

Abstract
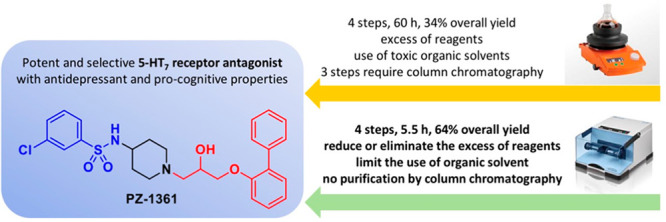
A mechanochemical procedure was developed to obtain PZ-1361, a potent and selective 5-HT7 receptor antagonist, with antidepressant properties in rodents. The elaborated protocol offered several advantages over classical batch synthesis, including improvement of the overall yield (from 34% to 64%), reduction of reaction time (from 60 to 5.5 h), limitation of the use of toxic solvents, and the formation of byproducts. This approach represents a rare example of the synthesis of biologically active compounds exclusively performed using mechanochemical reactions.
Synthetic organic chemistry represents a key component of many drug discovery programs. In the last decades, different techniques have been developed, for example, microwave-assisted organic chemistry and flow chemistry, which might reduce the time required to generate compound libraries for biological screening or might ensure more efficient production of active pharmaceutical ingredients (APIs).1−4 Recently, mechanochemical synthesis has also been recognized as an innovative methodology,5 with a wide range of practical applications in both academic and industrial research. In particular, mechanochemistry has been used to produce various families of compounds.6−10 The primary driving force underlying the rediscovery of mechanochemistry is green chemistry,11−13 in particular, the need of chemical and pharmaceutical industries for the development of more sustainable synthetic protocols characterized by the energy efficiency of chemical transformations and reduction of solvent use. The use of such approaches offers additional advantages of mechanosynthesis over classical organic chemistry, in terms of excellent selectivity and the possibility to perform previously unknown reactions.14−16 Interestingly, an increasing number of mechanochemical procedures for generating pharmaceutically relevant fragments and functionalities have been reported thus far.17−19 This novel mechanochemical application led to coining the term “medicinal mechanochemistry.”20,21
We have recently developed a novel class of a potent and selective 5-HT7 receptor (5-HT7R) antagonist, namely, an arylsulfonamide derivative of (aryloxy)alkyl alicyclic amine, and identified several lead structures that exhibit significant in vivo antidepressant and pro-cognitive properties in rodents (Figure 1).22−26
Figure 1.

Chemical structure of the potent and selective 5-HT7R antagonist PZ-1361 belonging to the class of arylsulfonamides of (aryloxy)alkyl alicyclic amines.
The classical “in batch” synthetic pathway of this class of derivatives consists of four steps involving the alkylation of commercially available phenols in biphasic conditions, nucleophilic substitution of Boc-protected alicyclic amines, removal of the protecting group, and sulfonylation of the resulting primary amine in an alkaline environment (Scheme 1). The critical step of the entire process is the alkylation of phenol, as this reaction should be performed in the presence of a large excess of halogeno-alkanes (from 3 to 6 equiv) to avoid unwanted dimerization or opening of the epoxide ring. Additionally, apart from the deprotection of amine function, column chromatography purification is required in all of the remaining steps together with the use of a large amount of organic solvents (in particular, the highly toxic dichloromethane).27,28 To overcome these issues and simultaneously extend the concept of medicinal mechanochemistry, we adapted the synthetic pathway by using a mechanochemical approach for the synthesis of the potent and selective 5-HT7R antagonist PZ-1361.29 To demonstrate the versatility of this method, we subsequently increased the diversity of building blocks by conducting experiments using 2-substituted phenols, different central amine cores (e.g., piperazine, 3-amino-tropane, 3-aminopyrrolidine), and differently substituted arylsulfonyl chlorides. This allowed proposing mechanochemistry as a promising synthetic strategy in medicinal chemistry, which would enable the preparation of lead compounds for preclinical development in a more sustainable and greener manner.30
Scheme 1. In-Solution Synthesis of the Compound PZ-1361.

The optimization of the synthetic pathway started with the alkylation of commercially available 2-phenylphenol with racemic epichlorohydrin (1 equiv). The reaction was initially performed in a 10 mL PTFE jar with a 1 cm diameter stainless steel ball by using a vibratory ball mill (vbm) operated at 30 Hz. A thorough study of the different parameters was performed and is summarized in Table 1 (for more information, see Tables S3–5).
Table 1. Optimization of Milling Conditions for the Alkylation of 2-Phenylphenola.

| conversion
(%)b |
||||
|---|---|---|---|---|
| entry | base (equiv) | time (min) | 1a | 2a |
| 1 | Na2CO3 (3) | 80 | 1 | 0 |
| 2 | K2CO3 (3) | 80 | 35 | 0 |
| 3 | Cs2CO3 (3) | 80 | 29 | 10 |
| 4 | KOH (3) | 80 | 36 | 10 |
| 5 | K2CO3 (3) | 140 | 43 | 2 |
| 6 | K2CO3 (3) | 180 | 48 | 5 |
| 7 | K2CO3 (3) | 220 | 52 | 10 |
| 8c | K2CO3 (3) | 140 | 61 | 2.5 |
| 9c,d | K2CO3 (3) | 140 | 90 | 2 |
Reaction conditions: 2-phenylphenol (1 equiv), epichlorohydrin (1 equiv), vbm 30 Hz, 10 mL PTFE jar, ϕball = 1 cm, total mass of reagents = 100 mg.
Conversions of 2-phenylphenol into 1a and 2a were determined by UPLC/MS analysis.
The ball used was 1.5 cm in diameter.
1.2 equiv of epichlorohydrin was used.
Next, to investigate the kinetics of the alkylation, the experiment was performed using potassium carbonate and different time periods ranging from 40 to 220 min. A reaction time of approximately 2 h was sufficient to obtain around 40% conversion (Table 1, entry 5), while a prolonged milling time slightly accelerated the formation of desired product 1a without reaching full conversion (Table 1, entries 6 and 7). Increasing the milling time promoted the formation of the unwanted side products, 2a, resulting from nucleophilic addiction of 2-phenylphenol to the oxirane derivative 1a. Of note, a similar epoxide ring opening with carboxylic acids was previously reported.31 On the basis of the assumption that more energetic collisions might be achieved by increasing the diameter of the milling ball, the use of a 1.5 cm diameter stainless steel ball significantly improved the formation of the monoalkylated product 1a, while restricting the formation of 2a to 2.5% (Table 1, entry 8). Additionally, the use of a slight excess of epichlorohydrin (Table 1, entry 9) enabled to achieve a 90% conversion of 2-phenylphenol into 1a (88% isolated yield) without increasing the formation of 2a. After identifying the optimal reaction conditions for alkylation on a small scale, the influence of different substituents at the phenol was subsequently determined using a 35 mL PTFE jar. The choice of substituent (e.g., 2-isopropyl and 2-iodo) was based on the biological data for this group of derivatives, wherein it was confirmed that only compounds bearing a sterically hindered substituent in 2-position preferentially bind to 5-HT7R.23,24 All of the tested substituted phenols showed higher conversion rates under milling than the unsubstituted one, with the following reactivity rank order: 2-Ph ≈ 2-I > 2-iPr > H (Table 2). These results are in line with yields and purities obtained from the classical organic synthesis pathway using biphasic conditions (for more information, see Supporting Information).
Table 2. Effect of Liquid-Assisted Grinding on the Alkylation of Different 2-Substituted Phenolsa.

