

Abstract
We have determined the fluid bilayer structure of palmitoyl sphingomyelin (PSM) and stearoyl sphingomyelin (SSM) by simultaneously analyzing small-angle neutron and X-ray scattering data. Using a newly developed scattering density profile (SDP) model for sphingomyelin lipids, we report structural parameters including the area per lipid, total bilayer thickness, and hydrocarbon thickness, in addition to lipid volumes determined by densitometry. Unconstrained all-atom simulations of PSM bilayers at 55°C using the C36 CHARMM force field produced a lipid area of 56 Å2, a value that is 10% lower than the one determined experimentally with the SDP analysis (61.9 Å2). Furthermore, scattering form factors calculated from the unconstrained simulations were in poor agreement with experimental form factors, even though segmental order parameter (SCD) profiles calculated from the simulations were in relatively good agreement with SCD profiles obtained from NMR experiments. Conversely, constrained area simulations at 61.9 Å2 resulted in good agreement between the simulation and experimental scattering form factors, but not with SCD profiles from NMR. We discuss possible reasons for the discrepancies between these two important types of data that are frequently used as validation metrics for molecular dynamics force fields.
Graphical Abstract
INTRODUCTION
Membranes play a central role in the life of a cell. Long thought to serve as a simple barrier, cellular membranes are now known to exhibit complex behaviors. The vast array of protein machinery found in membranes hints at a remarkable functionality, as nearly one third of the human genome encodes for membrane proteins.1 Membranes are the sites of transport of ions and small molecules, where the chemical gradients that supply energy to the cell are established and maintained. Importantly, the amphiphilic lipids are responsible for providing the characteristic bilayer structure and serve as the solvent for membrane proteins. Here too, what was initially thought to be a relatively simple role is giving way to a more complicated picture. Cells have evolved a remarkable array of different lipid species,2 and to a large extent the function of a membrane protein is determined by the particular coterie of lipids in its immediate environment. A major current challenge in membrane biophysics is to understand the extent to which nonrandom mixing of these various lipid species is coupled to protein function.3 At one extreme, discrete membrane domains enriched in some lipids and depleted in others can selectively incorporate certain proteins, providing a functional environment that is distinct from the globally averaged membrane environment.4 Detailed pictures of the lipid compositions of these domains are paving the way for well-controlled studies of chemically simplified model systems that nevertheless capture the salient details of a protein’s functional membrane environment.
Sphingomyelin (SM) is one of the most abundant lipids in mammalian plasma membranes, comprising nearly 50 mol% of the total phospholipid content of the outer leaflet.2 Biologically occurring SMs are typically highly ordered, high-melting lipids that increase the structural integrity of cell membranes and reduce their permeability to water and small molecules. In addition to modifying membrane structural properties, sphingomyelin, together with cholesterol, is a major component of plasma membrane rafts, which are thought to serve as functional platforms for transmembrane signaling.4 Unlike saturated phosphatidylcholine (PC) lipids that are often used as substitutes for SM in model membrane studies of rafts, the interfacial region of SM possesses both hydrogen bond donors and acceptors, rendering it capable of forming both inter- and intra-molecular hydrogen bonds.5 The hydrogen bonding potential of SM may contribute to the exceptionally high order of these lipids. Indeed, hydrogen bonding with cholesterol has been proposed to play an important role both in raft formation6 and in modifying the properties of the liquid-ordered phase.7 Despite the clear importance of SM in biological processes, structural studies of this class of lipids are underrepresented compared to other membrane lipids.
One of the most important parameters required to accurately determine bilayer structure, lipid-lipid, and lipid-protein interactions in biomembranes is lipid packing quantified by the average lipid lateral area. In addition to playing a key role in describing membrane structure and its associated functions, knowledge of lateral lipid area is central to Molecular Dynamics (MD) simulations. The most robust determination of lipid bilayer structure, including lateral lipid area, is obtained from a joint analysis of neutron and X-ray diffraction or scattering data.8–9 In combination with MD simulations, scattering data have been used to accurately and unambiguously determine the lipid areas of biologically relevant systems, thus reconciling long-standing differences found in the lipid literature.9 Here, we report experimentally determined structural parameters of palmitoyl sphingomyelin (PSM) and stearoyl sphingomyelin (SSM) from a broad array of techniques including differential scanning calorimetry (DSC), densitometry, small-angle neutron (SANS) and X-ray (SAXS) scattering, nuclear magnetic resonance (NMR), and electron spin resonance (ESR). PSM at 55 °C is analyzed further with unconstrained and area-constrained MD simulations using the CHARMM36 lipid force field. We emphasize a model-free comparison between simulation and experiment, which reveals a discrepancy between PSM bilayer structure determined by small-angle scattering and NMR.
MATERIALS AND METHODS
Materials.
N-palmitoyl-D-erythro-spingosylphosphorylcholine (palmitoyl sphingomyelin, PSM), N-stearoyl-D-erythro-spingosylphosphorylcholine (stearoyl sphingomyelin, SSM), N-palmitoyl-d31-D-erythro-spingosylphosphorylcholine (palmitoyl-d31 sphingomyelin, PSM-d31), egg sphingomyelin (eSM), 1-palmitoyl-2-stearoyl-(7-doxyl)-sn-glycero-3-phosphocholine (7 Doxyl PC, 7-PC), and 1-palmitoyl-2-stearoyl-(16-doxyl)-sn-glycero-3-phosphocholine (16 Doxyl PC, 16-PC) were purchased from Avanti Polar Lipids (Alabaster, AL) and used as received. D2O (99.96% D) was from Cambridge Isotope Laboratories (Andover, MA) and deuterium-depleted H2O (< 1 ppm D) was from Sigma Aldrich (St. Louis, MO).
Preparation of LUVs for scattering measurements.
Phospholipid films were prepared by transferring the desired volumes of stock solutions to a glass scintillation vial with a syringe (Hamilton USA, Reno, NV). Organic solvent was removed with a gentle argon stream and mild heating, followed by further solvent removal under vacuum overnight (~12 h). Dry lipid films were hydrated to a concentration of 40 mg/mL with D2O and then incubated at 60°C for ~ 2 h with intermittent vigorous vortexing to generate multilamellar vesicles (MLVs), followed by 5 freeze/thaw cycles between −80 °C and 60 °C. LUVs were prepared using a miniextruder (Avanti Polar Lipids) assembled with a single 50 nm pore size polycarbonate filter and heated to 50 °C. After extrusion, SANS samples were prepared by diluting 0.3 mL aliquots of the LUVs with H2O and D2O to produce separate 15–22 mg/mL samples in 100%, 70%, and 50% D2O. SAXS samples were prepared separately as described above, with the exception that the dry film was hydrated with H2O to a concentration of 20 mg/mL.
Small-angle neutron scattering (SANS).
Small-angle neutron scattering (SANS) experiments were conducted at Oak Ridge National Laboratory (ORNL), using both the CG-3 BioSANS instrument of the High Flux Isotope Reactor (HFIR) and the BL-6 extended Q-range small-angle neutron scattering (EQ-SANS) instrument of the Spallation Neutron Source (SNS). LUV suspensions were loaded into 2 mm path-length quartz banjo cells (Hellma USA, Plainview, NY) and mounted in a temperature-controlled cell holder with ~ 1°C accuracy. BioSANS data were collected at a 1.7 m sample-to-detector distance (SDD) using 6 Å wavelength neutrons (FWHM 12%), resulting in a total scattering vector of 0.04 < q < 0.4 Å−1. EQ-SANS data were taken at a 2.0 m SDD with a 2.5–6.0 Å wavelength band for a total scattering vector of 0.03 < q < 0.85 Å−1. Scattered neutrons were collected with a two-dimensional (1×1 m) 3He position-sensitive detector (ORDELA, Inc., Oak Ridge, TN) with 192×192 pixels (BioSANS) or 256×192 pixels (EQ-SANS). The 2D data were reduced using the Mantid software package.10 During reduction, data were corrected for detector pixel sensitivity, dark current and sample transmission, as well as background scattering from water. The one-dimensional scattering intensity as a function of the scattering vector q [q = 4π sin (θ)/λ, where λ is the neutron wavelength and θ is the scattering angle relative to the incident beam] was obtained by radial averaging of the corrected 2D data. The absolute form factor used in data analysis is given by:11
| (1) |
For both BioSANS and EQ-SANS data, the signal for q > 0.3 Å−1 was dominated by incoherent scattering and was not used in subsequent analysis.
Small-angle X-ray scattering (SAXS).
X-ray scattering data for joint structural analyses were collected at the Cornell High Energy Synchrotron Source (CHESS) G-1 station. A tightly collimated (0.21×0.21 mm2) beam of 1.17 Å wavelength X-rays was detected using a 1024×1024 pixel array FLICAM charge coupled device with 71 μm linear dimension pixels. The SDD was 426.7 mm, as determined from a powder sample of silver behenate (The Gem Dugout, State College, PA). Samples were loaded in 1.5 mm quartz capillaries and held in a temperature-controlled sample rack during measurement. The X-ray scattering form factors were calculated from corrected experimental intensities using the same relationship as for neutrons (Eq. 1). An additional 4th order polynomial was used to correct for a rise in background scattering with increasing q.12
Additionally, SAXS data from PSM MLV samples at 20 mg/mL were measured using a Rigaku BioSAXS-2000 home source system (Rigaku Americas, The Woodlands, TX) equipped with a HF007 copper rotating anode, a Pilatus 100K 2D detector, and an automatic sample changer. SAXS data were collected at a fixed sample-to-detector distance (SDD) using a silver behenate calibration standard. The one-dimensional scattering intensity I(q) was obtained by radial averaging of the corrected 2D image data, an operation that was performed automatically using Rigaku SAXSLab software. Data were collected in 5-minute frames with each frame processed separately to assess radiation damage; there were no significant changes in the scattering curves over time. Background scattering from water or buffer collected at the same temperature was subtracted from each frame, and the background-corrected I(q) from the individual frames was then averaged, with the standard deviation taken to be the measurement uncertainty.
Scattering Density Profile (SDP) model for SM lipids.
To jointly model SANS and SAXS data, we used a modified Scattering Density Profile (SDP) analysis appropriate for sphingomyelin structure. Briefly, the SDP model describes the underlying matter distribution in the transverse direction (i.e., normal to the plane of the bilayer) in terms of volume probability profiles of lipid component groups comprising several atoms. The Fourier transform of these volume probability profiles, when scaled by the scattering power of the group (i.e., the total neutron scattering length for SANS or atomic X-ray form factor for SAXS) and summed over all groups, gives a predicted scattering form factor that can be refined against experimental scattering data to determine the bilayer structure as described in previous publications8–9, 13–14 and reviews.15–16 The atomic groupings that define the components are not arbitrary, but rather are chosen to satisfy three criteria: (1) the time-averaged matter distribution of the group should be closely approximated by an analytic function whose Fourier transform is also analytic (e.g., a Gaussian); (2) the centers of electron density and neutron scattering length density should coincide to within ~ 1 Å; and (3) the number of groups should be limited by the quantity and resolution of the scattering data. In previous publications we and others have determined that, for a disordered fluid bilayer, approximately six component groups can be resolved using a combination of neutron and X-ray scattering data.8–9, 13 We used MD simulations to guide the grouping of atoms because it is trivial to evaluate criteria 1 and 2 from the simulation trajectory, as demonstrated in Fig. S1 for the CH component group of PSM. Tables S1 and S2 give additional information about the SDP component groups for PSM and SSM, respectively.
Sphingomyelin atoms were parsed into six component groups for SDP analysis as shown in Fig. S2. Similarly to the parsing scheme for PC lipids,17 the headgroup choline methyls (Chol-CH3) and N(CH2)PO4 moiety (PCN) are each represented by a Gaussian. In contrast to the glycerol backbone of PC, the sphingosine backbone C4H4X2NO2 (BB, also represented by a Gaussian) possesses two labile hydrogens (the amide and hydroxyl hydrogens, denoted with an X) that rapidly exchange with protium (1H) or deuterium (2H) in the water. Given the relatively low lipid concentration of samples used for scattering measurements (~ 2 wt% lipid), we assume that the time-averaged fraction of X sites occupied by 2H is equal to the fraction of D2O in the aqueous medium.18 The total hydrocarbon region (HC) is represented by an error function which is further subdivided into methine, methylene, and methyl groups. The trans double bond (CH) at the top of the sphingosine chain, as well as the combined terminal methyls (CH3) at the ends of the sphingosine and N-acyl chains, are modeled as separate Gaussians such that the difference between the hydrocarbon error function and the sum of the CH and CH3 groups gives the CH2 distribution. Fig. S3 shows model-free PSM component volume probabilities calculated from an MD simulation, demonstrating the validity of the chosen functional forms.
