure of EGFR in both inactive and activated conformations (Fig 5). V769 directly interacts with F856 of the conserved DFG loop, a key structural element that is involved in the activation of EGFR kinase activity. In the inactive conformation, the V769 residue is completely buried in the hydrophobic core of the protein globule where it has multiple noncovalent interactions with atoms of surrounding residues (Fig 5A). A substitution of the Val residue with a large Met residue will result in the destruction of molecular interaction and the destabilization of the inactive conformation of the kinase domain. In the activated conformation, the molecular surrounding of V769 undergoes significant changes, the total number of noncovalent interactions is reduced, and structural rearrangements that involve the DFG loop region facilitate the penetration and stabilization of ATP (Fig 5B). Thus, it is reasonable to hypothesize that the structural accommodation of Met residue in the activated conformation costs less energy than in the inactive conformation. The total effect of the V769M mutation is an activation of EGFR caused by a shift of the energy balance between activated and inactivated conformations as a result of significant destabilization of the inactivated state of the molecule. Moreover, the quantitative energy balance also favors the activated/open conformation in EGFR V769M and each of the comutant scenarios identified in this case (Appendix Table A1, online only). By contrast, the energy balance does not favor that activated state in the context of EGFR V769M plus L858R, which perhaps explains the absence of this combination in our patient or other reported cases.
Fig 5.

(A and B) The mutated residue V769 (green) is shown in two basic conformations of the kinase domain of epidermal growth factor receptor (EGFR): (A) closed/inactive (PDB ID: 2gs7) and (B) open/activated (PDB ID: 2gs6). In the closed/inactive conformation, V769 is tightly packed in hydrophobic surroundings formed by the residues of two alpha helices (752 to 768, 812 to 831), beta-strand (777 to 782) and two loop regions (769 to 776 and the conserved DFG loop region 855 to 857, a key structural element that regulates kinase activity). In addition, the closed conformation is stabilized by hydrogen bonds between amine groups of R831 and the main chain and side chain oxygens of S768. In the open/active conformation, the three-dimensional (3D) structure of the kinase domain changes drastically (for example, see flipping of 3D position of Glu865 shown in magenta in both panels) and the molecular packing of V769 is relaxed. The total number of 4 Ǻ atom-to-atom contacts is reduced from 34 in the closed conformation to 27 in the open conformation. The significant structural rearrangements involving the DFG loop region facilitate the penetration and stabilization of ATP molecule (B; ATP analog shown in brown). Red orange, alpha-helixes; yellow, beta-structure; and light gray, loop regions.
Mutant EGFR represents a paradigmatic targetable oncogene in lung cancers and is present in almost 20% of lung adenocarcinomas. Although the vast majority of EGFR mutations are somatic, reports of germline EGFR variants suggest that these abnormalities may contribute to lung cancer susceptibility. There have been three reports of germline EGFR V843I mutants—two distinct familial cohorts2,4 and one sporadic case.17 There have been several reports of patients with germline EGFR T790M mutations that have occurred both sporadically and in families.1,3,5,18,19 Much like the case we present here, the majority of germline EGFR variants that have been detected in lung cancers are associated with secondary, somatic activating mutations in EGFR (with some exceptions1,3,17,19). Consistent with the enhanced activation in the EGFR V769M plus G719C comutant model (Appendix Fig A2), it may be that germline variants in EGFR represent a predisposing first hit and that subsequent somatic mutations in EGFR further contribute to lung cancer oncogenesis.
In summary, we describe an EGFR V769M germline variant found in a patient with five synchronous lung cancers that represented several distinct primaries. Our structural modeling and functional studies demonstrate that EGFR V769M is activating and intermediately sensitive to erlotinib and support the proposed causal role of this variant in the development of multiple lung cancers in this patient. As paired tumor and germline sequencing is increasingly being integrated in the routine molecular evaluation of cancers,20 we suspect the identification of germline variants21 that are potentially associated with oncogenesis may become a frequent clinicopathologic conundrum. In this era, close collaborations between oncologists, clinical geneticists, molecular pathologists, and basic scientists will be critical for determining the biologic and clinical implications of these variants and guiding decisions for patients and their families.