| conversion
(%)c |
|||||
|---|---|---|---|---|---|
| entry | R | additiveb | 1a–d | 2a–d | % yield (1a–d)d |
| 1 | Ph | 84 | 2 | nde | |
| 2 | Ph | EtOAc | 87 | 3 | nde |
| 3 | Ph | iPrOH | 90 | 3 | 85 |
| 4 | H | 52 | 3 | nde | |
| 5 | H | EtOAc | 55 | 3 | nde |
| 6 | H | iPrOH | 63 | 3 | 70 |
| 7 | iPr | 64 | 3 | nde | |
| 8 | iPr | EtOAc | 66 | 3 | nde |
| 9 | iPr | iPrOH | 69 | 3 | 65 |
| 10 | I | 77 | 1 | nde | |
| 11 | I | EtOAc | 82 | 1 | nde |
| 12 | I | iPrOH | 88 | 1 | 84 |
Reaction conditions: phenol (1 equiv), epichlorohydrin (1.2 equiv), vbm 30 Hz, ϕball = 1.5 cm, time = 120–140 min, 35 mL PTFE jar; total mass of reagents, 330 mg.
50 μL, η = 0.15 μL mg–1.
Conversions of 2-substituted phenols into 1a–d and 2a–d were determined by UPLC/MS analysis.
Isolated yield of pure compounds 1a–d.
nd = not determined.
Because the conversion rate was slightly lower than that obtained in the 10 mL milling jar (Table 2, entry 1), ethyl acetate and isopropanol as nontoxic liquid assistants were evaluated to enhance the overall mixing and reaction kinetics.32−36 The results showed that, regardless of the nature of the phenol tested, liquid-assisted grinding (LAG) enabled to increase the conversions into 1a–d up to 11%, while limiting the formation of side products 2a–d, with isopropanol being the best assistant. A simple basic extraction allowed to remove unreacted substrates and side products and afforded intermediates 1a–d in high purity and yields (65–85%). As a comparison, the same reactions performed in the solution for 48 h provided a higher amount of side products (20–35%), which required column chromatography separation.
The next step involved the alkylation of Boc-4-amino-piperidine with the [1,1′-biphenyl] oxirane derivative 1a in a 35 mL PTFE jar using vbm at 30 Hz (Scheme 2). This reaction was originally performed in refluxing ethanol for 4 h, with an excess of reagents and required purification on silica gel (see Supporting Information). To our delight, the mechanochemical approach resulted in a complete conversion of the starting materials, reduced the reaction time to 70 min, limited the amount of solvent (from 15 mL to 33 μL, η = 0.1 μL mg–1), avoided the excess of an alkylating agent, and eliminated the purification step on silica gel. The desired compound 3a was obtained in 90% yield (Scheme 2). Encouraged by these findings, we extended this protocol to the alkylation of amine function by testing different Boc-protected alicyclic amines. The kinetics of the reaction strongly depended on the type of amine used as the alkylation of 3-Boc-amino-8-azabicyclooctane (tropane), and 4-Boc-piperazine required a higher milling time (140 and 120 min, respectively) to achieve high conversions. The corresponding intermediates 3b and 3c were isolated in 90% and 89% yields, respectively. When 3-Boc-amino-pyrrolidine was used as a nucleophile, a prolonged milling time afforded the desired compound 3d in 83% yield. However, 15% of the dehydrated byproduct 3e, which was not generated in solution, was observed. In the case of other alicyclic amines, a lower percentage of the respective dehydrated side products (5–10%) were observed when the milling time exceeded 160 min.
Scheme 2. Mechanochemical Alkylation of Different Boc-Protected Alicyclic Amines.

Reaction conditions: vbm 30 Hz, ϕball = 1.5 cm, total mass of reagents = 330 mg, 35 mL PTFE jar, EtOH (33 μL, η = 0.1 μL mg–1).
Various accessible methods to form a sulfonamide bond in conventional solution or on solid support have been extensively reported in literature.37−42 However, most of them involve the use of a strong organic base, excess of a sulfonylating agent, and low environmentally friendly solvents (in particular dichloromethane). To develop a more sustainable and efficient methodology, sulfonylation of intermediate 4 with 3-chlorobenzenesulfonyl chloride was optimized under milling conditions. Prior to the reaction, primary amine 4 was obtained by the treatment of Boc-derivative 3a in the presence of gaseous HCl (for more information, see Supporting Information).
Compound PZ-1361 (5a) was finally isolated in a high yield after milling of the substrates in the presence of nontoxic K2CO3 for 1 min (Table 3, entry 1). To illustrate the versatility of this mechanochemical approach, compounds 5b–e bearing electron-withdrawing or -donating substituents in different positions of the benzenesulfonamide moiety were prepared. It seems that the position of the substituent has more influence on sulfonylation rather than its electronic effect. All derivatives were obtained in good yields and purities comparable to those obtained in solution. In addition, the presence of a 2-fluoro or 3-methoxy substituent (Table 3, entries 3 and 4) led to higher conversions in a relatively shorter time than 4-nitro and unsubstituted analogues (Table 3, entries 2 and 5).
Table 3. Optimization of Milling Conditions for Sulfonylation of Primary Aminea.

| entry | R | product | time [min] | % yieldb |
|---|---|---|---|---|
| 1 | 3-Cl | 5a | 1 | 86 |
| 2 | H | 5b | 5 | 87 |
| 3 | 2-F | 5c | 2 | 85 |
| 4 | 3-OMe | 5d | 3 | 86 |
| 5 | 4-NO2 | 5e | 5 | 80 |
Reaction conditions: vbm 30 Hz, 35 mL PTFE jar, ϕball = 1.5 cm, total mass of reagents = 330 mg.
After the addition of EtOAc and washing with KHSO4 (aq).
In summary, mechanochemistry enabled the preparation of the biologically active compound PZ-1361 (5a) in 4 steps with an overall yield of 64%. This approach required a total milling time of 331 min; only a slight excess of the alkylating agent was required, and no chromatographic purification was necessary to isolate 5a in high purity (Figure 2). The versatility of the protocol was confirmed by introducing diversification at the aryloxy fragment (1a–d), central amine core (3a–d), and the benzenesulfonamide moiety (5a–e). Compared to the classical thermal methods in solution, the mechanochemical approach offered several advantages including (i) improvement of the overall yield (from 34% to 64%), (ii) reduction of reaction time (from 60 to 5.5 h), and (iii) limitation of the use of toxic solvents and the formation of byproducts. Moreover, reaction conditions and workup procedures were simplified because intermediates and final compounds were obtained by simple extraction without the need for column chromatography purification. To assess the sustainability of the newly developed approach, the green chemistry metrics E factor and Ecoscale score were calculated,43,44 which confirmed the advantage of the mechanochemical strategy over the classical organic chemistry pathway (for more information, see Supporting Information). The E factor calculated for the 4-step synthesis of PZ-1361 is reduced by 2.7 (from 1932 to 715) when switching from classical solution synthesis to mechanochemistry.
Figure 2.

In-solution vs solid-state synthesis of PZ-1361.
In addition, Ecoscale scores were found to be excellent for each synthetic step (>70) in the solid state, while some of them were inadequate in solution (<50). To the best of our knowledge, this approach represents the first example of organic synthesis of a CNS-acting agent exclusively performed using the mechanochemical methodology. Further studies aimed to confirm the suitability of the mechanochemical approach for an efficient and sustainable synthesis of a focused library of biologically active compounds are under development.
Experimental Section
General Remarks
The milling treatments were carried out in a vibratory ball-mill Retsch MM400 operated at 30 Hz. The milling load is defined as the sum of the mass of the reactants per free volume in the jar and was equal to 10 or 25 mg/mL. All of the reactions using vibratory ball mill were performed under air. 1H and 13C NMR spectra were recorded on a JEOL JNM-ECZR500 RS1 (ECZR version) at 500 and 126 MHz, respectively, and are reported in ppm using deuterated solvent for calibration (CDCl3 or CD3OD-d4). The J values are reported in hertz (Hz), and the splitting patterns are designated as follows: br s. (broad singlet), br d. (broad doublet), s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), dt (doublet of triplets), td (triplet of doublets), tt (triplet of triplets), m (multiplet). Mass spectra were recorded on a UPLC-MS/MS system consisting of a Waters ACQUITY UPLC (Waters Corporation, Milford, MA, USA) coupled to a Waters TQD mass spectrometer (electrospray ionization mode ESI-tandem quadrupole). Chromatographic separations were carried out using the Acquity UPLC BEH (bridged ethyl hybrid) C18 column; 2.1 mm × 100 mm, and 1.7 μm particle size, equipped with Acquity UPLC BEH C18 Van Guard precolumn; 2.1 mm × 5 mm, and 1.7 μm particle size. The column was maintained at 40 °C and eluted under gradient conditions from 95% to 0% of eluent A over 10 min, at a flow rate of 0.3 mL min–1. Eluent A: water/formic acid (0.1%, v/v), Eluent B: acetonitrile/formic acid (0.1%, v/v). HRMS analyses were performed on an UPLC Acquity H-Class from Waters hyphenated to a Synapt G2-S mass spectrometer with a dual ESI source from Waters.
