
Figure 3: Key functional pathways are perturbed in RMS.
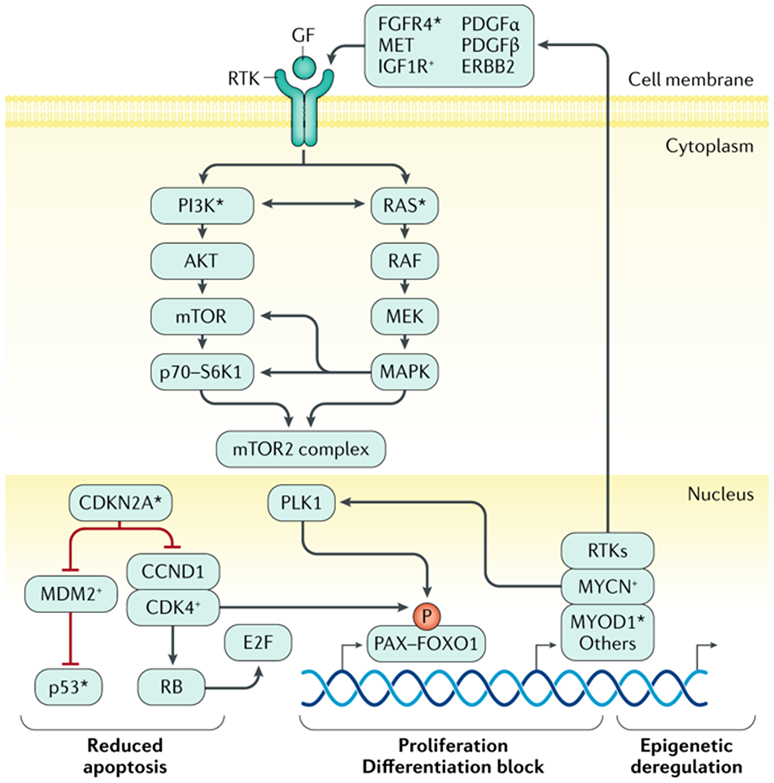
Key processes of apoptosis, cell proliferation, cellular differentiation, and epigenetic homeostasis are deregulated by mutation or gene copy-number and/or gene expression alterations in fusion negative (FN) or fusion positive (FP) rhabdomyosarcoma (RMS). In FP RMS, chromosomal translocations result in PAX3–FOXO1 or PAX7–FOXO1 fusion genes. The aberrant PAX3–FOXO1 fusion protein can synergize with loss of p16 or p53 functionality that is associated with CDKN2A gene loss and/or promoter methylation and TP53 mutation. The stability and subcellular localization of the PAX3–FOXO1 protein is dependent on phosphorylation of specific sites and it works in a complex that can include BRD4. The PAX3-FOXO1 containing complex acts as pioneer factor and drives expression of other transcription factors such as MYCN and MYOD1 via super-enhancers that lead to reprogramming of the transcriptional and epigenetic landscape of tumors. The genes encoding MYCN and MYOD1 transcription factors may themselves be genetically amplified or mutated, likely contributing to RMS formation or progression in a subset of cases, respectively. The fusion protein also drives expression of specific receptor tyrosine kinases (RTKs). Overexpression and activating mutations of genes encoding the same RTKs, and mutation of genes encoding downstream signaling components, are seen in FN RMS. Together this leads to frequent activation of PI3K and RAS pathway signaling in FP and FN RMS, which likely contribute to disease pathogenesis by altering cell proliferation, apoptosis, and other metabolic pathways in ways that are not yet precisely defined. Next-generation DNA sequencing and other molecular genetics tools have demonstrated deleterious mutations in genes encoding certain proteins involved in RMS pathogenesis (*). Exactly how these pathways driven RMS pathogenesis is not clear.