Refining the positions, widths, and volumes of the functional groups together with the area per lipid in a least squares analysis results in the most likely volume probability profile for the bilayer. A number of model parameters are either directly varied in the fit or are derived from fitted or constrained parameters. We follow the naming convention used by Kučerka et al.9 and subsequent SDP papers: AL is the lateral lipid area; VL, VHL, and Vwat are the molecular volumes of the total lipid, lipid headgroup, and water, respectively; DB and 2DC are the total (Luzzati) bilayer thickness and hydrocarbon thickness, respectively; DHH is the headgroup-headgroup separation as determined by the distance between electron density maxima; and DH1 is the difference between DHH/2 and DC For all Gaussian components (i.e., j = CH, CH3, BB, PCN, and CholCH3), zj and σj represent the center and width (with zCH3 = 0), while rj represents a volume ratio used to determine the appropriate scale factor (specifically, rCH = VCH/VCH2, rCH3 = VCH3/VCH2, rBB = VBB/VHL, rPCN = VPCN/VHL). Finally, σHC is the width of the error functions representing the total hydrocarbon chain region.
Lipid volumes are critical for the SDP analysis. The total lipid volume VL and the total bilayer (Luzzati) thickness DB are related through the average area per lipid AL (i.e., VL = AL × DB/2). Temperature-dependent lipid volumes for PSM and SSM were independently measured as described below and constrained in the SDP analysis. The sphingomyelin headgroup volume VHL is also a model parameter and was constrained to the value of 274 Å 3 obtained in MD simulations (this value was assumed to be independent of temperature). The difference between VL and VHL gives the total hydrocarbon volume VC. The partitioning of the headgroup and hydrocarbon volumes between their constituent fragments was allowed to vary in the fit subject to soft (quadratic) restraints as previously described.9
Electron spin resonance (ESR).
Samples for ESR were prepared by mixing 13 μmol of PSM in chloroform with 65 nmol of a lipid spin probe (16-PC or 7-PC) in chloroform. Bulk solvent was removed with a nitrogen stream for ~ 30 min, followed by dring under vacuum for ~ 1 h at 55°C. The dry film was hydrated with 520 μL of H2O and incubated for 1 h at ~ 50 °C, vortexing every 15 min. Samples were subjected to 5 freeze/thaw cycles between liquid nitrogen and a water bath at ~ 50 °C. Each sample was then divided into separate aliquots for MLV, LUV, and small unilamellar vesicle (SUV) samples. To generate LUVs, a 220 μL sample aliquot was extruded through a single 50 nm pore size polycarbonate filter 31 times at 50 °C. To generate SUVs, a 220 μL sample aliquot was sonicated in a bath sonicator at 50 °C for ~ 5 min and then gently centrifuged to bring down droplets from the sides of the sample tube. Sonication was then repeated for a total of ~ 20 min until the sample appeared translucent. Aliquots (20 μL) of the LUV and SUV samples were set aside for DLS. The LUVs, as well as 80 μL of the original MLV sample, were diluted to 200 μL with H2O and centrifuged (140,000×g) at 4 °C for 1 h. The SUVs were centrifuged (140,000×g) at 22 °C for 2 hrs. Supernatant was then removed from each sample, and the pellets were diluted with 3–6 μL of H2O. The concentrated samples were loaded into open-ended 1.5–1.8×100 mm glass capillaries (Kimble Chase Life Science, Rockwood, TN). After flame sealing one end of the capillary, the sample was spun down with low speed centrifugation, and the other end was then flame sealed. For measurement, the sealed capillary was placed into a 2 mm NMR tube containing light mineral oil to ensure proper temperature control. ESR spectra were recorded on a Bruker EleXsys-II E500 CW EPR spectrometer operating at X-band frequency (9.4 GHz). The spectrometer settings for all samples were as follows: center field = 3362.9 G, sweep width = 100 G, microwave power = 0.3170 mW, modulation frequency = 100 kHz, modulation amplitude = 0.8 G, resolution (points) = 1024. Reported spectra are the average of 4–16 scans depending on the signal intensity.
ESR first derivative spectra were integrated once to obtain the corresponding absorbance spectrum, and then a second time to obtain a total area which is proportional to the total amount of spin probe in the sample (Fig. S4); the areas were used as normalization factors for both the first derivative and absorbance spectra to correct for sample-to-sample differences in concentration. Normalized spectra were then re-centered to align the second absorbance peak at 3320 G. Model-free order parameters were determined from a lineshape analysis of normalized and centered ESR spectra which was automated using custom code written in Mathematica v. 11.0 (Wolfram Research, Inc.). As shown in Fig. S4, the inner and outer hyperfine splittings of the first derivative spectrum (2Amin and 2Amax, respectively) were determined by fitting the corresponding absorbance spectrum as a sum of Lorentzians:
| (2) |
where B is the magnetic field strength and the Ci, Γi and Bi are fit parameters. Typically, N = 50–70 produced a (purely phenomenological) analytical function A(B) that very closely approximated the raw data, as demonstrated by the residual plots in Fig. S4 (note that residuals are expanded by a factor of 105 relative to the corresponding data). Zeroes in the second derivative of A(B) were then used to determine the peak positions in the first derivative spectrum that define 2Amin and 2Amax. The uncorrected (apparent) order parameter Sapp is given by:
| (3) |
where Axx, Ayy and Azz are the principal components of the hyperfine tensor. These constants are obtained from the first derivative spectra of the dilute probe embedded in a crystal and are often referred to as “single crystal values”. Here, we use values for 7-PC and 16-PC previously reported in the literature,19 namely (Axx, Ayy, Azz) = (5.4, 5.4, 33.4 G) for 7-PC and (5.0, 5.0, 32.6 G) for 16-PC. We note that because these values are constants that appear in the denominator in Eq. 3, they act as a scale factor for the difference in the hyperfine splittings measured directly from the spectra; therefore their precise values do not affect the trends in order parameter that we use to interpret relative differences in the fluidity of vesicles as a function of size and temperature. Finally, the molecular-frame order parameter Szz is calculated as:20
| (4) |
Solid state 2H NMR.
Multilamellar dispersions of 50 wt% PSM-d31 were prepared as follows. A lipid film was prepared by dissolving 26 mg PSM-d31 powder in chloroform, followed by removal of the bulk solvent on a rotary evaporator and overnight vacuum. The film was hydrated with 2 mL degassed, deuterium depleted water (DDW) preheated to 50 °C and then incubated at 50 °C for 2 h with intermittent vortexing. The suspension was lyophilized, weighed, and rehydrated with 2 mL DDW. This cycle was repeated two additional times to reduce the amount of 2HHO from its naturally abundant level. The final rehydration was in 26 μL DDW, followed by five freeze/thaw cycles between a −80 °C and 50 °C. The sample was then transferred to a 5 mm NMR tube and sealed with a cap and Teflon tape for measurement. LUVs were prepared using a miniextruder (Avanti Polar Lipids) assembled with a single 50 nm pore size polycarbonate filter and heated to 50 °C. The LUVs were diluted to 200 μL with DDW and centrifuged (140,000×g) at 4 °C for 1 h. Supernatant was then removed from each sample, and the pellets were diluted with additional DDW to 50 wt%. During the experimental process, aliquots of the sample were tested using dynamic light scattering to verify LUV size integrity.
Solid state 2H NMR experiments were performed on a homebuilt NMR spectrometer operating at 46.0 MHz with a 7.05 T superconducting magnet (Oxford Instruments, Osney Mead, UK),21 equipped with an in-house assembled programmable pulse generator and a dual-channel digital oscilloscope (R1200 M, Rapid Systems, Seattle, WA) to acquire signals in quadrature. Sample temperature was regulated to within ± 0.5 °C with a temperature controller (1600 Series, Love Controls, Michigan City, IN). A phase-alternated quadrupolar echo sequence (90°x-τ−90°y-aquire-delay) was used to eliminate spectral distortion due to the receiver recovery time.22 The following instrument parameters were used: 90° pulse width = 3.7 μs; separation between pulses τ = 50 μs; delay between pulse sequences = 1.0 s; sweep width = ± 100 kHz; and number of scans = 8,192. The 2H NMR spectra acquired are a superposition of powder patterns from all positions of isotopic labeling in the N-acyl chain of PSM-d31.23 Each powder pattern has a pair of most intense peaks split in frequency by Δvr that relates to an order parameter SCD for the C-2H bond in a chain segment according to a standard expression.24 Further, spectra can be analyzed in terms of moments,23 which are defined as:
| (5) |
where Mn is moment of order n, ω is the angular frequency, and f(ω) is the spectral line shape. For MLV in the liquid crystalline state each powder pattern has a pair of most intense peaks split in frequency by Δvr that relates to an order parameter SCD for the C-2H bond in a chain segment according to a standard expression.24 The first moment corresponds to the average order parameter according to the expression ,23 where the quadrupolar coupling constant is, χQ = 1.68 kHz. Although the relationship to average order is inapplicable to MLV in the gel phase and to LUV in either gel or liquid crystalline phase, moments are still sensitive to phase transitions.
Densitometry.
Samples for densitometry were prepared by dispensing SM stock solution in chloroform/methanol (98/2 v/v) into a clean, preweighed glass vial. Bulk solvent was then removed with a nitrogen stream and gentle heating, followed by overnight vacuum pumping at room temperature. The vial was then weighed, and the lipid mass (20–40 mg) was calculated to within 0.1 mg. H2O (1.5 mL) was added to the vial and the weight recorded to within 0.1 mg. The sample was hydrated at 60 °C for 2 h with intermittent brief sonication in a Bransonic 5510 ultrasonic bath (Emerson, St. Louis, MO) to disperse the lipid. Density measurements were performed with an Anton Parr (Ashland, VA) DMA 5000 M vibrating tube density meter. Lipid molecular volume VL was calculated using the relationship:25
| (6) |
where ML is the molar mass of the lipid, ρS and ρW are the measured densities of the sample and H2O, respectively, mS is the mass of the dry lipid, and mW is the mass of H2O added to the dry lipid. Densitometry data for PSM and SSM are shown in Fig. S5.
Differential scanning calorimetry (DSC).
MLV samples for DSC were prepared by adding 0.8 mL H2O to ~ 4 mg dry lipid powder, then vortexing vigorously to disperse the lipid. The sample was hydrated at 60 °C for 2 h with intermittent vortexing, followed by 5 freeze/thaw cycles between a −80 °C and 60 °C. DSC measurements were performed using a TA Nano DSC (TA Instruments, USA, New Castle, DE). The sample was first annealed with two complete temperature cycles between 5 °C to 55 °C at a scan rate of 1 °C/min. A production cycle was then collected from 5 °C to 55 °C at a scan rate of 0.2 °C/min. DSC data for PSM and SSM are shown in Fig. S5.
Molecular Dynamics simulations.
PSM bilayers for MD simulations were constructed using the CHARMM-GUI Membrane builder.26–29 Each bilayer had a total of 200 lipids (100 lipids per leaflet) and was hydrated with 45 waters per lipid molecule. One system was constructed with the default area per lipid for PSM in CHARMM-GUI (55.4 Å2) and simulated in the NPT ensemble, allowing for the bilayer area to converge to its equilibrium value. Five additional simulations were run with constrained areas per lipid of 59, 61, 61.9, 63 and 65 Å2, and were simulated in an NPAT ensemble, keeping the lateral bilayer area fixed while allowing for the z-dimension of the simulation box to vary.
All simulations were performed with the NAMD software30 and the CHARMM36 force field for lipids.31–33 Each system was initially equilibrated with the CHARMM-GUI equilibration protocol, followed by a production run of 350 ns for the unconstrained simulation, and 180–200 ns for the constrained-area simulations. A cutoff of 10–12 Å was used for non-bonded and short-range electrostatic interactions with the VdwForceSwitching option turned on. Particle Mesh Ewald with grid spacing of 1 Å was used for long-range electrostatics. Constant temperature of 55 °C was maintained with a Langevin thermostat. Constant pressure of 1 atm was achieved with the Langevin piston Nose-Hoover method with a 200 fs period and 50 fs decay. All simulations were run with a 2 fs time-step and the rigidbonds option set to all.