Appendix
DNA Sequencing
DNAs from five tumor samples and blood were extracted and testing was performed by using a custom gene panel, MSK-AmpliSeq, which covers 94 cancer-related genes. Library construction was performed with 20 ng of formalin-fixed, paraffin-embedded tumor and matched blood DNA by multiplexed amplification of 891 regions tiled by 2,126 amplicons. Sequencing was performed on an Ion Torrent PGM (ThermoFisher, Waltham, MA), and data were analyzed with a custom pipeline developed at Memorial Sloan Kettering Cancer Center. Aligned BAM files generated by Torrent Suite (ThermoFisher, Waltham, MA) were primer trimmed and variant calling was performed to detect single nucleotide and INDEL variants using VarScan2, which were annotated using ANNOVAR. Single nucleotide and INDEL variants were filtered for variant allele read depth and variant allele frequency. Genotyping of historical (unmatched) normal blood BAM files at variant loci was used to remove recurrent systemic artifacts.
Functional Analyses of Mutant EGFRs
Mutations were introduced into full-length wild-type epidermal growth factor receptor (EGFR) cDNAs by using a QuikChange II XL Site-Directed Mutagenesis Kit. The following primers were used to generate the V769M mutation: forward: 5′-gggggttgtccatgatggccatcacgtag-3′, reverse: 5′-ctacgtgatggccatcatggacaaccccc-3′. The following primers were used to generate the G719C mutation: forward: 5′-cgaacgcaccggagcacagcactttgatctt-3′, reverse: 5′- aagatcaaagtgctgtgctccggtgcgttcg-3′. L858R and T790M mutant cDNAs were generated previously (Pao W, et al: PLoS Med 2:e73, 2005). All mutant clones were fully resequenced bidirectionally to ensure that no additional mutations were introduced. For transient transfections, 293T human embryonic kidney cells were transfected (2 × 105 cells per well in six-well plates) by using FuGENE6 and 0.8 μg of plasmid DNA. Cells were grown in DMEM with high glucose, 10% fetal bovine serum, 2 mM l-glutamine, 10 U/mL penicillin, and 10 μg/mL streptomycin at 37°C and 5% CO2. After 36 hours, cells were serum starved in media that contained 0.1% serum. Cells were treated with different concentrations of erlotinib (Selleckchem, Houston, TX) for 3 hours. Three independent experiments were performed for all analyses.
Immunoblotting
Cells were lysed in 50 mM Tris·HCl, pH 8.0, 150 mM sodium chloride, 5 mM magnesium chloride, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 40 mM sodium fluoride, 1 mM sodium orthovanadate, and complete protease inhibitors (Roche). After quantitation by Bio-Rad protein assays (Bio-Rad, Hercules, CA), approximately 50 μg of each sample was separated by gel electrophoresis on 4% to 12%Bis-Tris precast gels (Thermo Fisher Scientific Life Sciences, Waltham, MA) and transferred to 0.2 μm polyvinylidene difluoride membrane (Thermo Fisher Scientific Life Sciences) by using Wet/Tank Blotting Systems (Bio-Rad). Specific proteins were detected by Amersham ECL Western Blotting Detection Reagent (Thermo Fisher Scientific Life Sciences), and the following antibodies: anti–total EGFR 1:2,500 (BD Transduction Laboratories), anti–phospho-EGFR (Tyr-1068) 1:1,000 (Cell Signaling Technology, Danvers, MA), anti–total p44/42 mitogen-activated protein kinase (Erk1/2) 1:1000 (Cell Signaling Technology), anti–phospho-p44/42 mitogen-activated protein kinase (Erk1/2; Thr202/Tyr204; D13.14.4E) 1:1000 (Cell Signaling Technology), anti–total Akt 1:1000 (Cell Signaling Technology), anti–phospho-Akt (Ser473; D9E), anti–horseradish peroxidase–conjugated anti-rabbit Ig 1:10,000 (R&D Systems, Minneapolis, MN), and horseradish peroxidase–conjugated anti-mouse IgG 10,000 (R&D Systems). Quantification of protein expression was analyzed by using ImageJ 2.0.0-rc-30 (NIH, Bethesda, MD). Data on phospho-EGFR/total-EGFR in transfected 293T cells that were treated with different concentrations of erlotinib was fitted to log(inhibitor) versus normalized response (variable slope) by using GraphPad Prism 6.0f (GraphPad Software, La Jolla, CA). Three independent experiments were performed for all analyses.
