

Keywords: crypt fission, hyaluronic acid, Lgr5+ stem cell, PGE2, TLR4
Abstract
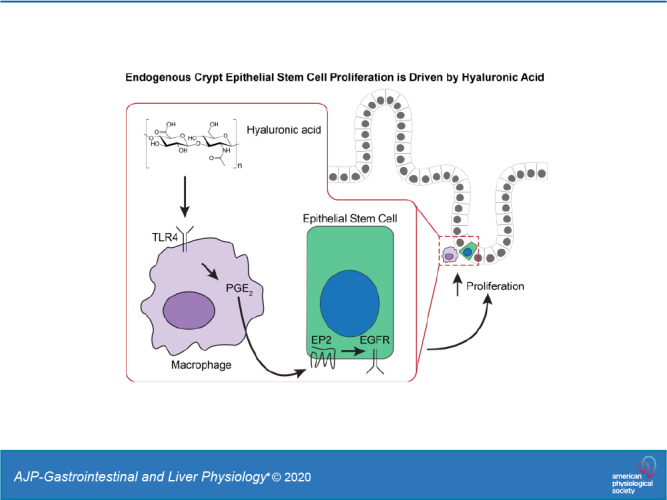
Hyaluronic acid (HA), a glycosaminoglycan in the extracellular matrix, binds to CD44 and Toll-like receptor 4 (TLR4). We previously demonstrated that both CD44 and TLR4, but predominately TLR4, mediated HA stimulation of Lgr5+ stem cell proliferation, crypt fission, and intestinal growth in postnatal mice. Here we address the questions of which cell type expresses the relevant TLR4 in driving intestinal growth and what are the downstream events from TLR4 activation. Studies were done in 14-day-old mice: wild type (WT), mice deficient in cyclooxygenase 2 (COX2), mice deficient in myeloid cell TLR4, and mice deficient in epithelial cell epidermal growth factor receptor (EGFR). Biological end points included crypt fission and Lgr5 cell proliferation. In WT mice, treatment with NS-398 (a COX2 inhibitor), clodronate (a macrophage-depleting agent), or tyrphostin (an EGFR inhibitor) resulted in 30% reductions in crypt fission and Lgr5+ stem cell proliferation compared with control mice. Mice deficient in COX2 or myeloid TLR4 or epithelial cell EGFR all had 30% reductions in crypt fission and Lgr5+ stem cell proliferation compared with WT mice. Administration of dimethyl PGE2, a stable PGE2 analog, increased crypt fission and Lgr5+ stem cell proliferation. Administration of dimethyl PGE2 reversed the effects of NS-398, clodronate, COX2 deficiency, and myeloid TLR4 deficiency but had no effect on mice treated with tyrphostin or mice deficient in epithelial cell EGFR. We conclude that, in postnatal mice, ~30% of intestinal growth as manifested by crypt fission and Lgr5+ stem cell proliferation is driven by a novel pathway: Extracellular HA binds TLR4 on pericryptal macrophages, inducing the production of PGE2 through COX2. PGE2 transactivates EGFR in Lgr5+ epithelial stem cells, resulting in Lgr5+ stem cell proliferation and crypt fission.
NEW & NOTEWORTHY This study, in newborn mice, describes a novel molecular pathway regulating Lgr5+ epithelial stem cell proliferation and normal intestinal elongation, as assessed by crypt fission. In this pathway, endogenous extracellular hyaluronic acid binds to Toll-like receptor 4 on pericryptal macrophages releasing PGE2 which binds to epidermal growth factor receptor on Lgr5+ stem cells resulting in proliferation. Lgr5+ stem cell proliferation leads to crypt fission and intestinal elongation. The demonstration that normal growth requires microbial-independent Toll-like receptor activation is novel.
INTRODUCTION
Hyaluronic acid (HA), a glycosaminoglycan polymer, is an important constituent of the extracellular matrix. In the intestine, extracellular HA is distributed in a thin layer adjacent to the basal surface of epithelial cells at the crypt base (46). The biologic effects of HA are mediated by binding to its receptors. The HA receptor CD44 is expressed on the plasma membrane of most cells, including fibroblasts, smooth muscle cells, epithelial cells, and immune cells (35). In addition to binding to CD44, HA binds to Toll-like receptor 4 (TLR4), a component of the innate immune system (16, 40). TLR4 is widely distributed in the gastrointestinal tract and participates in mediating the host response to commensal and pathogenic bacteria (25, 37). Although lipopolysaccharide produced by gram-negative bacteria is the ligand typically associated with TLR4, several host molecules including HA are also TLR4 ligands. HA binding to TLR4 promotes growth and wound repair in the intestine, but the mechanism by which this occurs is not clear (28, 29, 46).
In the recovery phase of the dextran sodium sulfate (DSS) colitis model, TLR4 activation promotes epithelial proliferation through PGE2 (3, 10, 25). DSS colitis mice deficient in Myd88, a TLR adaptor molecule, and mice deficient in COX2 exhibited a profound inhibition of epithelial proliferation in the rectal crypts (3). Exogenous addition of 16,16-dimethyl PGE2 (dmPGE2), a stable PGE2 analog, rescues the effects of this injury in both knockout mouse strains, indicating that Myd88 signaling is upstream of COX2 and PGE2. HA promotes epithelial proliferation in the recovery phase of DSS colitis through TLR4 activation (46). HA is sensed by macrophages in the gut, which induces the production of PGE2 through COX2 (46). In turn, PGE2 promotes epithelial proliferation. HA binding to macrophage TLR4 also induces the expression of HA synthases, resulting in increased HA synthesis and thus a feed-forward loop to enhance epithelial repair (22). Exogenous HA can be administered therapeutically in this model (46). Exogenous HA is protective against DSS colitis in wild-type (WT) mice but not in mice deficient in Myd88, TLR4, or COX2.