For analysis, the last 100 ns of each trajectory were first centered on the average position of the terminal methyl carbons of all PSM molecules. Number density profiles of all PSM and water atoms were calculated with the Density Profile tool in VMD with a resolution of 0.2 Å.34 Ensemble-averaged acyl chain order parameters for each carbon were obtained with in-house Tcl and MATLAB scripts using the formula:
| (7) |
where Θ represents the angle between a CH bond and the bilayer normal assumed to be the z-dimension of the simulation box.
Model-free comparison of experimental and simulation scattering data.
Unconstrained and constrained-area simulations were evaluated against experimentally determined scattering data sets in a model-free comparison as previously described.35 Briefly, the time-averaged real-space atomic number density profiles were directly converted to reciprocal space scattering data by Fourier transform, thereby avoiding assumptions associated with models. Calculated scattering form factors on an absolute scale were then compared with experimentally determined form factors through a chi-square goodness-of-fit criterion:
| (8) |
where Fs(q) and Fe(q) are the simulated and experimentally determined form factors, σe(q) is the experimental uncertainty, Nq is the number of experimental data points, and ke is a scale factor that minimizes χ2.12
For the model-free evaluation of MLV samples, the form factors in Eq. 8 were replaced with scattering intensities I(q). To compute Is(q), the simulation form factor was modified by a structure factor S(q) to account for Bragg scattering between stacked bilayers:
| (9) |
where S(q) depends on the average number of stacked bilayers, the distance between bilayers, and a parameter related to bilayer undulations.36 For each simulation/experiment comparison, the structure factor parameters were optimized to minimize χ2 as described in ref.37.
RESULTS
Structure of SM from joint SANS/SAXS analysis of LUVs.
Small-angle neutron and X-ray scattering have proven instrumental in the determination of bilayer structure.15–16 The quantification of structural parameters from the data requires a real-space bilayer model, where lipid component groups are represented by probability distributions, whose scattering profile is compared to the raw scattering form factor in an iterative fitting procedure. The fitting routine can be applied simultaneously to multiple differently-contrasted data sets of nominally the same bilayer. Since X-rays and neutrons interact differently with a sample, SANS and SAXS themselves represent different contrasts. Moreover, additional contrast conditions can be achieved with SANS by selectively deuterating part of the sample (water and/or lipids) due to the unique interactions of neutrons with 1H and 2H. In general, the more contrast conditions are used to refine the real-space bilayer model, the more robust are the structural parameters obtained in the analysis.
For both PSM and SSM, we measured X-ray scattering form factors in H2O and neutron scattering form factors in 100, 70, and 50% D2O. We also measured the SANS form factor of PSM-d31 in 100% D2O (not shown). Together, these data sets comprise two different radiation contrasts, three different external (solvent) contrasts, and in the case of PSM, one internal (bilayer) contrast. All of these data sets were refined jointly against a single model of the bilayer’s matter distribution to provide a robust determination of the bilayer structure, including the average area per lipid and various bilayer thicknesses. The modeling approach assumes volume incompressibility and derives the average area per lipid from the lipid volume (see Table 1 and Fig. S5) and leaflet thickness calculated as half the bilayer (Luzzati) thickness (DB in Table 1). Because of the relatively high main transition temperatures of PSM (42°C) and SSM (45°C), we collected data at only two fluid phase temperatures for each. Figure 1 illustrates the determination of PSM bilayer structure at 45°C: Fig. 1a shows SANS data for different external and internal contrasts, Fig. 1b shows SAXS data, Fig. 1c and 1d show electron density and neutron scattering length density profiles, respectively, and Fig. 1e shows the underlying volume probability profile.
Table 1.
Structural parameters of PSM and SSM obtained from joint analysis of SAXS and SANS data, and those from MD simulation with a constrained area per lipid of 61.9 Å2. Parameter symbols are defined in Materials and Methods. Uncertainty of free parameters is estimated to be 2%.9
| Parameter | SSM | PSM | |||
|---|---|---|---|---|---|
| Experiment | Experiment | Simulation | |||
| T [°C] | 55 | 65 | 45 | 55 | 55 |
| Vwat [Å3]** | 30.2 | 30.3 | 30.2 | 30.3 | 30.5 |
| VL [Å3]** | 1226.8 | 1237.1 | 1151.6 | 1161.7 | 1172.4 |
| VHL [Å3]** | 274 | 274 | 274 | 274 | 275.1 |
| rBB* | 0.34 | 0.33 | 0.36 | 0.37 | 0.37 |
| rPCN* | 0.33 | 0.33 | 0.4 | 0.4 | 0.35 |
| rCH3* | 2.14 | 2.06 | 1.99 | 2 | 2.12 |
| rCH* | 1.12 | 1.09 | 1.14 | 1.13 | 1.03 |
| DB [Å] | 39.3 | 38.1 | 38.4 | 37.5 | 37.9 |
| DHH [Å] | 40 | 39.4 | 38.9 | 37.8 | 38 |
| 2DC [Å] | 30.5 | 29.7 | 29.3 | 28.7 | 29 |
| DH1 [Å] | 4.77 | 4.85 | 4.8 | 4.55 | 4.5 |
| AL [Å2] | 62.5 | 64.9 | 60.0 | 61.9 | 61.9 |
| zBB [Å] | 14.7 | 14.2 | 14.6 | 14.1 | 15.8 |
| σBB [Å] | 2.1 | 2.03 | 2.55 | 2.36 | 2.44 |
| zPCN [Å] | 20.1 | 19.7 | 19.5 | 18.9 | 19.2 |
| σPCN [Å] | 2.5 | 2.48 | 2.24 | 1.89 | 2.52 |
| zCholCH3 [Å] | 23.7 | 23.1 | 21.7 | 22 | 20.5 |
| σCholCH3 [Å]** | 2.95 | 2.95 | 2.95 | 2.95 | 2.94 |
| zCH [Å] | 15.8 | 15.3 | 14.8 | 14.9 | 14 |
| σCH [Å]** | 2.54 | 2.54 | 2.54 | 2.54 | 2.48 |
| σHC [Å]* | 2.79 | 2.76 | 2.81 | 2.8 | 2.51 |
| σCH3 [Å] | 3.05 | 3.38 | 2.83 | 3.159 | 3.19 |
constrained parameter;
restrained parameter
Figure 1. Joint SDP analysis of PSM scattering data at 45°C.
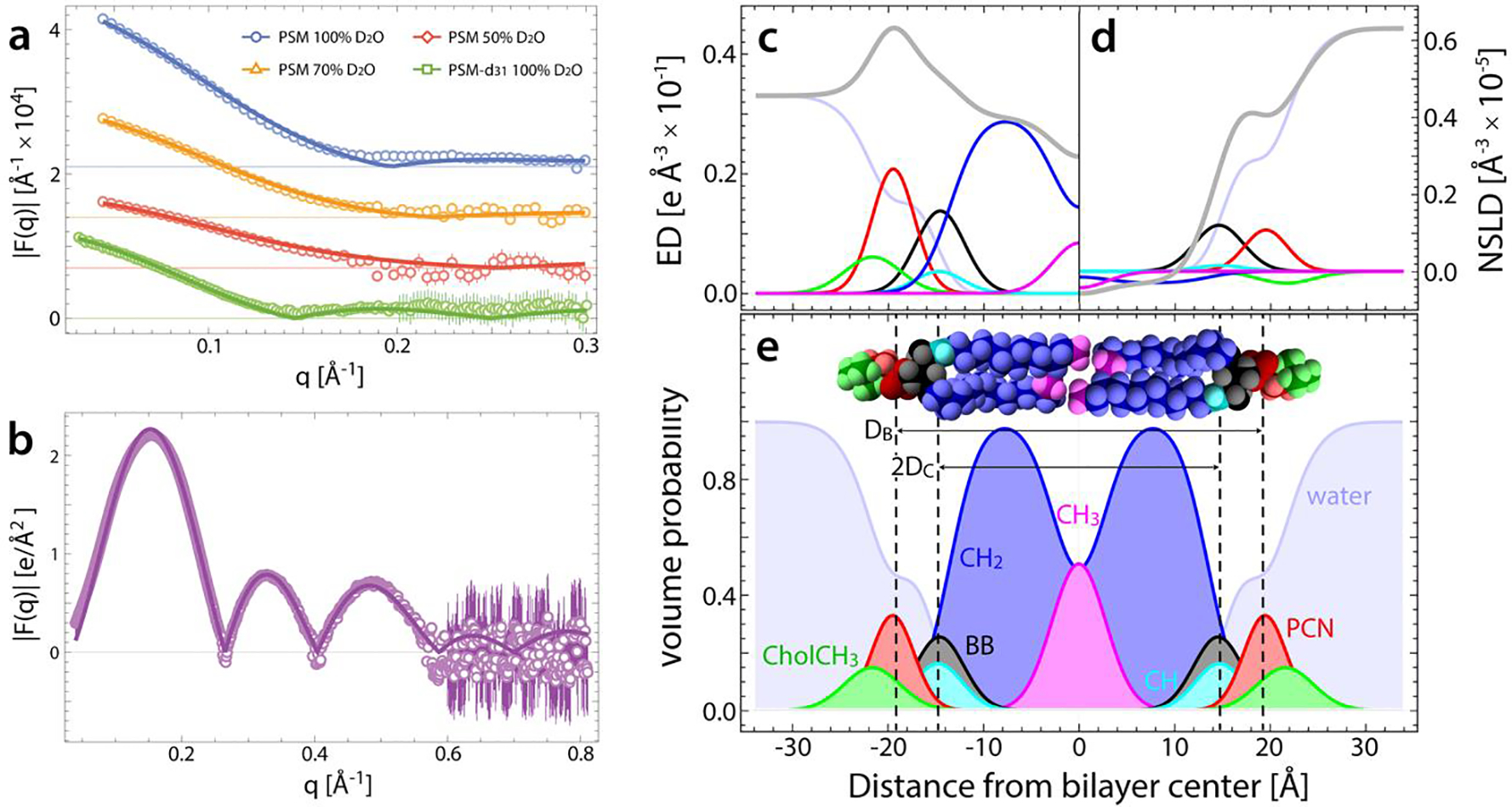
(a,b) Small-angle neutron (a) and X-ray (b) scattering data from 50 nm LUVs (open symbols), and corresponding fits (lines) to the SDP model (see text for details). SANS data were obtained at three external contrasts and one internal contrast (not shown). (c-e) Electron density (c) and neutron scattering length density (d) distributions (ED and NSLD, respectively) for PSM component groups, calculated from volume probability distributions shown in (e). The total SLD profiles in c and d (gray lines) are the sum of the component SLD profiles (colored lines). Fourier transform of the total SLD profiles yields the analytical scattering curves for 100% D2O SANS data in panel a and SAXS data in panel b.
Table 1 gives a complete list of the structural parameters for PSM and SSM. At 55°C, SSM is thicker than PSM, as expected from the two additional carbons in the N-linked chain in SSM. With increasing temperature, both bilayers become thinner and their areas per lipid increase.
Order parameters from 2H NMR of PSM MLVs and LUVs.
2H NMR spectra for deuterated PSM (PSM-d31), prepared as MLVs and LUVs, were obtained over a range of temperatures from 30 °C to 65 °C. Below 38 °C, spectra for both types of preparation are broad and relatively featureless (Fig. S6). This shape reflects the slow rotational diffusion undergone by the rigid, all-trans palmitic acid chains on PSM-d31 in the gel state. Above 38 °C, the spectra (Fig. S6) are dramatically reduced in width by fast axial rotation associated with rapid isomerization about C-C bonds in melted chains. The resulting shape in the case of MLVs is a signature for the lamellar liquid crystalline phase23 where spectral components for some of the methylene groups (and the methyl group) become identifiable. Although spectral narrowing also occurs in LUVs above 38 °C (Fig. S6), distinct spectral components are not observed. This is likely due to the curvature of LUVs, where lateral diffusion and vesicle tumbling move lipids between bilayer regions at different orientations on the NMR time scale, smearing out individual spectral components. Although the LUV line shape is not directly amenable to segmental analysis, moment analysis nevertheless provides a sensitive indication of bilayer phase.