Fig A1.

Representative images of multifocal lung nodules that were resected. Arrows identify individual nodules that were resected.
Fig A2.

Activation of 293T cells transfected with EGFR WT, EGFR V769M, EGFR V719C, or EGFR V769M plus G719C. EGFR activation quantified by pERK/ERK and p-AKT/AKT protein expression, relative to expression in WT EGFR setting.
Fig A3.
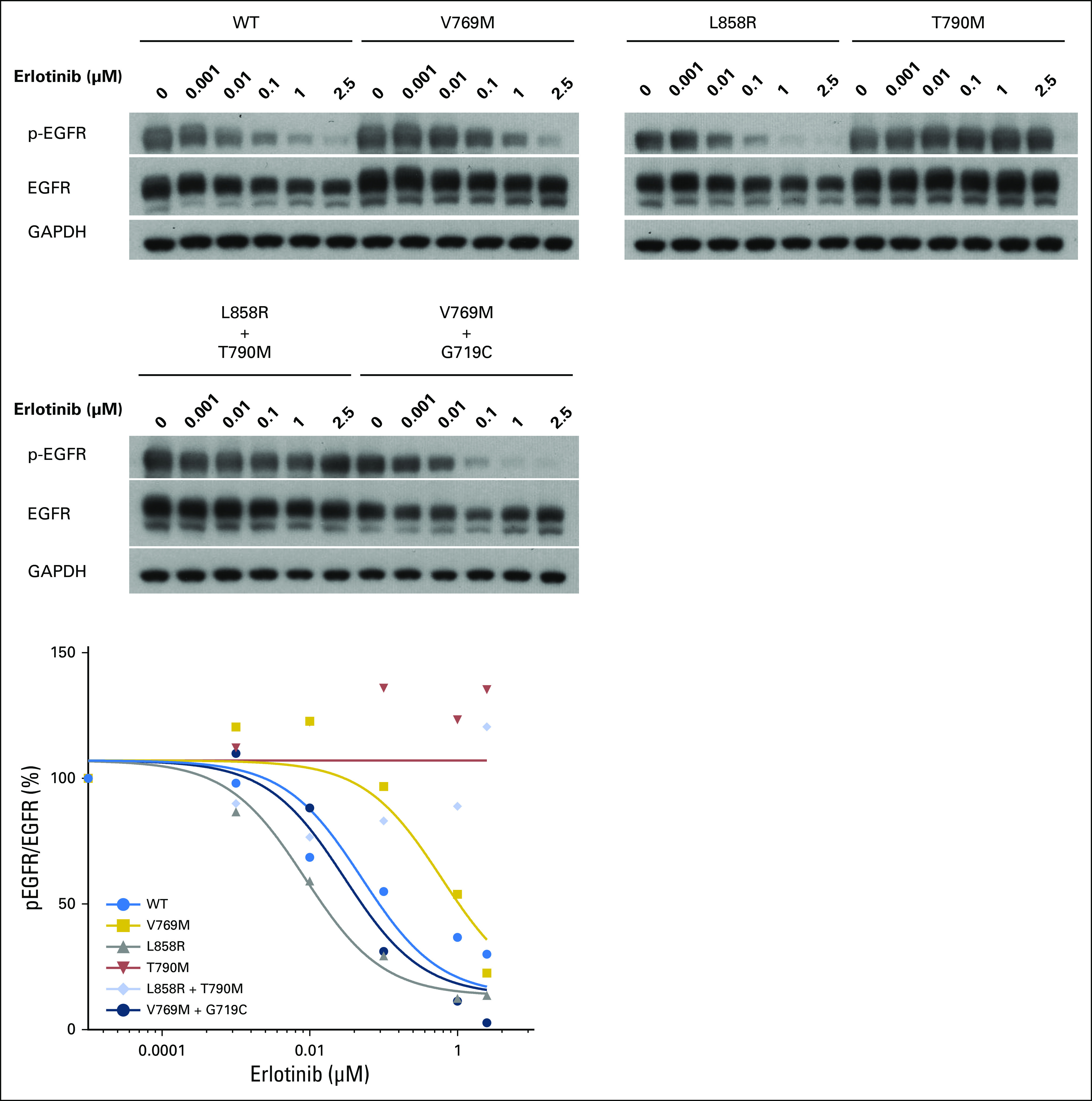
Sensitivity to erlotinib in 293T cells transfected with EGFR wild-type or mutant variants shown below. Sensitivity to erlotinib quantified by pEGFR/EGFR protein expression with increasing concentrations of erlotinib. EGFR, epidermal growth factor receptor; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Table A1.