Alkylation of 2-Phenylphenol in Ball Mill (General Procedure A)
2-Phenylphenol (24.5 mg, 0.144 mmol, 1 equiv) and previously grinded K2CO3 (59.58 mg, 0.431 mmol, 3.0 equiv) were introduced in a 10 mL PTFE jar (milling load 10 mg/mL) with one stainless steel ball (ϕball = 1.5 cm) followed by the addition of epichlorohydrin (13.5 μL, 0.172 mmol, 1.2 equiv). The reaction was carried out for 120–140 min at rt. The mixture was then solubilized in CH2Cl2 (15 mL), and the organic phase was washed with 2 N NaOH aqueous solution (3 × 5 mL) and saturated NaCl solution (1 × 5 mL), dried over Na2SO4, and finally filtered and concentrated under reduced pressure, yielding intermediate 1a as a white powder (29.0 mg, yield 88%).
Alkylation of Phenol in Ball Mill (General Procedure B)
Different substituted phenol (1 equiv) and previously grinded K2CO3 (3 equiv) were introduced in a 35 mL PTFE jar (milling load 10 mg/mL) with one stainless steel ball (ϕball = 1.5 cm). Then epichlorohydrin (1.2 equiv) was added. The reaction was carried out for 120–140 min at rt. The mixture was solubilized in CH2Cl2 (15 mL), and the organic phase was washed with 2 N NaOH aqueous solution (3 × 5 mL) and saturated NaCl solution (1 × 5 mL), dried over Na2SO4, and finally filtered and concentrated under reduced pressure.
2-(([1,1′-Biphenyl]-2-yloxy)methyl)oxirane (1a) [CAS 7144-65-2]
General Procedure B was followed with 2-phenylphenol (80.7 mg, 0.474 mmol, 1 equiv), previously grinded K2CO3 (196.6 mg, 1.42 mmol, 3 equiv), epichlorohydrin (44.6 μL, 0.569 mmol, 1.2 equiv), and 2-propanol (50 μL, η = 0.15 μL mg–1) to afford intermediate 1a as a white powder (81 mg, yield 85%).
C15H14O2, MW: 226.27. Monoisotopic mass: 226.10. UPLC/MS: tR = 7.20 min, purity = 99%, [M + MeCN + H]+ 268.1. 1H NMR (500 MHz, CDCl3): δ 7.58 (dt, J = 8.0, 1.1 Hz, 1H), 7.55–7.60 (m, 1H), 7.41–7.46 (m, 2H), 7.34–7.38 (m, 2H), 7.32 (td, J = 8.0, 1.7 Hz, 1H), 7.08 (td, J = 7.4, 1.1 Hz, 1H), 6.98–7.03 (m, 1H), 4.22 (dd, J = 11.5, 2.9 Hz, 1H), 3.99 (dd, J = 10.9, 5.2 Hz, 1H), 3.25–3.29 (m, 1H), 2.81 (t, J = 4.6 Hz, 1H), 2.68 (dd, J = 5.2, 2.9 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 155.5, 138.4, 131.4, 131.2, 129.7, 128.7, 128.1, 127.1, 121.8, 113.3, 69.1, 50.4, 44.7. HRMS (ESI-TOF): m/z [M + MeCN + H]+ calcd for C17H18NO2, 268.1338; found, 268.1346. Data in agreement with lit.29
2-(([1,1′-Biphenyl]-2-yloxy)methyl)oxirane (1a) [CAS 7144-65-2] (Synthesis on a mmol Scale)
2-Phenylphenol (195.7 mg, 1.15 mmol, 1 equiv) and previously grinded K2CO3 (476.7 mg, 3.45 mmol, 3.0 equiv) were introduced in a 35 mL PTFE jar (milling load 25 mg/mL) with one stainless steel ball (ϕball = 1.5 cm) followed by the addition of epichlorohydrin (108.2 μL, 1.38 mmol, 1.2 equiv). The reaction was carried out for 140 min at rt. The mixture was then solubilized in CH2Cl2 (25 mL), and the organic phase was washed with 2 N NaOH aqueous solution (3 × 10 mL) and saturated NaCl solution (1 × 10 mL), dried over Na2SO4, and finally filtered and concentrated under reduced pressure, yielding intermediate 1a as a white powder (215.8 mg, yield 83%).
2-(Phenoxymethyl)oxirane (1b) [CAS 122-60-1]
General Procedure B was followed with phenol (50.1 mg, 0.532 mmol, 1 equiv), previously grinded K2CO3 (220.8 mg, 1.597 mmol, 3 equiv), epichlorohydrin (50.1 μL, 0.639 mmol, 1.2 equiv), and 2-propanol (50 μL, η = 0.15 μL mg–1) to afford intermediate 1b as a white powder (55 mg, yield 70%).
C9H10O2, MW: 150.17. Monoisotopic mass: 150.07. UPLC/MS: tR = 5.44 min, purity = 97%, [M + MeCN + H]+ 192.1. 1H NMR (500 MHz, CDCl3): δ 7.30 (tt, J = 8.6, 1.7 Hz, 2H), 6.96–7.01 (m, 1H), 6.91–6.95 (m, 2H), 4.20 (dd, J = 11.2, 3.2 Hz, 1H), 3.90 (dd, J = 10.9, 5.7 Hz, 1H), 3.30–3.35 (m, 1H), 2.84–2.88 (m, 1H), 2.72 (dd, J = 4.9, 2.6 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 158.6, 129.7, 121.3, 114.8, 68.8, 50.3, 44.7. HRMS (ESI-TOF): m/z [M + MeCN + H]+ calcd for C11H14NO2, 192.1025; found, 192.1032. Data in agreement with lit.45
2-((2-Isopropylphenoxy)methyl)oxirane (1c) [CAS 5904-89-2]
General Procedure B was followed with 2-isopropylphenol (65.2 mg, 0.479 mmol, 1 equiv), previously grinded K2CO3 (198.4 mg, 1.436 mmol, 3 equiv), epichlorohydrin (45.1 μL, 0.575 mmol, 1.2 equiv), and 2-propanol (50 μL, η = 0.15 μL mg–1) to afford intermediate 1c as a colorless oil (59.8 mg, yield 65%).
C12H16O2, MW: 192.25. Monoisotopic mass: 192.12. UPLC/MS: tR = 7.33 min, purity = 100%, [M + MeCN + H]+ 234.2. 1H NMR (500 MHz, CDCl3): δ 7.25–7.29 (m, 1H), 7.18 (td, J = 7.4, 1.7 Hz, 1H), 6.99 (t, J = 7.4 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 4.26 (dd, J = 10.9, 2.9 Hz, 1H), 4.00 (dd, J = 10.9, 5.2 Hz, 1H), 3.37–3.45 (m, 2H), 2.92 (dd, J = 5.2, 4.0 Hz, 1H), 2.80 (dd, J = 4.9, 2.6 Hz, 1H), 1.29 (dd, J = 6.9, 1.1 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3): δ 155.8, 137.4, 126.7, 126.4, 121.4, 111.7, 68.9, 50.5, 44.7, 27.0, 22.9. HRMS (ESI-TOF): m/z [M + MeCN + H]+ calcd for C14H20NO2, 234.1494; found, 234.1502. Data in agreement with lit.29
2-((2-Iodophenoxy)methyl)oxirane (1d) [CAS 75746-33-7]
General Procedure B was followed with 2-iodophenol (93.9 mg, 0.427 mmol, 1 equiv), previously grinded K2CO3 (176.9 mg, 1.28 mmol, 3 equiv), epichlorohydrin (40.1 μL, 0.512 mmol, 1.2 equiv), and 2-propanol (50 μL, η = 0.15 μL mg–1) to afford intermediate 1d as a white powder (100 mg, yield 84%).