Figure 2 shows the first moments obtained by 2H NMR spectroscopy for LUVs and MLVs. The data shows that the phase transition (identified as the mid-point of an accompanying dramatic drop in value of moment) of PSM in the MLVs and LUVs is 38.0 ± 0.5 °C. The lower melting temperature of the PSM-d31 vesicles relative to their protiated counterparts measured with DSC (Fig. S5) is consistent with previous observations of the effect of different isotopes on bilayer state transitions.38–39 The LUV phase transition is broadened due to the lack of cooperativity between bilayer stacks in MLV phases.40 However, the effects of cooperativity near the phase transition are found to diminish above 40 °C for both systems. We emphasize that the reduction in moments for LUVs compared to MLVs is due to motional averaging associated with vesicle curvature, and not a result of substantially greater disorder.41
Figure 2.
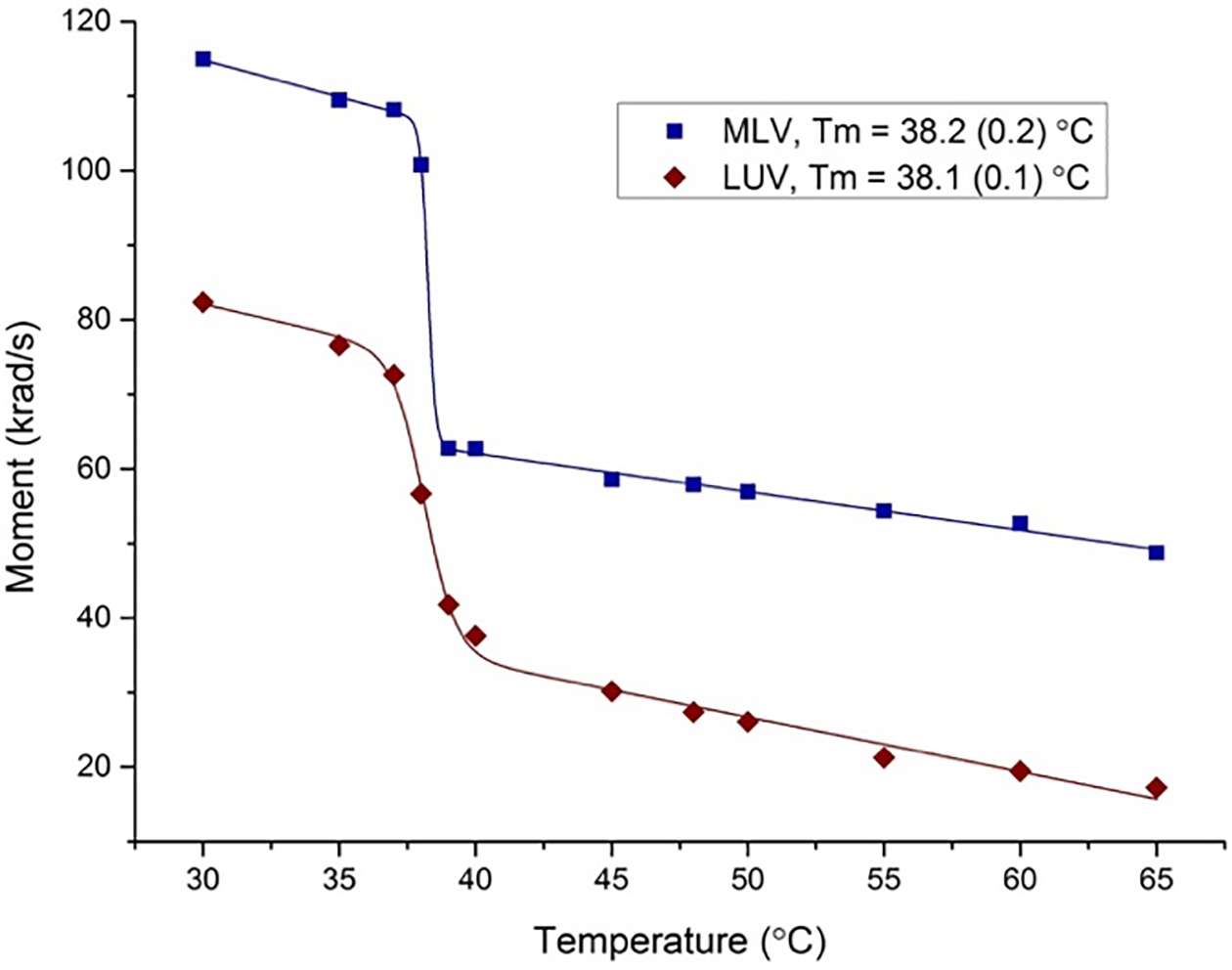
First moment plot of PSM MLV (blue) and LUV (red) samples determined from NMR as a function of temperature. We estimate a reproducibility of ± 1% for moments and ± 0.5 °C for temperature, respectively.
For PSM-d31 in MLVs, distinct quadrupolar splittings were observed for many of the acyl chain segments at temperatures above 40 °C (Fig. S6). Using a FFT depaking algorithm,42 we calculated the order parameter profile for PSM-d31 at 55 °C (Fig. 3c, solid gray squares). Experimentally, order parameters are traditionally plotted from the highest order to the lowest order unless specific assignments are known. It is assumed that carbon segments towards the aqueous interface are more ordered and become increasingly disordered towards the end of the chain at the center of the bilayer. This assumption is valid and easily applied to the terminal methyl group and nearby methylene groups in the lower portion of the chain (C12-C16). However, tight packing in the vicinity of the aqueous interface can constrain orientation and result in reduced order parameters, i.e., a conformation placing a C-2H bond closer to the magic angle (θ = 54°54′) relative to the bilayer normal will reduce the time averaged second order Legendre polynomial that defines the order parameter irrespective of the amount of angular fluctuation. The second carbon segment on the palmitoyl chain of PSM-d31, where distinct order parameters are observed for the two deuterons in the methylene segment, is an example.43 These individual deuterons are identified in the 2H NMR spectra for the perdeuterated chain on the basis of integrated intensity (i.e., they have half the area of a methylene peak and a third of the area of the methyl peak) and their order parameters are included in Fig. S7A. The remaining methylene groups in the upper portion of the chain (C3-C11) are indistinguishable by area considerations and are assigned according to decreasing order. Where multiple methylene groups form a composite peak, in particular, the peak is deconvoluted into signals attributed to separate methylene groups. For comparison with MD simulations, experimental order parameters are plotted in decreasing order (i.e. as a function of sort index).
Figure 3. Model-free comparison of experimental and simulated data.
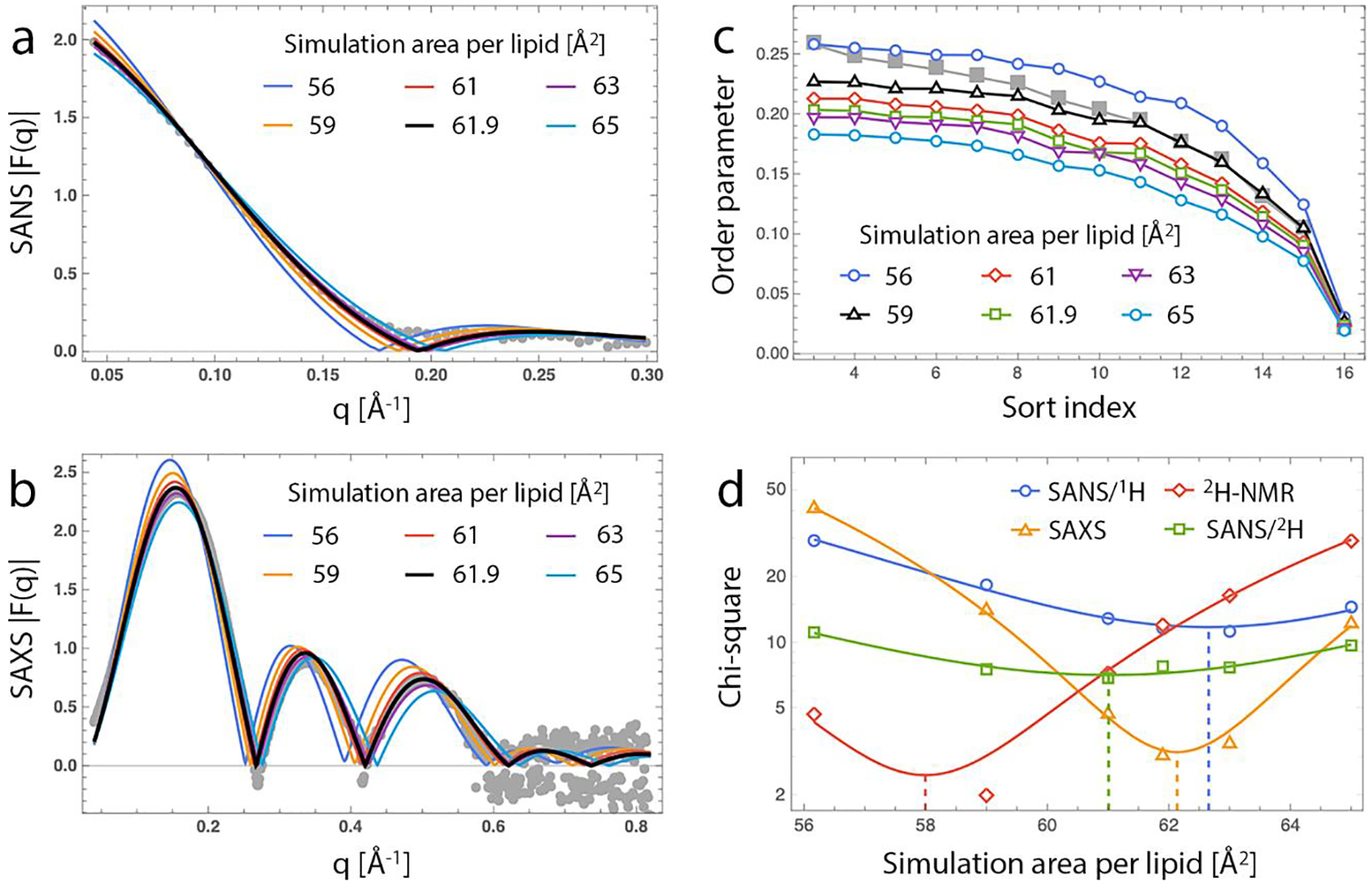
Experimentally determined data sets (solid gray symbols) are compared with corresponding data calculated from unconstrained and area-constrained MD simulations (solid lines) with areas per lipid shown in the legend: SANS form factor for PSM in 100% D2O (a), SAXS form factor (b); NMR segmental order parameter profile (c). In each plot the simulation data that most closely matches the experimental data (as judged from a chi-square metric, see panel d) is shown in black. (d) Chi-square values plotted on a log scale for the model-free comparison of simulated and experimental data. Each set of chi-square values was fit to a parabola; the solid dashed line shows the minimum of each fit. SANS/1H and SANS/2H refer to PSM and PSM-d31, respectively, both in 100% D2O. Errors for NMR order parameters are estimated to be ± 1%.
Model-free validation of simulated PSM bilayers.
Small-angle scattering and NMR data are routinely used in the refinement and validation of force field parameters for MD simulations of lipid bilayers. To facilitate the comparison between simulation and experiment and evaluate the robustness of the sphingomyelin parameters in one of the most widely used force fields, CHARMM36, we performed all-atom MD simulations of a PSM bilayer at 55°C. In the absence of any constraints the bilayer reached an equilibrium area per lipid of 56.2 Å2, 10% lower than the area per lipid obtained from joint analysis of the SANS and SAXS data (Table 1). To compare simulation and experiment more directly, we calculated the 100% D2O SANS and SAXS scattering form factors of the simulated bilayer from the bilayer’s number density profile as described in Materials and Methods. Consistent with the observed trend in the area per lipid, the simulated form factors showed scattering minima at lower q values compared to the experimental data (Fig. 3a and Fig. 3b, solid dark blue lines), indicating a thicker bilayer and tighter lipid packing in the simulation.
A point-by-point comparison of the simulation and experimental form factors provides a model-free approach for quantifying the similarity between simulation and experiment.35 We thus sought to use this model-free analysis to identify the lipid packing at which the two techniques agree. To this end, we ran a series of simulations of the same PSM bilayer at 55 °C but with the average area per lipid fixed to either 59, 61, 61.9, 63 or 65 Å2. The SANS and SAXS form factors of each simulated bilayer (Fig. 3a and Fig. 3b, solid lines) were compared to the raw experimental data (Fig. 3a and Fig. 3b, gray circles) and the similarity between them was quantified by the corresponding chi-square or goodness-of-fit parameter (Fig. 3d). Taking the minimum of a parabola fit to the chi-square vs. simulated area data (Fig. 3d, solid lines) to be the implied best area, the model-free analysis indicated best agreement for SAXS LUV data at a simulated area per lipid of 62.1 Å2, and a best agreement for SANS data from PSM and PSM-d31 at simulated areas per lipid of 62.7 Å2 and 61.0 Å2, respectively. Additionally, two independent SAXS data sets for PSM in H2O at 55 °C showed best agreement with the 63 Å2 constrained-area simulation, and areas per lipid of 62.8 Å2 and 63.9 Å2 implied by quadratic fits to the chi-square data (Fig. S9). In other words, all scattering data sets had best agreement with simulated bilayers over a relatively narrow range of 61–64 Å2. Indeed, the structural parameters from the fixed-area simulation at 61.9 Å2 were in excellent agreement with the results from the joint analysis of the SAXS/SANS data (Table 1).
