Figure 2. Mutations that alter the nucleosome DNA entry-exit site cause widespread 3’ extension of snoRNAs.
(A, B) 3’ extension index in (A) the H3 T45A mutant and (B) the H3 R52A mutant. The ratio (mutant/WT) of spike-in normalized RNA-seq read counts between mutant and wild-type strains produced by plasmid shuffling of strain KY812 was determined in a window 150 bp downstream of each annotated snoRNA 3’ end. Ratios equal to or greater than 1.5 (dotted line) are highlighted in purple. (C, D) Browser tracks visualized in IGV (Thorvaldsdóttir et al., 2013) showing de novo transcript annotations across (C) the SNR48 locus and (D) the SNR47 locus. The browser tracks represent spike-in normalized RNA-seq reads in a wild-type strain and the H3 T45A and H3 R52A mutants. Lines of matching color beneath correspond to the de novo transcript annotations for each dataset. Arrows below gene names indicate directionality of transcription.

Figure 2—figure supplement 1. Agreement between biological replicates of RNA-seq datasets.

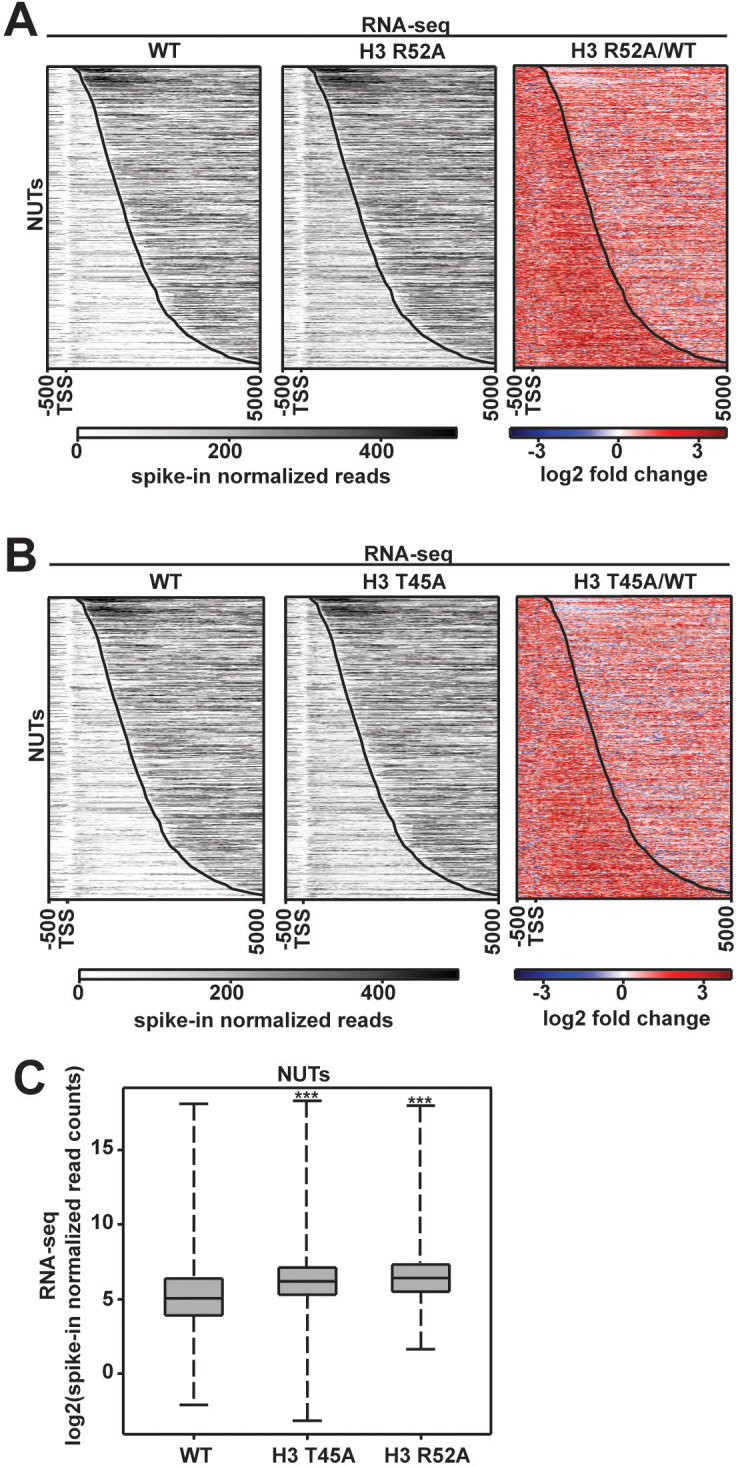
Figure 2—figure supplement 2. Levels of the Nrd1-unterminated transcripts (NUTs) change in DNA entry-exit site mutants.