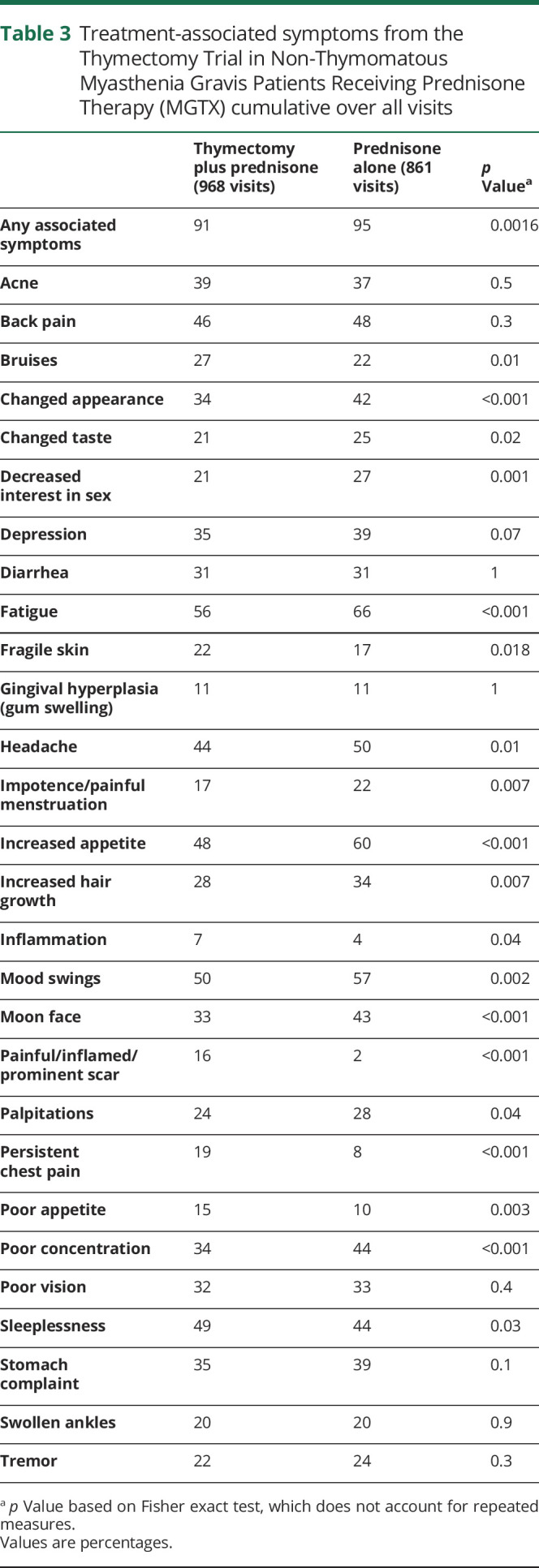
Ikjae Lee
Ikjae Lee, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,✉,
Hui-Chien Kuo
Hui-Chien Kuo, MS
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Inmaculada B Aban
Inmaculada B Aban, PhD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Gary R Cutter
Gary R Cutter, PhD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Tarrant McPherson
Tarrant McPherson, MA
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Henry J Kaminski
Henry J Kaminski, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Jon Sussman
Jon Sussman, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Philipp Ströbel
Philipp Ströbel, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Joel Oger
Joel Oger, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Gabriel Cea
Gabriel Cea, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Jeannine M Heckmann
Jeannine M Heckmann, MBChB
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Amelia Evoli
Amelia Evoli, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Wilfred Nix
Wilfred Nix, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Emma Ciafaloni
Emma Ciafaloni, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Giovanni Antonini
Giovanni Antonini, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Rawiphan Witoonpanich
Rawiphan Witoonpanich, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
John O King
John O King, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Said R Beydoun
Said R Beydoun, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Colin H Chalk
Colin H Chalk, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Alexandru C Barboi
Alexandru C Barboi, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Anthony A Amato
Anthony A Amato, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Aziz I Shaibani
Aziz I Shaibani, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Bashar Katirji
Bashar Katirji, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Bryan RF Lecky
Bryan RF Lecky, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Camilla Buckley
Camilla Buckley, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Angela Vincent
Angela Vincent, MBBS
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Elza Dias-Tosta
Elza Dias-Tosta, MD, PhD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Hiroaki Yoshikawa
Hiroaki Yoshikawa, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Marcia Waddington-Cruz
Marcia Waddington-Cruz, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Michael T Pulley
Michael T Pulley, MD, PhD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Michael H Rivner
Michael H Rivner, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Anna Kostera-Pruszczyk
Anna Kostera-Pruszczyk, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Robert M Pascuzzi
Robert M Pascuzzi, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Carlayne E Jackson
Carlayne E Jackson, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Jan JG Verschuuren
Jan JG Verschuuren, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Janice M Massey
Janice M Massey, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
John T Kissel
John T Kissel, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Lineu C Werneck
Lineu C Werneck, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Michael Benatar
Michael Benatar, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Richard J Barohn
Richard J Barohn, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Rup Tandan
Rup Tandan, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Tahseen Mozaffar
Tahseen Mozaffar, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Robin Conwit