We then investigated how well the simulation force fields capture the structure of PSM determined by NMR by calculating the N-acyl chain order parameters from the simulated bilayers. Figure 3c shows the respective order parameters sorted from highest to lowest (see Fig. S7 for the data vs. carbon number from the simulations). The experimentally determined lipid order was lower than that calculated from the unconstrained simulation, indicating a looser lipid packing in the experimental system, similar to the scattering data. Interestingly however, a model-free comparison between the NMR data and the order profile of each constrained MD simulation revealed best agreement between NMR and a lipid area of 58 Å2, rather than the 61.9 Å2 obtained from the scattering experiments (Fig. 3d). Possible reasons for this discrepancy are outlined in the Discussion section.
ESR of PSM for vesicles of different curvature.
While vesicles used in scattering experiments were extruded through 50 nm pores, the MLVs measured with NMR were micron-sized. We therefore sought to examine the effects of curvature on the order parameter of the lipids. To this end, we used ESR spectroscopy, which provides information about lipid order through the spectral characterization of a paramagnetic probe incorporated into the bilayer. The probe—a PC lipid with a fully saturated 16C sn-1 chain and nitroxide radical attached to either the 7th (7-PC) or 16th (16-PC) carbon of the 18C sn-2 chain (structures shown in Fig. 4)—absorbs energy when placed in a magnetic field at a specified frequency. Subsequent analysis of the spectral distances in the resulting profile quantifies the probe’s motions, thereby informing on lipid motions in the probe’s immediate environment (see Methods). Moreover, depending on the location of the nitroxide label, information about the lipid order at different depths within the bilayer can be obtained.
Figure 4. Average order parameter of PSM in vesicles of increasing curvature determined by ESR as a function of temperature.
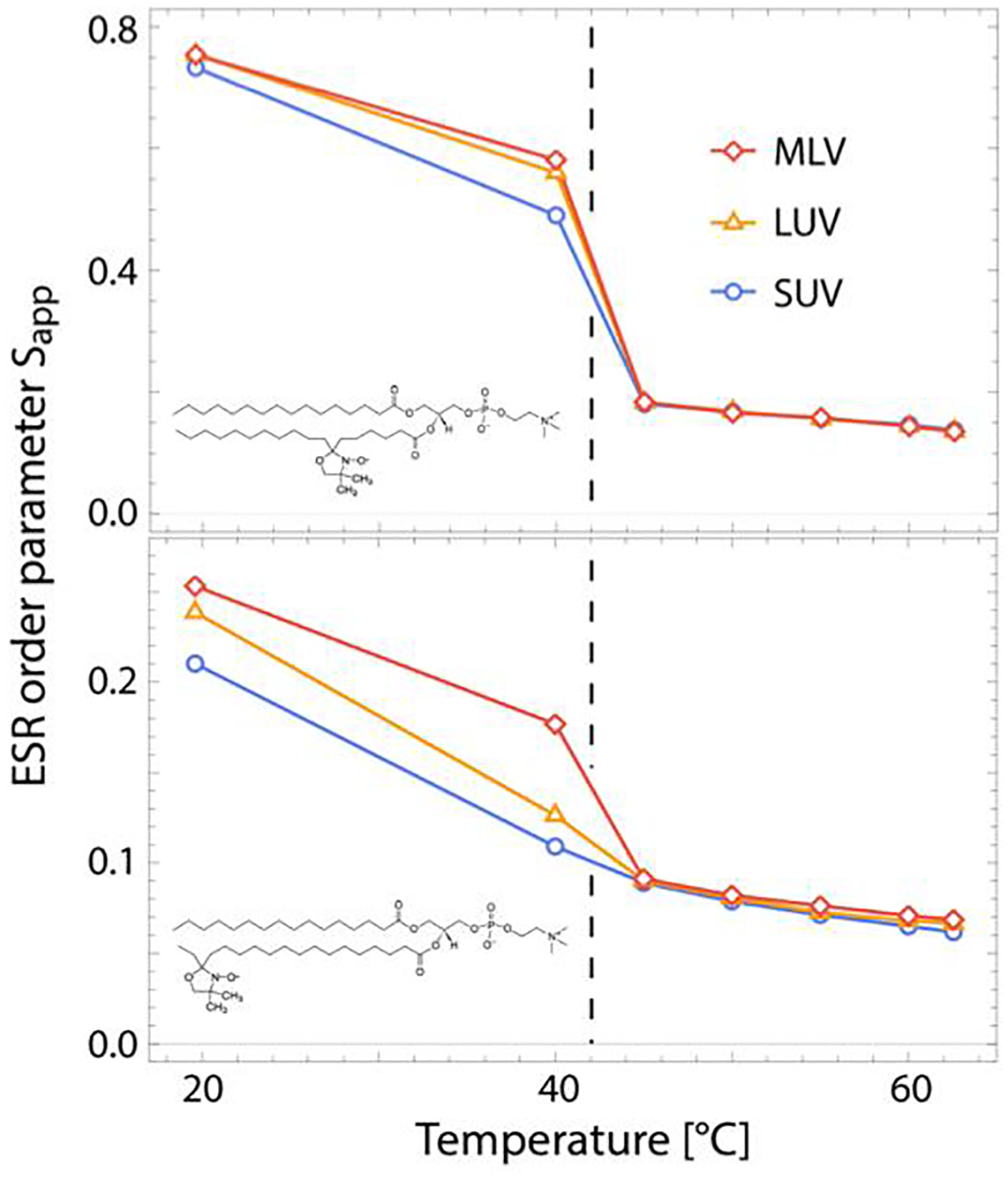
Shown are data for two different probes, 7-PC (top) and 16-PC (bottom), in micron-sized MLVs, extruded LUVs with a diameter of ~ 85 nm, and sonicated SUVs with a diameter of ~ 50 nm. Schematics of the structure of each probe are shown in the lower left corner of the respective plots. Reproducibility of ESR order parameters are < 1%.
Figure 4 shows the apparent order parameter of 16-PC and 7-PC in PSM vesicles of different size (and hence, bilayer curvature) as a function of temperature (sonicated SUVs had a diameter of ~ 50 nm as determined by DLS, while extruded LUVs had a diameter of ~ 85 nm). Below the bilayer’s melting temperature large differences are observed between the three samples, with the order of both probes decreasing with increasing curvature. In contrast, in the fluid state the probes showed a similar degree of order in all vesicles (small differences following the same trend as in the gel state can be seen only in 16-PC at higher temperatures).
DISCUSSION
The SDP model has its roots in the pioneering work of Wiener and White, who first described the joint analysis of neutron and X-ray diffraction data for determining bilayer structure.8, 13–14 Kučerka and coworkers9 modified Wiener and White’s composition space method to utilize the continuous form factor obtained from small-angle scattering experiments (rather than the discrete structure factors obtained from diffraction experiments), allowing for data collection on unilamellar vesicles at full hydration and thus circumventing the inherent difficulties in making fully hydrated oriented bilayers for diffraction measurements.44 Variations of the SDP model have been developed for analysis of neutron and X-ray reflectometry data from supported bilayers,45–46 multilamellar vesicles,47 asymmetric bilayers,48 and membranes containing proteins,49–50 small molecules,51 or mixtures of lipids.52.
In developing the SDP model, an important innovation of Kučerka et al.9 was the use of MD simulations to optimize the parsing of the lipid molecule into quasimolecular fragments, which increased the accuracy of the modeling approach. For each class of lipids, a parsing scheme must be designed that satisfies certain criteria (outlined in Materials and Methods). SDP models have now been constructed for PC,17 PG,11, 25, 53 PE,12 PS,11 cardiolipin,54 ether-linked lipids,55 and PC lipids with polyunsaturated chains.56 Here we report the SDP model (i.e., parsing scheme) for sphingomyelins (Fig. S2), which we used to determine the structure of fluid phase PSM and SSM through joint analysis of SANS and SAXS data combined with independently determined lipid volumes (Fig. S5). Complete results from the analysis are summarized in Table 1.
The structure of fluid phase sphingomyelin.
Structurally, sphingomyelin (SM) has the same choline headgroup as phosphatidylcholine (PC) lipids but possesses a sphingosine rather than a glycerol backbone. Both SM and PC lipids are present in the outer leaflet of mammalian plasma membranes in similar amounts.2 While biologically relevant rafts are thought to be enriched in SM and cholesterol,4 in vitro studies of raft mixtures often use high-melting, fully saturated PC lipids (e.g., DPPC or DSPC) as stand-ins for sphingomyelin.57–58 However, in raft-mimicking mixtures with the same low-melting lipid and cholesterol, there are reported differences between high-melting saturated PC lipids and sphingomyelins in their interactions with cholesterol,59 ternary phase behavior,60–61 and domain size.61–62 It is therefore informative to compare the properties of the two lipid classes.
Figure 5 shows a comparison of the average chain length dependence of sphingomyelin structural parameters with those of various fully saturated or monounsaturated PC lipids17 with values either measured at, interpolated at, or (in the case of PSM) extrapolated to 60 °C, where all lipids are fluid phase. For all lipids, both the total volume and the bilayer thickness increase with increasing average chain length. This is also the case for PG18 and PE12 lipids, suggesting that the increase in bilayer thickness upon addition of a CH2 group is universal (that is, it does not depend on the lipid headgroup, or the nature of the chemical linkages in the backbone). In contrast to the thickness, the trend in AL depends on chain saturation.
Figure 5. Comparison of structural parameters of PSM and SSM to phosphatidylcholine bilayers.