C9H9IO2, MW: 276.07. Monoisotopic mass: 275.96. UPLC/MS: tR = 6.49 min, purity = 99%, [M + MeCN + H]+ 318.0. 1H NMR (500 MHz, CDCl3): δ 7.71 (dd, J = 8.0, 1.7 Hz, 1H), 7.22 (td, J = 7.7, 1.7 Hz, 1H), 6.76 (dd, J = 8.3, 1.4 Hz, 1H), 6.67 (td, J = 8.0, 1.1 Hz, 1H), 4.28 (dd, J = 11.5, 2.9 Hz, 1H), 3.93 (dd, J = 10.9, 5.2 Hz, 1H), 3.29–3.33 (m, 1H), 2.80–2.85 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3): δ 157.1, 139.7, 129.8, 123.3, 112.8, 86.8, 69.5, 50.0, 44.8. HRMS (ESI-TOF): m/z [M + MeCN + H]+ calcd for C11H13INO2, 317.9991; found, 317.9998. Data in agreement with lit.46
Alkylation of Boc-N-Protected Alicyclic Amines (General Procedure C)
Intermediate 1a (1 equiv) and Boc-protected alicyclic amine (1 equiv) were introduced in a 35 mL PTFE jar (milling load 10 mg/mL) with one stainless steel ball (ϕball = 1.5 cm), followed by the addition of EtOH (33 μL, η = 0.1 μL mg–1) as a liquid assistant. The reaction was carried out for 70–160 min at rt. Then, the product was solubilized in ethyl acetate (20 mL), and the organic phase was washed with KHSO4 aqueous solution at pH = 3.5 (3 × 7 mL) and saturated NaCl solution (1 × 7 mL), dried over Na2SO4, and finally filtered and concentrated under reduced pressure.
tert-Butyl ((1-(3-([1,1′-Biphenyl]-2-yloxy)-2-hydroxy propyl)piperidin-4-yl)carbamate (3a) [CAS 2095849-45-7]
General Procedure C was followed with compound 1a (175.1 mg, 0.774 mmol, 1 equiv), 4-Boc-amino-piperidine (154.9 mg, 0.774 mmol, 1 equiv), and EtOH (33 μL, η = 0.1 μL mg–1) to afford intermediate 3a as a white powder (297 mg, yield 90%). C25H34N2O4, MW: 426.55. Monoisotopic mass: 426.25. UPLC/MS: tR = 5.46 min, purity = 97%, [M + H]+ 427.3. 1H NMR (500 MHz, CDCl3): δ 7.47 (d, J = 7.4 Hz, 2H), 7.27–7.41 (m, 5H), 7.05 (t, J = 7.4 Hz, 1H), 6.94–6.98 (m, 1H), 4.51 (br s, 1H), 4.13 (br s, 1H), 3.97–4.10 (m, 1H), 3.88 (t, J = 7.4 Hz, 1H), 3.42–3.58 (m, 2H), 2.86–3.10 (m, 2H), 2.50–2.65 (m, 2H), 2.44 (t, J = 11.5 Hz, 1H), 2.05–2.25 (m, 1H), 1.87–2.05 (m, 2H), 1.62–1.76 (m, 2H), 1.43 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ 155.2, 138.6, 131.3, 130.9, 129.8, 128.9, 128.0, 127.0, 121.8, 113.2, 79.8, 70.4, 65.4, 61.6, 51.7, 31.1, 29.8, 28.5. HRMS (ESI-TOF): m/z [M + H]+ calcd for C25H35N2O4, 427.2591; found, 427.2601. Data in agreement with lit.29
tert-Butyl ((1-(3-([1,1′-Biphenyl]-2-yloxy)-2-hydroxy propyl)piperidin-4-yl)carbamate (3a) [CAS 2095849-45-7] (Synthesis on a mmol Scale)
Intermediate 1a (424.5 mg, 1.88 mmol, 1 equiv) and Boc-N-4-aminopiperidine (375.5 mg, 1.88 mmol, 1 equiv) were introduced in a 35 mL PTFE jar (milling load 25 mg/mL) with one stainless steel ball (ϕball = 1.5 cm) followed by the addition of EtOH (80 μL, η = 0.1 μL mg–1) as a liquid assistant. The reaction was carried out for 70 min at rt. Then, the product was solubilized in ethyl acetate (30 mL), and the organic phase was washed with KHSO4 aqueous solution at pH = 3.5 (3 × 10 mL) and saturated NaCl solution (1 × 10 mL), dried over Na2SO4, and finally filtered and concentrated under reduced pressure, yielding intermediate 3a as a white powder (704.3 mg, yield 88%).
tert-Butyl ((1R,5S)-8-(3-([1,1′-Biphenyl]-2-yloxy)-2-hydroxypropyl)-8-azabicyclo[3.2.1]octan-3-yl)carbamate (3b)
General Procedure C was followed with compound 1a (165.0 mg, 0.729 mmol, 1 equiv), 3-Boc-amino-tropane (165.0 mg, 0.729 mmol, 1 equiv), and EtOH (33 μL, η = 0.1 μL mg–1) to afford intermediate 3b as a white powder (302.6 mg, yield 90%).
C27H36N2O4, MW: 452.59. Monoisotopic mass: 452.27. UPLC/MS: tR = 5.73 min, purity = 95%, [M + H]+ 453.3. 1H NMR (500 MHz, CDCl3): δ 7.43–7.49 (m, 2H), 7.33–7.38 (m, 2H), 7.27–7.32 (m, 3H), 7.04 (t, J = 6.9 Hz, 1H), 6.94–6.98 (m, 1H), 4.53 (br d, J = 7.4 Hz, 1H), 4.17 (br s, 1H), 4.08–4.14 (m, 1H), 3.82–3.89 (m, 1H), 3.75–3.81 (m, 1H), 3.41–3.46 (m, 1H), 3.31–3.37 (m, 1H), 2.65 (d, J = 12.6 Hz, 1H), 2.40–2.61 (m, 1H), 1.73–1.95 (m, 5H), 1.60–1.71 (m, 2H), 1.39–1.46 (m, 9H), 0.75–0.91 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3): δ 155.4, 155.1, 138.6, 131.1, 130.9, 129.7, 128.9, 127.9, 127.1, 121.7, 112.8, 79.7, 69.7, 66.1, 62.7, 60.9, 56.5, 41.3, 36.9, 29.8, 29.7, 28.5, 25.9, 25.2. HRMS (ESI-TOF): m/z [M + H]+ calcd for C27H37N2O4, 453.2753; found, 453.2760.
tert-Butyl (4-(3-([1,1′-Biphenyl]-2-yloxy)-2-hydroxy propyl)piperazine-1-carboxylate (3c)
General Procedure C was followed with compound 1a (181.0 mg, 0.800 mmol, 1 equiv), Boc-piperazine (149.0 mg, 0.800 mmol, 1 equiv), and EtOH (33 μL, η = 0.1 μL mg–1) to afford intermediate 3c as a white powder (292.2 mg, yield 89%).
C24H32N2O4, MW: 412.52. Monoisotopic mass: 412.24. UPLC/MS: tR = 5.4 min, purity = 97%, [M + H]+ 413.2.
