
Fig. 3.
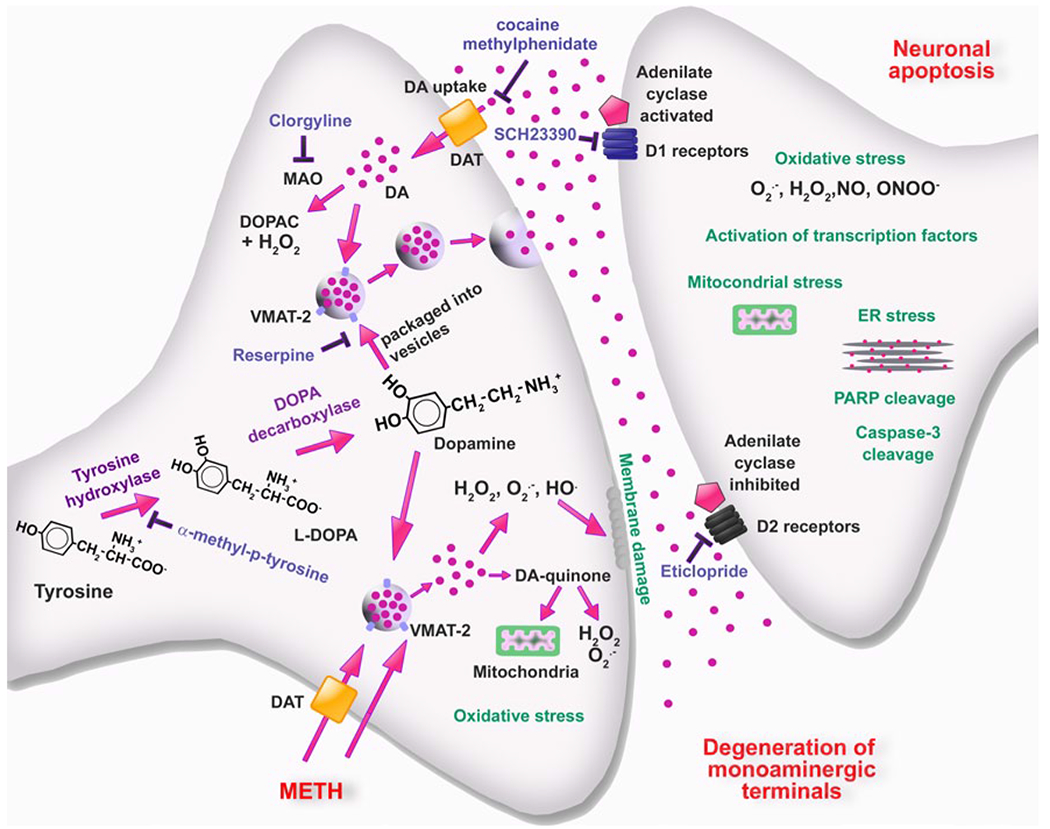
Schematic representation of cellular and molecular events involved in METH-induced DA neurotoxicity within the striatum. METH enters dopaminergic neurons via DAT and passive diffusion. Within these neurons, METH enters synaptic vesicles through VMAT-2 and causes DA release into the cytoplasm by disturbing pH balance. In the cytoplasm, DA auto-oxidizes to form toxic DA quinones followed by generation of superoxide radicals and hydrogen peroxides. Subsequent formation of hydroxyl radicals through interactions of superoxides and hydrogen peroxide with transition metals leads to oxidative stress, mitochondrial dysfunctions, and peroxidative damage to pre-synaptic membranes. The involvement of endogenous DA in METH toxicity is supported by findings that the TH inhibitor, α-methyl-p-tyrosine, which blocks DA synthesis, affords protection against METH toxicity. Also, the role of DA is supported by observations that use of the MAO inhibitor, clorgyline, and of the irreversible inhibitor of vesicular transport, reserpine, which results in increases in cytoplasmic DA levels can exacerbate METH-induced toxicity. These events are thought to be partly responsible for the loss of DA terminals. Toxic effects of released DA appear to depend on activation of DA receptors, because DA receptor antagonists block degeneration of DA terminals. Interactions of DA with D1 receptors on post-synaptic membrane cause activation of various transcription factors and subsequent up-regulation of death cascades in post-synaptic neurons. Modified from Krasnova and Cadet [93]