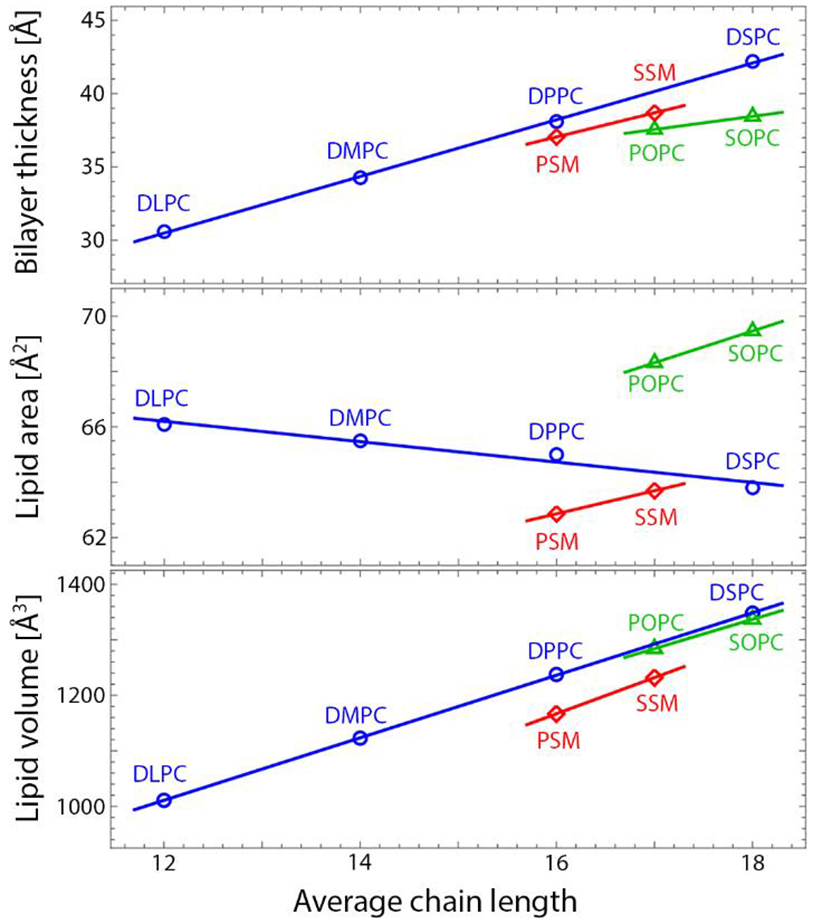
Shown are bilayer thickness (top), area per lipid (middle), and lipid volume (bottom) as a function of average chain length. The comparison is done at 60 °C; PSM is extrapolated and SSM is interpolated from values reported in Table 1. We estimate a 2% uncertainty for lipid area and bilayer thickness9 and 0.1% for lipid volumes.67
A comparison of PSM and DPPC is instructive, as these lipids have the same average chain length and nearly identical main transition temperature. In particular, AL is 3 Å2 smaller for PSM compared to DPPC, suggesting that the sphingosine backbone is conducive to stronger lipid-lipid interactions (and hence, tighter packing) as was shown in a recent study.63 For saturated PC lipids, decreased AL with increasing chain length suggests that the largest contribution to lipid-lipid interactions is van der Waals attraction in the acyl chains; over a limited range, AL decreases approximately linearly with increasing chain length. However, the introduction of a single cis double bond in the sn-2 chain of mixed-chain lipids (i.e., POPC and SOPC) reverses this trend: the addition of two CH2 groups to the saturated sn-1 chain now causes an increase in AL. Interestingly, a similar effect is observed for sphingomyelins upon increasing the length of the fully saturated N-acyl chain. For POPC and SOPC, the increase in AL has been attributed to rotational isomerization counteracting the attractive van der Waals interactions.17 This is unlikely to be true for sphingomyelins: although they possess a double bond in their sphingosine chain, it is the trans isomer, it is located near the top of the chain, and it has an ordering rather than disordering effect on the lipid chains. Instead, the increase in AL may be due to increasing interdigitation of the N-acyl chain with increasing length. Using X-ray diffraction, Maulik et al. reported AL values of 47 Å2,64 55 Å2,65 and 61.3 Å2 66 for PSM, SSM, and C24:0-SM, respectively. Although the first two values are much smaller than those reported here (a discrepancy that is discussed below), the trend of increasing AL with increasing N-acyl chain length is similar to our observations for PSM and SSM. Those authors speculated that the trend may be due to the increasing length mismatch between the N-acyl and sphingosine chains. Interestingly, even though the sphingosine chain possesses 18 carbons, we find that in PSM simulations, the number density distribution of its terminal methyl carbon overlaps with the terminal methyl carbon of the 16-carbon N-acyl chain (Fig. S8). This suggests that due to the geometry of the sphingosine backbone, the sphingosine and N-acyl chain lengths are precisely matched for PSM. It is therefore reasonable that any further addition of carbons to the N-acyl chain will result in increased mismatch with the sphingosine chain. Although the SDP model combines the terminal methyls of the sphingosine and N-acyl chain into a single Gaussian distribution and therefore does not directly inform on interdigitation, it does report indirect effects such as increased AL.
Comparison of area per lipid with literature values.
Experimentally determined bilayer structure plays a crucial role in the refinement and validation of MD force fields. Because it is directly related to bilayer thickness and indirectly related to many other bilayer properties including bending rigidity,68 AL often serves as a reference metric for the bilayer structure itself, and is the most frequently reported point of comparison between different experiments or between simulation and experiment.
Table 2 compares all literature reports (to our knowledge) of the major structural parameters for PSM and SSM at various temperatures determined from experiment or simulation. A few important points emerge from the data in Table 2. First, experimental reports of sphingomyelin structure are rather sparse. A recent study by Arsov et al.69 analyzed PSM at 45 °C using diffuse low-angle X-ray scattering (LAXS), but the only other available scattering data for PSM and SSM is from X-ray diffraction (XRD) measurements of MLVs at 60 wt% water.64–66 Second, a large discrepancy is observed in AL obtained with XRD compared to LAXS or SAXS, with XRD values being much smaller both for PSM and SSM. For PSM in particular, the XRD value of 47 Å2 suggests tight chain packing inconsistent with a fluid phase, and is indeed remarkably similar to values reported for saturated PC lipids in the gel phase.70 Since AL for PSM obtained from LAXS of oriented bilayer stacks is also much higher than the XRD result, curvature effects are unlikely to account for the observed inconsistency. We note that at 45 °C the PSM AL obtained from LAXS (64 Å2) is also larger than the AL we measured with SANS/SAXS (60 Å2). However, this difference is much smaller compared to the XRD values and can be explained in part by the different lipid volume used by Arsov et al., which accounts for about 1 Å2 of the discrepancy.
Table 2.
Comparison of sphingomyelin structural parameters obtained from experiment and simulation.
| Lipid | T [°C] | VL | AL | DB | DHH | Sample | Hydration | Technique | Reference |
|---|---|---|---|---|---|---|---|---|---|
| PSM | 45 | 1151.6 | 60.0 | 38.4 | 38.9 | LUV | full | SANS/SAXS | This work |
| 1171 | 64(2) | 36.6 | 38 | OMB | full* | Diffuse LAXS | 69 | ||
| 48 | 55.4 | 40.7 | 72 lipids | MD (CHARMM) | 31 | ||||
| 56.5 | 39.6 | 288 lipids | MD (ANTON) | 31 | |||||
| 50 | 1180(10) | 52 | 45.4 | 128 lipids | MD (GROMACS) | 71 | |||
| 1168(12) | 53.3(0.4) | 43.8 | 43.7 | 200 lipids | MD (GROMACS) | 72 | |||
| 1126 | 54.1 | 41.6 | 128 lipids | MD (Slipids) | 73 | ||||
| 55 | 1161.7 | 61.9 | 37.5 | 37.8 | LUV | full | SANS/SAXS | This work | |
| 1199 | 47 | 51 | 41 | MLV | 60% | XRD | 64 | ||
| SSM | 45 | 54.5 | 43.0(0.1) | 72 lipids | MD (CHARMM) | 31 | |||
| 50 | 55.4 | 42.7(0.1) | 72 lipids | MD (CHARMM) | 31 | ||||
| 1182 | 53 | 44.6 | 42.4 | 400 lipids | MD (GROMACS) | 74 | |||
| 1181 | 54.0 | 43.7 | 128 lipids | MD (Slipids) | 73 | ||||
| 55 | 1226.8 | 62.5 | 39.3 | 40.0 | LUV | full | SANS/SAXS | This work | |
| 1232a | 55 | 45 | 41 | MLV | 60% | XRD | 65 | ||
| 65 | 1237.1 | 64.9 | 38.1 | 39.4 | LUV | full | SANS/SAXS | This work |
Hydrated from vapor phase at 100% relative humidity
Owing to their biological relevance, sphingomyelins are frequently used in MD simulation studies.31,71–75 Table 2 also shows that our experimentally determined AL values for PSM and SSM are significantly larger than those obtained from MD simulations using the C36 CHARMM lipid force field for sphingolipids,31 which was modified specifically to obtain agreement with available 2H-NMR order parameter data.43 Other force fields result in AL values similar to, or even smaller than, those obtained with CHARMM36. We speculate on possible explanations for the seemingly contradictory structural information revealed by NMR and scattering techniques in the last section of the Discussion.
Effects of curvature on SM structure.
Different techniques used to study bilayer structure such as wide-angle X-ray diffraction,76 small-angle X-ray and neutron scattering,77 2H NMR,78 and MD simulations,79 differ not only in their methodological details and probed structural aspects of the membrane, but also in the nature of the sample being examined. For example: X-ray diffraction requires stacked bilayers (either oriented on a substrate or as MLVs); SAXS and SANS utilize unilamellar free-floating liposomes typically with a diameter of 50–150 nm; 2H NMR is usually obtained from micron-sized MLVs; and MD simulations are routinely performed on flat bilayer patches of at most a few hundred lipids. When comparing results from different techniques, the underlying assumption is that bilayer structure is not influenced by the size or geometry of the system. However, one fundamental difference between the samples is the varying extents of membrane curvature. Indeed, our ESR measurements were designed to directly probe the effect of curvature on the order parameter—and by extension, lipid packing—of PSM.
As shown in Fig. 4, we found that membrane order reported by two nitroxide probes clearly decreases with increasing curvature below the main transition temperature of PSM, but not above it. One potential explanation is the accumulated curvature frustration in vesicles of small diameter, causing bilayer stress and potential packing defects in the gel-like membrane environment. A bilayer in the gel state is much harder to compress and bend relative to the fluid state, making the effects of curvature frustration more pronounced. It is worth noting that a recent study showed a pre-transition for PSM at 24°C indicating the presence of a possible ripple phase between 24°C and the main transition at 41°C.69 While our study was not designed to characterize the full thermotropic behavior of PSM, we cannot exclude the possibility of differential partitioning of the ESR probes in the ordered and disordered regions of a rippled bilayer. Similarly, we cannot claim with certainty that the probes are equally distributed in the two bilayer leaflets. It has been reported that in SUVs the two leaflets of the bilayer can have substantially different packing densities due to the high curvature,80 accompanied by a potential asymmetric interleaflet lipid distribution when multiple lipid species are present.81 Since both ESR probes contain a bulky nitroxide attached to their chains, it is possible that they preferentially reside in one or the other leaflet when in smaller vesicles. However, that ESR order cannot distinguish between PSM vesicles of different curvature at temperatures above the main transition strongly suggests that curvature is not a likely effector of changes in bilayer structure in the fluid state. This conclusion is further supported by the best-fit AL values implied by model-free comparisons of simulations with experimental SAXS data for PSM LUVs (Fig. 3b and Fig. 3d) and MLVs (Fig. S9), as well as the relative agreement in AL obtained from LUVs in the current work and published data from flat bilayer stacks69 (Table 2).
Fluid bilayer structure determination: small-angle scattering compared to NMR.
The accuracy of MD simulations depends to a large extent on the set of force field parameters that define the interaction and bond energies between the lipid (and water) atoms. The development of these parameter sets relies on quantum mechanical calculations and refinement based on robust comparisons with experimental data. The latest CHARMM36 parameters for sphingomyelin were validated against available NMR data,43 and showed very good agreement in the order parameter profile of the N-linked chain of PSM obtained from simulation and experiment at 48 °C.31 However, as we report here, the same set of parameters fails to reproduce the lipid packing of PSM measured with SANS and SAXS at 55 °C. In particular, the average area per lipid in an unconstrained NPT simulation of PSM at 55 °C (56.2 Å2) is 10% lower than that obtained from analysis of the scattering data (61.9 Å2).
One potential argument for this discrepancy is the inability of the scattering analysis to produce a reliable packing density of the bilayer from the scattering form factors. As mentioned earlier, the analysis is based on a real space bilayer model whose parameters are adjusted in an iterative procedure by fitting the bilayer’s scattering profile to the raw SAXS/SANS data in Fourier space. Since theoretically multiple real space models can produce the same scattering form factor, how likely is it that the resulting parameters faithfully represent the bilayer structure? A few different facts argue against the validity of this concern. First, the SDP modeling approach combines multiple sources of information (i.e., SAXS, SANS, and volumetric measurements) to increase the robustness of the recovered structural parameters. This approach has been shown to yield reproducible results for a wide range of lipids. Second, a direct model-free comparison of PSM scattering form factors from simulations to the raw scattering data shows in an unbiased way that the best agreement between the two is precisely at the area per lipid obtained from the joint refinement of the experimental data (Fig. 3). For that constrained area simulation at 61.9 Å2, both the neutron (Fig. 3a) and X-ray (Fig. 3b) scattering form factors and all structural parameters are remarkably similar to the experimentally determined values (Table 1). This result supports the reliability of the scattering analysis and further confirms the deviation of the structure of the unconstrained simulated bilayer from that of the LUV membrane.
Having established the validity of the scattering results, we expected that the constrained-area bilayer at 61.9 Å2 would also show best agreement with NMR data that we collected at the simulation temperature of 55 °C. Surprisingly however, the model-free comparison with the experimental order parameter profile implied best overall agreement with a constrained area simulation at 58 Å2 (Fig. 3c and Fig. 3d). Interestingly, none of the simulations resulted in good agreement for all carbons along the N-acyl chain. Specifically, the order of the first few carbons on the N-linked chain matched those of the PSM lipids in the unconstrained simulation, while the order of the remaining carbons was almost identical to the 59 Å2 constrained-area simulation (Fig. 3c). Still, the PSM order parameter profile from the unconstrained simulation at 55 °C shows a much better agreement with the NMR order parameter profiles that we obtained at 45 °C and 48 °C (Table S3). While this observation cannot explain the discrepancy with scattering results (since even at 45 °C the SDP analysis indicates an AL of 60 Å2), it does call into question the reliability of temperature scaling in MD simulations of sphingomyelin. While the CHARMM36 force field has been shown to reliably capture the temperature dependence of hydrocarbons and a range of lipids,67,82–84 sphingolipids may pose challenges because they have a higher melting temperature and are often close to the main phase transition.