1H NMR (500 MHz, CDCl3): δ 7.48–7.53 (m, 2H), 7.35–7.41 (m, 2H), 7.27–7.34 (m, 3H), 7.04 (td, J = 7.4, 1.1 Hz, 1H), 6.98 (d, J = 8.0 Hz, 1H), 3.92–4.02 (m, 3H), 3.35–3.43 (m, 4H), 2.93 (br s, 1H), 2.41–2.52 (m, 4H), 2.25–2.32 (m, 2H), 1.46 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ 155.6, 154.8, 138.5, 131.4, 130.9, 129.7, 128.8, 128.0, 127.0, 121.6, 113.2, 79.9, 71.0, 65.9, 60.9, 53.3, 28.5. HRMS (ESI-TOF): m/z [M + H]+ calcd for C24H33N2O4, 413.2440; found, 413.2446.
tert-Butyl ((1-(3-([1,1′-Biphenyl]-2-yloxy)-2-hydroxy propyl)pyrrolidin-3-yl)carbamate (3d)
General Procedure C was followed with compound 1a (181.0 mg, 0.800 mmol, 1 equiv), 3-Boc-amino-pyrrolidine (149.0 mg, 0.800 mmol, 1 equiv), and EtOH (33 μL, η = 0.1 μL mg–1) to afford intermediate 3d as a white powder (268.7 mg, yield 83%). In this case, crystallization in EtOH was required to obtain the pure compound.
C24H32N2O4, MW: 412.52. Monoisotopic mass: 412.24. UPLC/MS: tR = 5.52 min, purity = 95%, [M + H]+ 413.2.
1H NMR (500 MHz, CDCl3): δ 7.50 (d, J = 7.6 Hz, 2H), 7.35–7.40 (m, 2H), 7.32 (dd, J = 7.4, 1.7 Hz, 1H), 7.26–7.30 (m, 2H), 7.03 (td, J = 7.4, 1.1 Hz, 1H), 6.94–7.00 (m, 1H), 5.05–5.13 (m, 1H), 4.09–4.17 (m, 1H), 3.13–3.42 (m, 1H), 2.80–2.89 (m, 1H), 2.67–2.75 (m, 1H), 2.55–2.64 (m, 2H), 2.42–2.52 (m, 1H), 2.34–2.40 (m, 1H), 2.28–2.34 (m, 1H), 2.11–2.23 (m, 2H), 1.48–1.60 (m, 1H), 1.43 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ 155.6, 155.5, 138.5, 131.3, 130.9, 129.7, 128.8, 128.0, 127.0, 121.6, 113.3, 79.3, 71.2, 71.2, 67.7, 61.7, 61.1, 58.8, 52.8, 49.8, 32.6, 28.5. HRMS (ESI-TOF): m/z [M + H]+ calcd for C24H33N2O4, 413.2440; found, 413.2446.
Deprotection of Boc Function in Solid State (General Procedure D)
Intermediate 3a (0.5 g, 1.16 mmol) was submitted to HClgas for 2 h at rt to afford the primary amine 4 as a white hydrochloride salt (0.41 g, yield 98%).
1-([1,1′-Biphenyl]-2-yloxy)-3-(4-aminopiperidin-1-yl) propan-2-ol (4)
White powder (0.41 g, yield 98%). C20H26N2O2·HCl, MW: 362.90. Monoisotopic mass: 326.43. UPLC/MS: tR = 3.62 min, purity = 100%, [M + H]+ 327.2. 1H NMR (500 MHz, CD3OD): δ 7.46–7.51 (m, 2H), 7.38–7.44 (m, 2H), 7.29–7.33 (m, 2H), 7.25–7.28 (m, 1H), 7.07 (d, J = 8.2 Hz, 1H), 7.01–7.05 (m, 1H), 4.30–4.36 (m, 1H), 4.04 (dd, J = 9.9, 4.4 Hz, 1H), 3.97 (dd, J = 9.7, 5.6 Hz, 1H), 3.58 (q, J = 7.0 Hz, 2H), 3.44–3.53 (m, 1H), 3.09–3.19 (m, 3H), 2.18–2.28 (m, 2H), 1.96–2.12 (m, 2H), 1.12–1.18 (m, 4H). 13C{1H} NMR (126 MHz, CD3OD): δ 155.4, 138.6, 131.3, 130.5, 129.5, 128.7, 127.9, 126.9, 121.7, 113.4, 70.6, 64.2, 57.0, 50.4, 45.3, 17.2. HRMS (ESI-TOF): m/z [M + H]+ calcd for C20H27N2O2, 327.2073; found, 327.2079.
Sulfonylation of Primary Amine (General Procedure E)
Intermediate 4 (1 equiv), selected benzenesulfonyl chloride (1 equiv), and previously grinded K2CO3 (1 equiv) were introduced in a 35 mL PTFE jar (milling load 10 mg/mL) with one stainless steel ball (ϕball = 1.5 cm). The reaction was carried out for 1–5 min at rt. Then, the crude mixture was solubilized in ethyl acetate (20 mL), and the organic phase was washed with KHSO4 aqueous solution at pH = 3.5 (3 × 7 mL), saturated NaCl solution (1 × 7 mL), dried over Na2SO4, and finally filtered and concentrated under a vacuum.
3-Chloro-N-(1-(3-([1,1′-biphenyl]-2-yloxy)-2-hydroxy propyl)piperidin-4-yl)-benzenesulfonamide PZ-1361 (5a) [CAS 2095849-69-5]
General Procedure E was followed with primary amine 4 (168.2 mg, 0.463 mmol, 1 equiv), 3-chloro-benzenesulfonyl chloride (65.24 μL, 0.463 mmol, 1 equiv), and previously grinded K2CO3 (64.0 mg, 0.463, 1 equiv) to afford final compound 5a as a white powder (200.1 mg, yield 86%).
C26H29ClN2O4S, MW: 501.04. Monoisotopic mass: 500.15. UPLC/MS: tR = 5.89 min, purity = 98%, [M + H]+ 501.2.
1H NMR (500 MHz, CDCl3): δ 7.89 (t, J = 1.9 Hz, 1H), 7.77 (dt, J = 7.7, 0.9 Hz, 1H), 7.53 (dq, J = 8.0, 0.9 Hz, 1H), 7.50 (dt, J = 8.3, 1.4 Hz, 2H), 7.43 (t, J = 8.0 Hz, 1H), 7.36–7.39 (m, 2H), 7.26–7.33 (m, 3H), 7.03 (td, J = 7.5, 1.0 Hz, 1H), 6.96 (dd, J = 8.2, 0.7 Hz, 1H), 5.27 (s, 1H), 3.84–4.00 (m, 3H), 3.16 (spt, J = 4.3 Hz, 1H), 2.72–2.78 (m, 1H), 2.56–2.62 (m, 1H), 2.34–2.40 (m, 1H), 2.30 (dd, J = 15.0, 3.8 Hz, 1H), 2.16–2.23 (m, 1H), 1.91–1.99 (m, 1H), 1.68–1.76 (m, 2H), 1.39–1.52 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3): δ 155.6, 143.2, 138.5, 135.3, 132.8, 131.3, 130.9, 130.6, 129.7, 128.8, 127.9, 127.1, 127.0, 126.1, 121.6, 113.2, 70.9, 66.0, 60.5, 53.2, 51.3, 50.8, 33.0, 32.9, 29.7. HRMS (ESI-TOF): m/z [M + H]+ calcd for C26H30ClN2O4S, 501.1615; found, 501.1620. Data in agreement with lit.29
3-Chloro-N-(1-(3-([1,1′-biphenyl]-2-yloxy)-2-hydroxy propyl)piperidin-4-yl)-benzenesulfonamide PZ-1361 (5a) [CAS 2095849-69-5] (Synthesis on a mmol Scale)
Intermediate 4 (407.7 mg, 1.12 mmol, 1 equiv), 3-chlorobenzenesulfonyl chloride (158.2 μL, 1.12 mmol, 1 equiv), and previously grinded K2CO3 (155.3 mg, 1.12 mmol, 1 equiv) were introduced in a 35 mL PTFE jar (milling load 25 mg/mL) with one stainless steel ball (ϕball = 1.5 cm). The reaction was carried out for 1 min at rt. Then, the crude mixture was solubilized in ethyl acetate (25 mL), and the organic phase was washed with KHSO4 aqueous solution at pH = 3.5 (3 × 10 mL) and saturated NaCl solution (1 × 10 mL), dried over Na2SO4, and finally filtered and concentrated under a vacuum, yielding compound 5a as a white powder (472.2 mg, yield 84%).