Energy Scores Produced by Modeller Software for EGFR Mutations

Footnotes
M.D.H. and T.H. contributed equally to this work.
Supported by Memorial Sloan Kettering Cancer Center Support Grant/Core Grant No. P30-CA008748 and Memorial Sloan Kettering Cancer Center Lung Cancer Grant No. P01-CA129243.
AUTHOR CONTRIBUTIONS
Conception and design: Matthew D. Hellmann, Helena A. Yu, Mark G. Kris, Mark E. Robson, Maria E. Arcila, Paul K. Paik
Financial support: Mark G. Kris
Administrative support: Mark G. Kris
Provision of study materials or patients: Matthew D. Hellmann, William D. Travis, Mark G. Kris, Paul K. Paik
Collection and assembly of data: Matthew D. Hellmann, Takuo Hayashi, Olivia Wilkins, Nicole Bramletta, Megan Harlan Fleischut, Mark G. Kris, Mark E. Robson, Paul K. Paik, Marc Ladanyi
Data analysis and interpretation: Matthew D. Hellmann, Takuo Hayashi, Boris Reva, Gregory J. Riely, Prasad S. Adusumilli, William D. Travis, Raghu Chandramohan, Mark G. Kris, Mark E. Robson
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Identification and Functional Characterization of EGFR V769M, a Novel Germline Variant Associated With Multiple Lung Adenocarcinomas
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or po.ascopubs.org/site/ifc.
Matthew D. Hellmann
Consulting or Advisory Role: Bristol-Myers Squibb, Merck, Genentech, AstraZeneca, MedImmune, Novartis, Janssen
Research Funding: Bristol-Myers Squibb, Genentech
Takuo Hayashi
No relationship to disclose
Boris Reva
No relationship to disclose
Helena A. Yu
Consulting or Advisory Role: AstraZeneca, Boehringer Ingelheim, Lilly
Research Funding: Clovis Oncology, AstraZeneca, Astellas Pharma, Incyte Pharmaceuticals, Lilly
Gregory J. Riely
Consulting or Advisory Role: Novartis, Genentech
Research Funding: Novartis (Inst), Genentech (Inst), Millennium Pharmaceuticals (Inst), GlaxoSmithKline (Inst), Pfizer (Inst), Infinity Pharmaceuticals (Inst), ARIAD Pharmaceuticals (Inst)
Patents, Royalties, Other Intellectual Property: Patent application submitted covering pulsatile use of erlotinib to treat or prevent brain metastases (Inst)
Travel, Accommodations, Expenses: Novartis
Prasad S. Adusumilli
No relationship to disclose
William D. Travis
No relationship to disclose
Olivia Wilkins
No relationship to disclose
Nicole Bramletta
Travel, Accommodations, Expenses: AstraZeneca
Raghu Chandramohan
No relationship to disclose
Megan Harlan Fleischut
No relationship to disclose
Mark G. Kris
Consulting or Advisory Role: AstraZeneca, ARIAD Pharmaceuticals, Genentech
Research Funding: Puma Biotechnology (Inst), Genentech (Inst)
Mark E. Robson
Honoraria: AstraZeneca
Consulting or Advisory Role: McKesson, AstraZeneca
Research Funding: AstraZeneca (Inst), AbbVie (Inst), Biomarin (Inst), Medivation (Inst), Tesaro (Inst)
Travel, Accommodations, Expenses: AstraZeneca
Maria E. Arcila
Consulting or Advisory Role: AstraZeneca
Travel, Accommodations, Expenses: AstraZeneca, Invivoscribe, Raindance Technologies
Paul K. Paik
Honoraria: Celgene, Bristol-Myers Squibb, Lilly, ARIAD Pharmaceuticals
Consulting or Advisory Role: Celgene, Lilly, ARIAD Pharmaceuticals, Bristol-Myers Squibb
Research Funding: Celgene, EMD Serono
Travel, Accommodations, Expenses: EMD Serono
Marc Ladanyi
Honoraria: Merck (I)
Consulting or Advisory Role: NCCN/Boehringer Ingelheim Afatinib Targeted Therapy Advisory Committee, NCCN/AstraZeneca Tagrisso RFP Advisory Committee
Research Funding: Loxo Pharmaceuticals (Inst)
REFERENCES