Because the NMR experiments were performed with PSM-d31, it is possible that the discrepancy with scattering data could be explained by a structural difference between the deuterated lipid and its protiated counterpart. Indeed, we observe a 4 °C lowering of the main transition temperature of PSM-d31 determined from NMR (Fig. 2) compared to PSM measured with DSC (Fig. S5), implying that the deuterated bilayer is slightly more disordered at a given fluid phase temperature. However, a chi-square comparison of the simulations with the PSM-d31 SANS data set yields a best match with the constrained-area simulations at 61 Å2 (Fig. 3d), slightly smaller than the 61.9 Å2 found in the joint analysis, but 3 Å larger than the 58 Å2 implied by NMR data. This suggests that isotope effects cannot fully explain the discrepancy between scattering and NMR data. Similarly, as discussed above, the different degrees of curvature in the MLVs measured with NMR and the LUVs measured with SANS/SAXS is also unlikely to play a role at 55 °C where the bilayer is in the fluid state.
One major difference between the bilayer models probed with the different experimental techniques is the amount of water in the samples. While scattering is performed on liposomes in excess water, unlike in the present study NMR measurements on PSM-d31 have often been performed at < 50 wt% water.43, 85 The XRD studies of Maulik et al. reported the effect of water content on the structural parameters of PSM64 and SSM65 using MLV preparations similar to those used in NMR experiments. Comparing the trends at hydration levels up to 60%, they observed a plateau of the fluid-phase lipid area and bilayer thickness at 20–35 wt% water. However, the resulting plateaued AL is lower than the value we obtained from analysis of SANS and SAXS data (Table 2). Similarly, the PSM AL from XRD on MLVs at 50 wt% water is much lower than that obtained either from SANS/SAXS or diffuse LAXS measurements (Table 2). Thus, while bilayer structure does not appear to change for > 40 wt% water, XRD and NMR both executed under the same sample conditions (i.e., MLVs at nominally 50 wt% water) produce structural parameters indicative of a substantially more ordered and tightly packed bilayer relative to results obtained from oriented bilayer stacks at full hydration69 or unilamellar vesicles at 98 wt% water (this work). One possible explanation is the potential difficulty in controlling bilayer hydration during prolonged periods of time at an elevated temperature. NMR measurements take ~1 hour per sample with the sample often measured at multiple temperatures consecutively (for example, in our NMR experiments we measured the sample at 12 different consecutive temperatures) while XRD data collection in the 1990s could take many hours on a home source instrument. Even though the samples were placed in sealed capillaries, we speculate that water condensation on the capillary walls could lead to a lower effective water content of the MLVs and consequently tighter lipid packing.
CONCLUSION
We report the structural parameters of palmitoyl and stearoyl sphingomyelin bilayers in the fluid state obtained from joint analysis of small-angle neutron and X-ray scattering data, including the ever-important area per lipid that is directly related to bilayer thickness and indirectly related to other bilayer properties. The results comprise an important addition to the scarce literature on SM structure as they provide robustly determined parameters of bilayer structure in fully hydrated free-floating liposomes. We further report order parameters of the N-linked chain of PSM obtained with 2H NMR as well as experimentally measured lipid volumes for both PSM and SSM. Through the use of all-atom MD simulations, a clear discrepancy is observed between the lipid packing density measured with scattering and the lipid order parameter obtained from NMR. As these two techniques provide crucial structural information used for testing and validation of simulation force field parameters, we discuss possible sources of the differences between the experimentally determined bilayer structures and emphasize the importance of considering sample conditions when interpreting structural results and designing experiments.
Supplementary Material
ACKNOWLEDGMENTS
We thank: Venky Pingali, Shuo Qian, and Chris Stanley for assistance with SANS measurements; Arthur Woll for assistance with SAXS measurements; and Boris Dzikovski for assistance with ESR data collection. FAH and HLS were supported by National Science Foundation (NSF) Grant No. MCB-1817929 (to FAH). MD was supported by National Institutes of Health (NIH) Grant No. 1F32GM134704–01. The computational work used the Extreme Science and Engineering Discovery Environment (XSEDE), accounts TG-MCB160016, TG-MCB160018 and TG-MCB130010. A portion of this research used resources from the CG3 BioSANS instrument at the High Flux Isotope Reactor, and the EQ-SANS instrument at the Spallation Neutron Source, both Department of Energy (DOE) Office of Science user facilities operated by Oak Ridge National Laboratory (ORNL). Synchrotron SAXS data were collected at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the NSF and the NIH/National Institute of General Medical Sciences under NSF award DMR-0936384. BioSAXS measurements were supported by DOE scientific user facilities at ORNL. Additional measurements were supported by the Biophysical Characterization Suite of the Shull Wollan Center at ORNL. ESR measurements were conducted at the National Biomedical Center for Advanced ESR Technology at Cornell University.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website.
Scattering parameters for PSM and SSM and NMR; order parameters for PSM; details of SM parsing schemes for scattering analysis; volume probability profiles, NMR order para profiles, and number density distributions from PSM simulation; details of ESR lineshape analysis; DSC and densitometry data for PSM and SSM; NMR spectra of PSM; SAXS data from PSM MLVs.
REFERENCES
- 1.Von Heijne G, The membrane protein universe: what’s out there and why bother? J. Intern. Med 2007, 261, 543–557. [DOI] [PubMed] [Google Scholar]
- 2.Lorent JH; Levental KR; Ganesan L; Rivera-Longsworth G; Sezgin E; Doktorova M; Lyman E; Levental I, Plasma membranes are asymmetric in lipid unsaturation, packing and protein shape. Nat. Chem. Biol 2020, 16, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levental I; Levental KR; Heberle FA, Lipid rafts: controversies solved, mysteries remain. Trends Cell Biol. 2020, 30, 341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lingwood D; Simons K, Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [DOI] [PubMed] [Google Scholar]
- 5.Talbott CM; Vorobyov I; Borchman D; Taylor KG; DuPré DB; Yappert MC, Conformational studies of sphingolipids by NMR spectroscopy. II. Sphingomyelin. Biochim. Biophys. Acta 2000, 1467, 326–337. [DOI] [PubMed] [Google Scholar]
- 6.Simons K; van Meer G, Lipid sorting in epithelial cells. Biochemistry 1988, 27, 6197–6202. [DOI] [PubMed] [Google Scholar]
- 7.Sodt AJ; Pastor RW; Lyman E, Hexagonal Substructure and Hydrogen Bonding in Liquid-Ordered Phases Containing Palmitoyl Sphingomyelin. Biophys. J 2015, 109, 948–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiener MC; White SH, Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. III. Complete structure. Biophys. J 1992, 61, 434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kučerka N; Nagle JF; Sachs JN; Feller SE; Pencer J; Jackson A; Katsaras J, Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data. Biophys. J 2008, 95, 2356–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnold O; Bilheux JC; Borreguero JM; Buts A; Campbell SI; Chapon L; Doucet M; Draper N; Leal RF; Gigg MA et al. , Mantid-Data analysis and visualization package for neutron scattering and mu SR experiments. Nucl. Instrum. Meth. A 2014, 764, 156–166. [Google Scholar]
- 11.Pan J; Cheng X; Monticelli L; Heberle FA; Kučerka N; Tieleman DP; Katsaras J, The molecular structure of a phosphatidylserine bilayer determined by scattering and molecular dynamics simulations. Soft Matter 2014, 10, 3716–3725. [DOI] [PubMed] [Google Scholar]
- 12.Kučerka N; van Oosten B; Pan J; Heberle FA; Harroun TA; Katsaras J, Molecular structures of fluid phosphatidylethanolamine bilayers obtained from simulation-to-experiment comparisons and experimental scattering density profiles. J. Phys. Chem. B 2015, 119, 1947–1956. [DOI] [PubMed] [Google Scholar]
- 13.Wiener MC; White SH, Fluid bilayer structure determination by the combined use of x-ray and neutron diffraction. II. “Composition-space” refinement method. Biophys. J 1991, 59, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiener MC; White SH, Fluid bilayer structure determination by the combined use of x-ray and neutron diffraction. I. Fluid bilayer models and the limits of resolution. Biophys. J 1991, 59, 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heberle FA; Pan J; Standaert RF; Drazba P; Kučerka N; Katsaras J, Model-based approaches for the determination of lipid bilayer structure from small-angle neutron and X-ray scattering data. Eur. Biophys. J 2012, 41, 875–890. [DOI] [PubMed] [Google Scholar]
- 16.Kučerka N; Heberle FA; Pan J; Katsaras J, Structural Significance of Lipid Diversity as Studied by Small Angle Neutron and X-ray Scattering. Membranes 2015, 5, 454–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kučerka N; Nieh MP; Katsaras J, Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochim. Biophys. Acta 2011, 1808, 2761–2771. [DOI] [PubMed] [Google Scholar]
- 18.Pan J; Marquardt D; Heberle FA; Kučerka N; Katsaras J, Revisiting the bilayer structures of fluid phase phosphatidylglycerol lipids: Accounting for exchangeable hydrogens. Biochim. Biophys. Acta 2014, 1838, 2966–2969. [DOI] [PubMed] [Google Scholar]
- 19.Smith AK; Freed JH, Dynamics and ordering of lipid spin-labels along the coexistence curve of two membrane phases: an ESR study. Chem. Phys. Lipids 2012, 165, 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schorn K, Marsh D, Extracting order parameters from powder EPR lineshapes for spin-labelled lipids in membranes. Spectrochim. Acta A 1997, 53, 2235–2240. [Google Scholar]
- 21.Williams JA; Batten SE; Harris M; Rockett BD; Shaikh SR; Stillwell W; Wassall SR, Docosahexaenoic and eicosapentaenoic acids segregate differently between raft and nonraft domains. Biophys. J 2012, 103, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis J; Jeffrey K; Bloom M; Valic M; Higgs T, Quadrupolar Echo Deuteron Magnetic Resonance Spectroscopy in Ordered Hydrocarbon Chains In Magnetic Resonance in Colloid and Interface Science; Resing HA, Wade CG, Eds.; ACS Symposium Series 34; American Chemical Society: Washigton, DC, 1976; pp 70–77. [Google Scholar]
- 23.Davis JH, The description of membrane lipid conformation, order and dynamics by 2H-NMR. Biochim. Biophys. Acta 1983, 737, 117–171. [DOI] [PubMed] [Google Scholar]
- 24.Seelig J, Deuterium magnetic resonance: theory and application to lipid membranes. Q. Rev. Biophys 1977, 10, 353–418. [DOI] [PubMed] [Google Scholar]
- 25.Pan J; Heberle FA; Tristram-Nagle S; Szymanski M; Koepfinger M; Katsaras J; Kučerka N, Molecular structures of fluid phase phosphatidylglycerol bilayers as determined by small angle neutron and X-ray scattering. Biochim. Biophys. Acta 2012, 1818, 2135–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu EL; Cheng X; Jo S; Rui H; Song KC; Davila-Contreras EM; Qi Y; Lee J; Monje-Galvan V; Venable RM et al. , CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem 2014, 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J; Cheng X; Swails JM; Yeom MS; Eastman PK; Lemkul JA; Wei S; Buckner J; Jeong JC; Qi Y et al. , CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput 2016, 12, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jo S; Lim JB; Klauda JB; Im W, CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J 2009, 97, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jo S; Kim T; Iyer VG; Im W, CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- 30.Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K, Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Venable RM; Sodt AJ; Rogaski B; Rui H; Hatcher E; MacKerell AD Jr.; Pastor RW; Klauda JB, CHARMM all-atom additive force field for sphingomyelin: elucidation of hydrogen bonding and of positive curvature. Biophys. J 2014, 107, 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klauda JB; Monje V; Kim T; Im W, Improving the CHARMM force field for polyunsaturated fatty acid chains. J. Phys. Chem. B 2012, 116, 9424–9431. [DOI] [PubMed] [Google Scholar]