N-(1-(3-([1,1′-Biphenyl]-2-yloxy)-2-hydroxypropyl)piperidin-4-yl)benzenesulfonamide (5b)
General Procedure E was followed with primary amine 4 (176.7 mg, 0.487 mmol, 1 equiv), benzenesulfonyl chloride (62.14 μL, 0.487 mmol, 1 equiv), and previously grinded K2CO3 (67.3 mg, 0.487 mmol, 1 equiv) to afford final compound 5b as a colorless oil (197.4 mg yield 87%).
C26H30N2O4S, MW: 466.59. Monoisotopic mass: 466.19. UPLC/MS: tR = 5.23 min, purity = 98%, [M + H]+ 467.2. 1H NMR (500 MHz, CDCl3): δ 7.87–7.90 (m, 2H), 7.56 (tt, J = 6.3, 1.4 Hz, 1H), 7.47–7.52 (m, 4H), 7.33–7.39 (m, 2H), 7.26–7.32 (m, 3H), 7.03 (td, J = 7.5, 1.0 Hz, 1H), 6.96 (dd, J = 8.3, 0.9 Hz, 1H), 5.28 (s, 1H), 5.04 (br. s, 1H), 3.86–3.94 (m, 3H), 3.15 (spt, J = 4.3 Hz, 1H), 2.74 (d, J = 11.5 Hz, 1H), 2.58 (d, J = 11.2 Hz, 1H), 2.36 (dd, J = 12.3, 8.6 Hz, 1H), 2.29 (dd, J = 12.6, 4.0 Hz, 1H), 2.19 (t, J = 10.2 Hz, 1H), 1.95 (t, J = 10.6 Hz, 1H), 1.67–1.75 (m, 2H), 1.36–1.50 (m, 2H).
13C{1H} NMR (126 MHz, CDCl3): δ 155.6, 141.3, 138.5, 132.7, 131.3, 130.9, 129.7, 129.2, 128.8, 128.0, 126.9, 121.6, 113.2, 70.9, 66.0, 60.5, 51.3, 50.6, 33.0, 32.9. HRMS (ESI-TOF): m/z [M + H]+ calcd for C26H31N2O4S, 467.2005; found, 467.2007.
2-Fluoro-N-(1-(3-([1,1′-biphenyl]-2-yloxy)-2-hydroxy propyl)piperidin-4-yl)-benzenesulfonamide (5c)
General Procedure E was followed with primary amine 4 (172.1 mg, 0.474 mmol, 1 equiv), 2-fluoro-benzenesulfonyl chloride (62.80 μL, 0.474 mmol, 1 equiv), and previously grinded K2CO3 (65.6 mg, 0.474 mmol, 1 equiv) to afford final compound 5c as a white powder (190.5 mg, yield 85%).
C26H29FN2O4S, MW: 484.58. Monoisotopic mass: 484.18. UPLC/MS: tR = 5.35 min, purity = 97%, [M + H]+ 485.2. 1H NMR (500 MHz, CDCl3): δ 7.91 (td, J = 7.6, 1.7 Hz, 1H), 7.52–7.58 (m, 1H), 7.50 (dt, J = 6.9, 1.4 Hz, 1H), 7.34–7.39 (m, 2H), 7.23–7.33 (m, 4H), 7.15–7.21 (m, 1H), 7.03 (td, J = 7.5, 1.0 Hz, 1H), 6.97 (dd, J = 8.3, 0.9 Hz, 1H), 5.23–5.28 (m, 1H), 3.86–3.95 (m, 3H), 3.23 (spt, J = 4.8 Hz, 1H), 2.74 (d, J = 11.7 Hz, 1H), 2.58 (d, J = 12.0 Hz, 1H), 2.37 (dd, J = 12.0, 8.0 Hz, 1H), 2.30 (dd, J = 12.0, 4.0 Hz, 1H), 2.18 (t, J = 10.5 Hz, 1H), 1.94 (t, J = 10.7 Hz, 1H), 1.68–1.76 (m, 2H), 1.40–1.54 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3): δ 158.8 (d, J = 254.1 Hz), 155.7, 138.5, 135.1, 135.0, 131.2, 130.9, 130.1, 129.7, 128.8, 127.9, 127.0, 124.7, 124.6, 121.6, 117.2, 117.0, 113.2, 70.9, 66.1, 60.5, 53.2, 51.4, 50.9, 32.9, 32.8. HRMS (ESI-TOF): m/z [M + H]+ calcd for C26H30FN2O4S, 485.1910; found, 485.1914.
3-Methoxy-N-(1-(3-([1,1′-biphenyl]-2-yloxy)-2-hydroxypropyl)piperidin-4-yl)-benzenesulfonamide (5d)
General Procedure E was followed with primary amine 4 (169.2 mg, 0.466 mmol, 1 equiv), 3-methoxybenzenesulfonyl chloride (65.99 μL, 0.466 mmol, 1 equiv), and previously grinded K2CO3 (64.4 mg, 0.466 mmol, 1 equiv) to afford final compound 5d as a white powder (190.5 mg, yield 86%).
C27H32N2O5S, MW: 496.62. Monoisotopic mass: 496.2. UPLC/MS: tR = 5.45 min, purity = 95%, [M + H]+ 497.2. 1H NMR (500 MHz, CDCl3): δ 7.48–7.51 (m, 2H), 7.45–7.48 (m, 1H), 7.43 (t, J = 2.0 Hz, 1H), 7.40 (d, J = 8.3 Hz, 1H), 7.34–7.38 (m, 2H), 7.26–7.33 (m, 3H), 7.08 (ddd, J = 8.0, 2.6, 0.6 Hz, 1H), 7.03 (td, J = 7.4, 1.1 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 5.27 (s, 1H), 3.87–3.95 (m, 3H), 3.83 (s, 3H), 3.11–3.19 (m, 1H), 2.74 (d, J = 11.2 Hz, 1H), 2.57 (d, J = 11.2 Hz, 1H), 2.36 (dd, J = 12.6, 8.9 Hz, 1H), 2.29 (dd, J = 12.9, 4.0 Hz, 1H), 2.19 (t, J = 10.2 Hz, 1H), 1.94 (t, J = 10.5 Hz, 1H), 1.72 (dd, J = 8.7, 3.9 Hz, 2H), 1.37–1.52 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3): δ 160.0, 155.6, 142.5, 138.5, 131.2, 130.9, 130.3, 129.7, 128.8, 128.0, 127.0, 121.6, 119.1, 119.0, 113.2, 111.8, 70.9, 66.0, 60.5, 55.8, 53.6, 51.4, 50.7, 33.0, 32.8. HRMS (ESI-TOF): m/z [M + H]+ calcd for C27H33N2O5S, 497.2110; found, 497.2115.
4-Nitro-N-(1-(3-([1,1′-biphenyl]-2-yloxy)-2-hydroxy propyl)piperidin-4-yl)-benzenesulfonamide (5e)
General Procedure E was followed with primary amine 4 (165.7 mg, 0.457 mmol, 1 equiv), 4-nitrobenzenesulfonyl chloride (101.2 mg, 0.457 mmol, 1 equiv), and previously grinded K2CO3 (63.1 mg, 0.457 mmol, 1 equiv) to afford final compound 5e as a yellow powder (187.1 mg, yield 80%).