- 1.Bell DW, Gore I, Okimoto RA, et al. : Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet 37:1315-1316, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Ikeda K, Nomori H, Mori T, et al. : Novel germline mutation: EGFR V843I in patient with multiple lung adenocarcinomas and family members with lung cancer. Ann Thorac Surg 85:1430-1432, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Prudkin L, Tang X, Wistuba II: Germ-line and somatic presentations of the EGFR T790M mutation in lung cancer. J Thorac Oncol 4:139-141, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohtsuka K, Ohnishi H, Kurai D, et al. : Familial lung adenocarcinoma caused by the EGFR V843I germ-line mutation. J Clin Oncol 29:e191-e192, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Oxnard GR, Miller VA, Robson ME, et al. : Screening for germline EGFR T790M mutations through lung cancer genotyping. J Thorac Oncol 7:1049-1052, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Noesel J, van der Ven WH, van Os TA, et al. : Activating germline R776H mutation in the epidermal growth factor receptor associated with lung cancer with squamous differentiation. J Clin Oncol 31:e161-e164, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto H, Higasa K, Sakaguchi M, et al. : Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. J Natl Cancer Inst 106:djt338, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen HY, Yu SL, Ho BC, et al. : R331W missense mutation of oncogene YAP1 is a germline risk allele for lung adenocarcinoma with medical actionability. J Clin Oncol 33:2303-2310, 2015 [DOI] [PubMed] [Google Scholar]
- 9.Adzhubei IA, Schmidt S, Peshkin L, et al. : A method and server for predicting damaging missense mutations. Nat Methods 7:248-249, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasuda H, Kobayashi S, Costa DB: EGFR exon 20 insertion mutations in non-small-cell lung cancer: Preclinical data and clinical implications. Lancet Oncol 13:e23-e31, 2012 [DOI] [PubMed] [Google Scholar]
- 11.Exome Aggregation Consortium : ExAC browser beta. http://exac.broadinstitute.org
- 12.Exome Variant Server : NHLBI GO Exome Sequencing Project (ESP): Exome variant server. http://evs.gs.washington.edu/EVS/
- 13.Huang SF, Liu HP, Li LH, et al. : High frequency of epidermal growth factor receptor mutations with complex patterns in non-small cell lung cancers related to gefitinib responsiveness in Taiwan. Clin Cancer Res 10:8195-8203, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Wu JY, Yu CJ, Chang YC, et al. : Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res 17:3812-3821, 2011 [DOI] [PubMed] [Google Scholar]
- 15.Jänne PA, Borras AM, Kuang Y, et al. : A rapid and sensitive enzymatic method for epidermal growth factor receptor mutation screening. Clin Cancer Res 12:751-758, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Santis G, Angell R, Nickless G, et al. : Screening for EGFR and KRAS mutations in endobronchial ultrasound derived transbronchial needle aspirates in non-small cell lung cancer using COLD-PCR. PLoS One 6:e25191, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demierre N, Zoete V, Michielin O, et al. : A dramatic lung cancer course in a patient with a rare EGFR germline mutation exon 21 V843I: Is EGFR TKI resistance predictable? Lung Cancer 80:81-84, 2013 [DOI] [PubMed] [Google Scholar]
- 18.Girard N, Lou E, Azzoli CG, et al. : Analysis of genetic variants in never-smokers with lung cancer facilitated by an Internet-based blood collection protocol: A preliminary report. Clin Cancer Res 16:755-763, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu HA, Arcila ME, Harlan Fleischut M, et al. : Germline EGFR T790M mutation found in multiple members of a familial cohort. J Thorac Oncol 9:554-558, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. doi: 10.1158/2159-8290.CD-16-1337. Jordan EJ, Kim HR, Arcila ME, et al: Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov 10.1158/2159-8290.CD-16-1337 [epub ahead of print on March 23, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. doi: 10.1001/jamaoncol.2015.5208. Schrader KA, Cheng DT, Joseph V, et al: Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol 2:104-111, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]