- 33.Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW, Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giorgino T, Computing 1-D atomic densities in macromolecular simulations: The density profile tool for VMD. Comput. Phys. Commun 2014, 185, 317–322. [Google Scholar]
- 35.Klauda JB; Kučerka N; Brooks BR; Pastor RW; Nagle JF, Simulation-based methods for interpreting x-ray data from lipid bilayers. Biophys. J 2006, 90, 2796–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pabst G; Koschuch R; Pozo-Navas B; Rappolt M; Lohner K; Laggner P, Structural analysis of weakly ordered membrane stacks. J. Appl. Crystallogr 2003, 36, 1378–1388. [Google Scholar]
- 37.Scott HL; Skinkle A; Kelley EG; Waxham MN; Levental I; Heberle FA, On the Mechanism of Bilayer Separation by Extrusion, or Why Your LUVs Are Not Really Unilamellar. Biophys. J 2019, 117, 1381–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guard-Friar D; Chen CH; Engle AS, Deuterium isotope effect on the stability of molecules: phospholipids. J. Phys. Chem 1985, 89, 1810–1813. [Google Scholar]
- 39.Katsaras J; Epand R; Epand R, Absence of chiral domains in mixtures of dipalmitoylphosphatidylcholine molecules of opposite chirality. Phys. Rev. E 1997, 55, 3751. [Google Scholar]
- 40.Heimburg T, Mechanical aspects of membrane thermodynamics. Estimation of the mechanical properties of lipid membranes close to the chain melting transition from calorimetry. Biochim. Biophys. Acta 1998, 1415, 147–162. [DOI] [PubMed] [Google Scholar]
- 41.Fenske DB; Cullis PR, Acyl chain orientational order in large unilamellar vesicles: comparison with multilamellar liposomes: a 2H and 31P nuclear magnetic resonance study. Biophys. J 1993, 64, 1482–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCabe MA; Wassall SR, Rapid deconvolution of NMR powder spectra by weighted fast Fourier transformation. Solid State Nucl. Magn. Reson 1997, 10, 53–61. [DOI] [PubMed] [Google Scholar]
- 43.Mehnert T; Jacob K; Bittman R; Beyer K, Structure and lipid interaction of N-palmitoylsphingomyelin in bilayer membranes as revealed by 2H-NMR spectroscopy. Biophys. J 2006, 90, 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katsaras J, Adsorbed to a rigid substrate, dimyristoylphosphatidylcholine multibilayers attain full hydration in all mesophases. Biophys. J 1998, 75, 2157–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shekhar P; Nanda H; Losche M; Heinrich F, Continuous distribution model for the investigation of complex molecular architectures near interfaces with scattering techniques. J. Appl. Phys 2011, 110, 102216–10221612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belicka M; Gerelli Y; Kučerka N; Fragneto G, The component group structure of DPPC bilayers obtained by specular neutron reflectometry. Soft Matter 2015, 11, 6275–6283. [DOI] [PubMed] [Google Scholar]
- 47.Heftberger P; Kollmitzer B; Heberle FA; Pan J; Rappolt M; Amenitsch H; Kučerka N; Katsaras J; Pabst G, Global small-angle X-ray scattering data analysis for multilamellar vesicles: the evolution of the scattering density profile model. J. Appl. Crystallogr 2014, 47, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eicher B; Heberle FA; Marquardt D; Rechberger GN; Katsaras J; Pabst G, Joint small-angle X-ray and neutron scattering data analysis of asymmetric lipid vesicles. J. Appl. Crystallogr 2017, 50, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doktorova M; Heberle FA; Kingston RL; Khelashvili G; Cuendet MA; Wen Y; Katsaras J; Feigenson GW; Vogt VM; Dick RA, Cholesterol Promotes Protein Binding by Affecting Membrane Electrostatics and Solvation Properties. Biophys. J 2017, 113, 2004–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nielsen JE; Bjornestad VA; Lund R, Resolving the structural interactions between antimicrobial peptides and lipid membranes using small-angle scattering methods: the case of indolicidin. Soft Matter 2018, 14, 8750–8763. [DOI] [PubMed] [Google Scholar]
- 51.Dupuy FG; Pagano I; Andenoro K; Peralta MF; Elhady Y; Heinrich F; Tristram-Nagle S, Selective Interaction of Colistin with Lipid Model Membranes. Biophys. J 2018, 114, 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosa R; Spinozzi F; Itri R, Hydroperoxide and carboxyl groups preferential location in oxidized biomembranes experimentally determined by small angle X-ray scattering: Implications in membrane structure. Biochim. Biophys. Acta 2018, 1860, 2299–2307. [DOI] [PubMed] [Google Scholar]
- 53.Kučerka N; Holland BW; Gray CG; Tomberli B; Katsaras J, Scattering density profile model of POPG bilayers as determined by molecular dynamics simulations and small-angle neutron and X-ray scattering experiments. J. Phys. Chem. B 2012, 116, 232–239. [DOI] [PubMed] [Google Scholar]
- 54.Pan J; Cheng X; Sharp M; Ho CS; Khadka N; Katsaras J, Structural and mechanical properties of cardiolipin lipid bilayers determined using neutron spin echo, small angle neutron and X-ray scattering, and molecular dynamics simulations. Soft Matter 2015, 11, 130–138. [DOI] [PubMed] [Google Scholar]
- 55.Pan J; Cheng X; Heberle FA; Mostofian B; Kučerka N; Drazba P; Katsaras J, Interactions between ether phospholipids and cholesterol as determined by scattering and molecular dynamics simulations. J. Phys. Chem. B 2012, 116, 14829–14838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marquardt D; Heberle FA; Pan J; Cheng X; Pabst G; Harroun TA; Kučerka N; Katsaras J, The structures of polyunsaturated lipid bilayers by joint refinement of neutron and X-ray scattering data. Chem. Phys. Lipids 2020, 229, 104892. [DOI] [PubMed] [Google Scholar]
- 57.Heberle FA; Feigenson GW, Phase separation in lipid membranes. Cold Spring Harb. Perspect. Biol 2011, 3, a004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heberle FA; Petruzielo RS; Goh SL; Konyakhina TM; Ackerman DG; Amazon JJ; Feigenson GW, Liposome-Based Models for Membrane Rafts Methodology and Applications. Liposomes, Lipid Bilayers and Model Membranes: From Basic Research to Application 2014, 143–165. [Google Scholar]
- 59.Nyholm TKM; Jaikishan S; Engberg O; Hautala V; Slotte JP, The Affinity of Sterols for Different Phospholipid Classes and Its Impact on Lateral Segregation. Biophys. J 2019, 116, 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Veatch SL; Keller SL, Miscibility phase diagrams of giant vesicles containing sphingomyelin. Phys. Rev. Lett 2005, 94, 148101. [DOI] [PubMed] [Google Scholar]
- 61.Petruzielo RS; Heberle FA; Drazba P; Katsaras J; Feigenson GW, Phase behavior and domain size in sphingomyelin-containing lipid bilayers. Biochim. Biophys. Acta 2013, 1828, 1302–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pathak P; London E, The Effect of Membrane Lipid Composition on the Formation of Lipid Ultrananodomains. Biophys. J 2015, 109, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yano Y; Hanashima S; Yasuda T; Tsuchikawa H; Matsumori N; Kinoshita M; Al Sazzad MA; Slotte JP; Murata M, Sphingomyelin Stereoisomers Reveal That Homophilic Interactions Cause Nanodomain Formation. Biophys. J 2019, 116, 1575–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maulik PR; Shipley GG, N-palmitoyl sphingomyelin bilayers: structure and interactions with cholesterol and dipalmitoylphosphatidylcholine. Biochemistry 1996, 35, 8025–8034. [DOI] [PubMed] [Google Scholar]
- 65.Maulik PR; Sripada PK; Shipley GG, Structure and thermotropic properties of hydrated N-stearoyl sphingomyelin bilayer membranes. Biochim. Biophys. Acta 1991, 1062, 211–219. [DOI] [PubMed] [Google Scholar]
- 66.Maulik PR; Shipley GG, X-ray diffraction and calorimetric study of N-lignoceryl sphingomyelin membranes. Biophys. J 1995, 69, 1909–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagle JF; Venable RM; Maroclo-Kemmerling E; Tristram-Nagle S; Harper PE; Pastor RW, Revisiting Volumes of Lipid Components in Bilayers. J. Phys. Chem. B 2019, 123, 2697–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Doktorova M; Harries D; Khelashvili G, Determination of bending rigidity and tilt modulus of lipid membranes from real-space fluctuation analysis of molecular dynamics simulations. Phys. Chem. Chem. Phys 2017, 19, 16806–16818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arsov Z; Gonzalez-Ramirez EJ; Goni FM; Tristram-Nagle S; Nagle JF, Phase behavior of palmitoyl and egg sphingomyelin. Chem. Phys. Lipids 2018, 213, 102–110. [DOI] [PubMed] [Google Scholar]
- 70.Sun WJ; Tristram-Nagle S; Suter RM; Nagle JF, Structure of gel phase saturated lecithin bilayers: temperature and chain length dependence. Biophys. J 1996, 71, 885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niemelä P; Hyvönen MT; Vattulainen I, Structure and Dynamics of Sphingomyelin Bilayer: Insight Gained through Systematic Comparison to Phosphatidylcholine. Biophys. J 2004, 87, 2976–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Metcalf R; Pandit SA, Mixing properties of sphingomyelin ceramide bilayers: a simulation study. J. Phys. Chem. B 2012, 116, 4500–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jambeck JP; Lyubartsev AP, Another Piece of the Membrane Puzzle: Extending Slipids Further. J. Chem. Theory Comput 2013, 9, 774–784. [DOI] [PubMed] [Google Scholar]
- 74.Chiu SW; Vasudevan S; Jakobsson E; Mashl RJ; Scott HL, Structure of sphingomyelin bilayers: a simulation study. Biophys. J 2003, 85, 3624–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kumari P; Kaur S; Sharma S; Kashyap HK, Impact of amphiphilic molecules on the structure and stability of homogeneous sphingomyelin bilayer: Insights from atomistic simulations. J. Chem. Phys 2018, 148, 165102. [DOI] [PubMed] [Google Scholar]
- 76.Lyatskaya Y; Liu Y; Tristram-Nagle S; Katsaras J; Nagle JF, Method for obtaining structure and interactions from oriented lipid bilayers. Phys. Rev. E 2001, 63, 011907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pabst G; Kučerka N; Nieh MP; Rheinstadter MC; Katsaras J, Applications of neutron and X-ray scattering to the study of biologically relevant model membranes. Chem. Phys. Lipids 2010, 163, 460–479. [DOI] [PubMed] [Google Scholar]
- 78.Brown MF, Theory of spin‐lattice relaxation in lipid bilayers and biological membranes. 2H and 14N quadrupolar relaxation. J. Chem. Phys 1982, 77, 1576–1599. [Google Scholar]
- 79.Marrink SJ; Corradi V; Souza PCT; Ingolfsson HI; Tieleman DP; Sansom MSP, Computational Modeling of Realistic Cell Membranes. Chem. Rev 2019, 119, 6184–6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chrzeszczyk A; Wishnia A; Springer CS Jr., The intrinsic structural asymmetry of highly curved phospholipid bilayer membranes. Biochim. Biophys. Acta 1977, 470, 161–169. [DOI] [PubMed] [Google Scholar]
- 81.Berden JA; Barker RW; Radda GK, NMR studies on phospholipid bilayers. Some factors affecting lipid distribution. Biochim. Biophys. Acta 1975, 375, 186–208. [DOI] [PubMed] [Google Scholar]
- 82.Zhuang X; Davila-Contreras EM; Beaven AH; Im W; Klauda JB, An extensive simulation study of lipid bilayer properties with different head groups, acyl chain lengths, and chain saturations. Biochim. Biophys. Acta 2016, 1858, 3093–3104. [DOI] [PubMed] [Google Scholar]
- 83.Zhuang X; Makover JR; Im W; Klauda JB, A systematic molecular dynamics simulation study of temperature dependent bilayer structural properties. Biochim. Biophys. Acta 2014, 1838, 2520–2529. [DOI] [PubMed] [Google Scholar]
- 84.Leonard AN; Simmonett AC; Pickard F. C. t.; Huang J; Venable RM; Klauda JB; Brooks BR; Pastor RW, Comparison of Additive and Polarizable Models with Explicit Treatment of Long-Range Lennard-Jones Interactions Using Alkane Simulations. J. Chem. Theory Comput 2018, 14, 948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bunge A; Müller P; Stöckl M; Herrmann A; Huster D, Characterization of the ternary mixture of sphingomyelin, POPC, and cholesterol: support for an inhomogeneous lipid distribution at high temperatures. Biophys. J 2008, 94, 2680–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.