Robin Conwit, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Greg Minisman
Greg Minisman, MA
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Joshua R Sonett
Joshua R Sonett, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
Gil I Wolfe
Gil I Wolfe, MD
1From the Departments of Neurology (I.L.) and Biostatistics (H.-C.K., I.B.A., G.R.C., T.M., G.M.), University of Alabama at Birmingham; Department of Neurology (H.J.K.), George Washington University School of Medicine and Health Sciences, Washington, DC; Department of Neurology (J.S.), Greater Manchester Neuroscience Center, Salford, Greater Manchester, UK; Institute of Pathology (P.S.), University Medical Center Göttingen; Division of Neurology (J.O.), University of British Columbia, Vancouver, Canada; Department of Neurology (G.C.), University of Chile, Santiago; Division of Neurology (J.M.H.), Department of Medicine, University of Cape Town, South Africa; Department of Neurology (A.E.), Catholic University, Rome, Italy; Department of Neurology (W.N.), Johannes Gutenberg University, Mainz, Germany; Department of Neurology (E.C.), University of Rochester Medical Center, NY; Department of Neurosciences (G.A.), Mental Health and Sensory Organs, Sapienza University of Rome, Italy; Division of Neurology (R.W.), Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; Department of Neurology (J.O.K.), Royal Melbourne Hospital, Victoria, Australia; Department of Neurology (S.R.B.), University of Southern California, Los Angeles; Department of Neurology (C.H.C.), McGill University, Montreal, Canada; Department of Neurology (A.C.B.), Medical College of Wisconsin, Milwaukee; Department of Neurology (A.A.A.), Harvard Medical School, Boston, MA; Nerve and Muscle Center of Texas (A.I.S.), Houston; Department of Neurology (B.K.), Case Western Reserve University, Cleveland, OH; Walton Centre for Neurology and Neurosurgery (B.R.F.L.), Liverpool; Nuffield Department of Clinical Neurosciences (C.B., A.V.), Oxford University, UK; Unit of Neurology (E.D.-T.), University of Brasilia, Brazil; Department of Neurology (H.Y.), Kanazawa University, Japan; Department of Neurology (M.W.-C.), Federal University, Rio de Janeiro, Brazil; Department of Neurology (M.T.P.), University of Florida, Jacksonville; Department of Neurology (M.H.R.), Augusta University, GA; Department of Neurology (A.K.-P.), Medical University of Warsaw, Poland; Department of Neurology (R.M.P.), Indiana School of Medicine, Indianapolis; Department of Neurology (C.E.J.), University of Texas Health Science Center, San Antonio; Department of Neurology (J.J.G.V.), Leiden University Medical Center, the Netherlands; Department of Neurology (J.M.M.), Duke University Medical Center, Durham, NC; Department of Neurology (J.T.K.), Ohio State University Wexner Medical Center, Columbus; Department of Neurology (L.C.W.), Universidade Federal do Parana, Curitiba, Brazil; Department of Neurology (M.B.), University of Miami, FL; Department of Neurology (R.J.B.), University of Kansas Medical Center, Kansas City; Department of Neurological Sciences (R.T.), University of Vermont College of Medicine, Burlington; Department of Neurology (T.M.), University of California Irvine Medical Center, Orange; Division of Extramural Research (R.C.), NIH, National Institute of Neurological Disorders and Stroke, Bethesda, MD; Section of General Thoracic Surgery (J.R.S.), Columbia University Medical Center, New York; and Department of Neurology (G.I.W.), University at Buffalo Jacobs School of Medicine and Biomedical Sciences, NY.
1,
on behalf of the MGTX study group