C26H29N3O6S, MW: 511.59. Monoisotopic mass: 511.18. UPLC/MS: tR = 5.67 min, purity 96%, [M + H]+ 512.2. 1H NMR (500 MHz, CDCl3): δ 8.27–8.33 (m, 2H), 8.01–8.07 (m, 2H), 7.46–7.53 (m, 2H), 7.33–7.38 (m, 2H), 7.24–7.33 (m, 3H), 7.00–7.06 (m, 1H), 6.95 (d, J = 7.7 Hz, 1H), 5.26–5.29 (m, 1H), 3.93–3.98 (m, 1H), 3.86–3.93 (m, 2H), 3.14–3.23 (m, 1H), 2.77 (d, J = 11.2 Hz, 1H), 2.61 (d, J = 11.2 Hz, 1H), 2.34–2.44 (m, 1H), 2.30 (dd, J = 12.6, 2.9 Hz, 1H), 2.19 (t, J = 11.2 Hz, 1H), 1.95 (t, J = 11.2 Hz, 1H), 1.67–1.75 (m, 2H), 1.37–1.52 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3): δ 155.6, 150.0, 147.4, 138.4, 131.2, 131.0, 129.7, 128.8, 128.2, 128.0, 127.0, 124.6, 121.7, 113.2, 70.8, 66.1, 60.3, 53.6, 51.3, 51.1, 33.2, 33.0. HRMS (ESI-TOF): m/z [M + H]+ calcd for C26H30N3O6S, 512.1855; found, 512.1863.
Acknowledgments
The project was financially supported by the Ministère de l’Europe et des Affaires Etrangères, le Ministère de l’Education Supérieure, de la Recherche et de l’Innovation and Polish Minister of Science and Higher Education (PHC Polonium program), by Jagiellonian University Medical College grant no N42/DBS/000018, by National Science Center, Poland grant no 2019/33/B/NZ7/02822, the University of Montpellier and the CNRS. V.F. thanks the Erasmus+ Program for a fellowship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c01044.
Green metrics calculation; MS, 1H NMR, and 13C NMR spectra for all synthesized compounds (PDF)
Author Contributions
V.C. and V.F. carried out the experiments; V.C. and X.B. conceived the experiments and wrote the manuscript with support from F.L. and P.Z. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Baumann M.; Baxendale I. R. The synthesis of Active Pharmaceutical Ingredients (APIs) using continuous flow chemistry. Beilstein J. Org. Chem. 2015, 11, 1194–1219. 10.3762/bjoc.11.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta R.; Benaglia M.; Puglisi A flow chemistry: recent developments in the synthesis of pharmaceutical products. Org. Process Res. Dev. 2016, 20, 2–25. 10.1021/acs.oprd.5b00325. [DOI] [Google Scholar]
- Tu N. P.; Searle P. A.; Sarris K. An automated microwave-assisted synthesis purification system for rapid generation of compound libraries. J. Lab. Autom. 2016, 21, 459–469. 10.1177/2211068215590580. [DOI] [PubMed] [Google Scholar]
- Berrino E.; Supuran C. T. Advances in microwave-assisted synthesis and the impact on novel drug discovery. Expert Opin. Drug Discovery 2018, 13, 861–873. 10.1080/17460441.2018.1494721. [DOI] [PubMed] [Google Scholar]
- Gomollón-Bel F. Ten chemical innovations that will change our world: IUPAC identifies emerging technologies in chemistry with potential to make our planet more sustainable. Chem. Int. 2019, 41, 12–17. 10.1515/ci-2019-0203. [DOI] [Google Scholar]
- James S. L.; Adams C. J.; Bolm C.; Braga D.; Collier P.; Friščić T.; Grepioni F.; Harris K. D.M.; Hyett G.; Jones W.; Krebs A.; Mack J.; Maini L.; Orpen A. G.; Parkin I. P.; Shearouse W. C.; Steed J. W.; Waddell D. C. Mechanochemistry: opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413–447. 10.1039/C1CS15171A. [DOI] [PubMed] [Google Scholar]
- Wang G. W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. 10.1039/c3cs35526h. [DOI] [PubMed] [Google Scholar]
- Howard J. L.; Cao Q.; Browne D. L. Mechanochemistry as an emerging tool for molecular synthesis: what can It offer?. Chem. Sci. 2018, 9 (12), 3080–3094. 10.1039/C7SC05371A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friščić T.; Mottillo C.; Titi H. M. Mechanochemistry for synthesis. Angew. Chem., Int. Ed. 2020, 59, 1018–1029. 10.1002/anie.201906755. [DOI] [PubMed] [Google Scholar]
- Beillard A.; Bantreil X.; Metro T. X.; Martinez J.; Lamaty F. Alternative technologies that facilitate access to discrete metal complexes. Chem. Rev. 2019, 119, 7529–7609. 10.1021/acs.chemrev.8b00479. [DOI] [PubMed] [Google Scholar]
- Horváth I. T.; Anastas P. T. Innovations and green chemistry. Chem. Rev. 2007, 107, 2169–2173. 10.1021/cr078380v. [DOI] [PubMed] [Google Scholar]
- Anastas P. T. Introduction: Green Chemistry. Chem. Rev. 2007, 107, 2167–2168. 10.1021/cr0783784. [DOI] [PubMed] [Google Scholar]
- Métro T. X.; Martinez J.; Lamaty F. 1,1′-Carbonyldiimidazole and mechanochemistry: a shining green combination. ACS Sustainable Chem. Eng. 2017, 5, 9599–9602. 10.1021/acssuschemeng.7b03260. [DOI] [Google Scholar]
- Tan D.; Mottillo C.; Katsenis A. D.; Strukil V.; Friščić T. Development of C-N coupling using mechanochemistry: catalytic coupling of arylsulfonamides and carbodiimides. Angew. Chem., Int. Ed. 2014, 53, 9321–9324. 10.1002/anie.201404120. [DOI] [PubMed] [Google Scholar]
- Hernandez J. G.; Bolm C. Altering product selectivity by mechanochemistry. J. Org. Chem. 2017, 82, 4007–4019. 10.1021/acs.joc.6b02887. [DOI] [PubMed] [Google Scholar]
- Howard J. L.; Brand M. C.; Browne D. L. Switching chemoselectivity: using mechanochemistry to alter reaction kinetics. Angew. Chem., Int. Ed. 2018, 57, 16104–16108. 10.1002/anie.201810141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira P. F. M.; Guidetti B.; Chamayou A.; Andre-Barres C.; Madacki J.; Kordulakova J.; Mori G.; Orena B. S.; Chiarelli L. R.; Pasca M. R.; Lherbet C.; Carayon C.; Massou S.; Baron M.; Baltas M. Mechanochemical synthesis and biological evaluation of novel isoniazid derivatives with potent antitubercular activity. Molecules 2017, 22, 1457. 10.3390/molecules22091457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirapegui C.; Acevedo-Fuentes W.; Dahech P.; Torrent C.; Barrias P.; Rojas-Poblete M.; Mascayano C. Easy and rapid preparation of benzoylhydrazides and their diazene derivatives as inhibitors of 15-lipoxygenase. Bioorg. Med. Chem. Lett. 2017, 27, 1649–1653. 10.1016/j.bmcl.2017.03.017. [DOI] [PubMed] [Google Scholar]
- Chakraborty B. Mechanochemical synthesis and cycloaddition reactions of fluoro nitrone under solvent-free conditions and potential antimicrobial activities of the cycloadducts. J. Heterocycl. Chem. 2019, 56, 3414–3422. 10.1002/jhet.3736. [DOI] [Google Scholar]
- Tan D.; Loots L.; Friščić T. Towards medicinal mechanochemistry: evolution of milling from pharmaceutical solid form screening to the synthesis of Active Pharmaceutical Ingredients (APIs). Chem. Commun. 2016, 52, 7760–7781. 10.1039/C6CC02015A. [DOI] [PubMed] [Google Scholar]
- Colacino E.; Porcheddu A.; Charnay C.; Delogu F. From enabling technologies to medicinal mechanochemistry: an eco-friendly access to hydantoin-based Active Pharmaceutical Ingredients. React. Chem. Eng. 2019, 4, 1179–1188. 10.1039/C9RE00069K. [DOI] [Google Scholar]
- Zajdel P.; Kurczab R.; Grychowska K.; Satała G.; Pawłowski M.; Bojarski A. J. The multiobjective based design, synthesis and evaluation of the arylsulfonamide/amide derivatives of aryloxyethyl- and arylthioethyl- piperidines and pyrrolidines as a novel class of potent 5-HT7 receptor antagonists. Eur. J. Med. Chem. 2012, 56, 348–360. 10.1016/j.ejmech.2012.07.043. [DOI] [PubMed] [Google Scholar]
- Zajdel P.; Canale V.; Partyka A.; Marciniec K.; Kurczab R.; Satała G.; Siwek A.; Jastrzębska-Więsek M.; Wesołowska A.; Kos T.; Popik P.; Bojarski A. J. Arylsulfonamide derivatives of (aryloxy)ethylpiperidines as selective 5-HT7 receptor antagonists and their psychotropic properties. MedChemComm 2015, 6, 1272–1277. 10.1039/C5MD00166H. [DOI] [Google Scholar]
- Canale V.; Kurczab R.; Partyka A.; Satała G.; Lenda T.; Jastrzębska-Więsek M.; Wesołowska A.; Bojarski A. J.; Zajdel P. Towards new 5-HT7 antagonists among arylsulfonamide derivatives of (aryloxy)ethylalkyl amines: multiobjective based design, synthesis, and antidepressant and anxiolytic properties. Eur. J. Med. Chem. 2016, 108, 334–346. 10.1016/j.ejmech.2015.11.040. [DOI] [PubMed] [Google Scholar]
- Canale V.; Kurczab R.; Partyka A.; Satała G.; Sloczyńska K.; Kos T.; Jastrzębska-Więsek M.; Siwek A.; Pękala E.; Bojarski A. J.; Wesołowska A.; Popik P.; Zajdel P. N-Alkylated arylsulfonamides of (aryloxy)ethyl piperidines: 5-HT7 receptor selectivity versus multireceptor profile. Bioorg. Med. Chem. 2016, 24, 130–139. 10.1016/j.bmc.2015.11.041. [DOI] [PubMed] [Google Scholar]
- Kurczab R.; Canale V.; Satała G.; Zajdel P.; Bojarski A. J. Amino acid hot spots of halogen bonding: a combined theoretical and experimental case study of the 5-HT7 receptor. J. Med. Chem. 2018, 61, 8717–8733. 10.1021/acs.jmedchem.8b00828. [DOI] [PubMed] [Google Scholar]
- Vidal S. Safety first: a recent case of a dichloromethane injection injury. ACS Cent. Sci. 2020, 6, 83–86. 10.1021/acscentsci.0c00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://echa.europa.eu/substance-information//substanceinfo/100.000.763.
- Canale V.; Partyka A.; Kurczab R.; Krawczyk M.; Kos T.; Satała G.; Kubica B.; Jastrzębska-Więsek M.; Wesołowska A.; Bojarski A. J.; Popik P.; Zajdel P. Novel 5-HT7R antagonists, arylsulfonamide derivatives of (aryloxy)propyl piperidines: add-on effect to the antidepressant activity of SSRI and DRI, and pro-cognitive profile. Bioorg. Med. Chem. 2017, 25, 2789–2799. 10.1016/j.bmc.2017.03.057. [DOI] [PubMed] [Google Scholar]
- An initial version of this work was deposited in ChemRxiv on April 15, 2020:; 10.26434/chemrxiv.12129390. [DOI] [Google Scholar]
- Ardila-Fierro K. J.; Pich A.; Spehr M.; Hernández J. G.; Bolm C. Synthesis of acylglycerol derivatives by mechanochemistry. Beilstein J. Org. Chem. 2019, 15, 811–817. 10.3762/bjoc.15.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowmaker G. A. Solvent-assisted mechanochemistry. Chem. Commun. 2013, 49, 334–348. 10.1039/C2CC35694E. [DOI] [PubMed] [Google Scholar]
- Bonnamour J.; Métro T. X.; Martinez J.; Lamaty F. Environmentally benign peptide synthesis using liquid-assisted ball-milling: application to the synthesis of Leu-enkephalin. Green Chem. 2013, 15, 1116–1120. 10.1039/c3gc40302e. [DOI] [Google Scholar]
- Hasa D.; Miniussi E.; Jones W. Mechanochemical synthesis of multicomponent crystals: one liquid for one polymorph? a myth to dispel. Cryst. Growth Des. 2016, 16, 4582–4588. 10.1021/acs.cgd.6b00682. [DOI] [Google Scholar]
- Porte V.; Thioloy M.; Pigoux T.; Métro T. X.; Martinez J.; Lamaty F. Peptide mechanosynthesis by direct coupling of N-protected α-amino acids with amino esters. Eur. J. Org. Chem. 2016, 2016, 3505–3508. 10.1002/ejoc.201600617. [DOI] [Google Scholar]
- Do J. L.; Friščić T. Mechanochemistry: a force of synthesis. ACS Cent. Sci. 2017, 3, 13–19. 10.1021/acscentsci.6b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonk J. D.; Amos D. T.; Olson S. J. Convenient one-pot synthesis of sulfonamides from thiols using trichloroisocyanuric acid. Synth. Commun. 2007, 37, 2039–2050. 10.1080/00397910701356942. [DOI] [Google Scholar]
- Zhu M.; Fujita K.; Yamaguchi R. Simple and versatile catalytic system for N-alkylation of sulfonamides with various alcohols. Org. Lett. 2010, 12, 1336–1339. 10.1021/ol1002434. [DOI] [PubMed] [Google Scholar]
- Zajdel P.; Masurier N.; Canale V.; Verdie P.; Amblard M.; Pawłowski M.; Martinez J.; Subra G. The pipecolic linker - an acid-labile handle for derivatization of secondary amines on a solid-support. Part 3. Tetrahedron Lett. 2013, 54, 998–1002. 10.1016/j.tetlet.2012.12.036. [DOI] [Google Scholar]
- Zajdel P.; Marciniec K.; Satała G.; Canale V.; Kos T.; Partyka A.; Jastrzębska-Więsek M.; Wesołowska A.; Basinska-Ziobron A.; Wójcikowski J.; Daniel W. A.; Bojarski A. J.; Popik P. N1-Azinylsulfonyl-1H-indoles: 5-HT6 receptor antagonists with procognitive and antidepressant-like properties. ACS Med. Chem. Lett. 2016, 7, 618–622. 10.1021/acsmedchemlett.6b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajdel P.; Kos T.; Marciniec K.; Satała G.; Canale V.; Kamiński K.; Hołuj M.; Lenda T.; Koralewski R.; Bednarski M.; Nowiński L.; Wójcikowski J.; Daniel W. A.; Nikiforuk A.; Nalepa I.; Chmielarz P.; Kuśmierczyk J.; Bojarski A. J.; Popik P. Novel multi-target azinesulfonamides of cyclic amine derivatives as potential antipsychotics with pro-social and pro-cognitive effects. Eur. J. Med. Chem. 2018, 145, 790–804. 10.1016/j.ejmech.2018.01.002. [DOI] [PubMed] [Google Scholar]
- Tsai A. S.; Curto J. M.; Rocke B. N.; Dechert-Schmitt A. M. R.; Ingle G. K.; Mascitti V. One-step synthesis of sulfonamides from N-tosylhydrazones. Org. Lett. 2016, 18, 508–511. 10.1021/acs.orglett.5b03545. [DOI] [PubMed] [Google Scholar]
- Roschangar F.; Sheldon R. A.; Senanayake C. H. Overcoming barriers to green chemistry in the pharmaceutical industry - the Green Aspiration Level concept. Green Chem. 2015, 17, 752–768. 10.1039/C4GC01563K. [DOI] [Google Scholar]
- Van Aken K.; Strekowski L.; Patiny L. EcoScale, a semi-quantitative tool to select an organic preparation based on economical and ecological parameters. Beilstein J. Org. Chem. 2006, 2, 3. 10.1186/1860-5397-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson C.; Musonda C. C.; Chibale K.; Campbell W. E.; Smith P. J. Synthesis of totarol amino alcohol derivatives and their antiplasmodial activity and cytotoxicity. Bioorg. Med. Chem. 2003, 11, 4417–4422. 10.1016/S0968-0896(03)00491-7. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Bao W. Copper-catalyzed tandem process: an efficient approach to 2-substituted-1,4-benzodioxanes. Org. Biomol. Chem. 2010, 8, 2700–2703. 10.1039/c003691a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.