Not only does HA promote epithelial protection and repair in the intestine and colon, it also regulates epithelial proliferation and small intestinal growth during normal development (28, 46). In the postnatal mouse, the intestine elongates through crypt fission (5a). In this process, there is proliferation of Lgr5+ epithelial stem cells leading to an increase in epithelial cell number. This is followed by an indentation at the base of the crypt that extends upward until the single crypt is divided into two crypts having a common crypt-villus junction. Elongation of the intestine in early postnatal life is driven by endogenous HA binding to both CD44 and TLR4 but primarily to TLR4 (29). PEP1 is a 12-mer peptide that binds to HA and blocks it from binding to its receptors (19). Intraperitoneal administration of PEP1 to mice from 7 to 14 days of age results in diminished Lgr5+ cell proliferation and diminished crypt fission (29). Administration of PEP1 reduces Lgr5+ cell proliferation and crypt fission in WT mice but not in TLR4 knockout mice, suggesting that the effects of endogenous HA on Lgr5+ proliferation and crypt fission are mediated primarily through TLR4. Although it is clear that the promotion of crypt fission and Lgr5+ stem cell proliferation by HA are mediated through TLR4 activation, the cellular location of the relevant TLR4 and downstream events from TLR4 activation are not known.
PGE2 promotes proliferation in the gastrointestinal epithelium and in colon cancer (3, 23, 41). Long-term administration of dmPGE2 in rats increases mucosal thickness in the stomach, duodenum, and colon (26). However, the mechanisms of these trophic actions remain unclear. Lgr5+ stem cells proliferate in response to β-catenin activation by wnts (2, 5, 7). However, activation of epidermal growth factor receptor (EGFR) also promotes Lgr5+ stem cell proliferation (32). In the intestinal epithelium EGFR activation promotes proliferation and blocks apoptosis and shedding (18, 23, 32, 39, 44). PGE2 promotes stem cell proliferation by modifying the wnt signaling cascade at the level of β-catenin degradation through cAMP/PKA-mediated stabilizing phosphorylation events (10). However, PGE2 also promotes colon cancer growth and gastrointestinal hypertrophy by transactivating EGFR (23).
In this study we address the mechanism by which HA promotes Lgr5+ stem cell proliferation and crypt fission. In particular, we address the following questions: 1) TLR4 activation mediates the effects of HA on crypt fission. Which cell types express the relevant TLR4? 2) PGE2 produced through COX2 promotes epithelial proliferation in the repair phase of DSS colitis. Does PGE2 promote Lgr5+ stem cell proliferation and crypt fission in normal intestinal growth? 3) What is the pathway by which PGE2 promotes Lgr5 stem cell proliferation and crypt fission? Is it the wnt/β-catenin pathway or EGFR activation?
METHODS
Chemicals and reagents.
Anionic liposomal clodronate (Clophosome-A, product no. F7010C-A) and control empty liposomes (F70101-N) were purchased from FormuMax Scientific (Sunnyvale, CA). dmPGE2 (catalog no. 14750), butaprost (catalog no. 13740), and NS-398 (catalog no. 70590) were purchased from Cayman Chemical (Ann Arbor, MI). Tamoxifen (T5648) and tyrphostin AG-1478 (T4182) were purchased from Millipore Sigma (St. Louis, MO).
Animals and experimental procedures.
EGFRf/f mice were a gift from Dr. Brad Warner (Washington University; see Ref. 38). Western blot of enterocyte cell fractions confirmed deletion of EGFR protein in EGFR-null mice (38). We purchased TLR4f/f and LysM-Cre mice, both on a C57BL/6J background, from Jackson Laboratories and performed out cross breeding for three generations to generate TLR4f/fLysM-Cre+/− mice (15). WT, COX2−/−, TLR4f/fLysM-Cre+/−, and EGFRf/fVil-Cre/ERT2 mice were on a C57BL/6J background and bred in-house. WT Lgr5/enhanced green fluorescent protein (EGFP), COX2−/−Lgr5/EGFP, TLR4f/fLysM-Cre+/−, Lgr5/EGFP, and EGFRf/fVil-Cre/ERT2-Lgr5/EGFP reporter mice were each generated by initially breeding nonreporter WT, COX2−/−, TLR4f/fLysM-Cre+/−, or EGFRf/fVil-Cre/ERT2 mice with WT Lgr5-EGFP-ires-creERT2 mice and by multiple-generation selective breedings obtaining mice that were WT, COX2−/−, TLR4f/fLysM-Cre+/−, or EGFRf/fVil-Cre/ERT2 and heterozygous for the Lgr5-EGFP-ires-creERT2 reporter. Mice were maintained on a 12:12-h light-dark schedule in a temperature-controlled specific pathogen-free facility and fed standard laboratory mouse chow. Animal procedures were carried out in accordance with the Washington University School of Medicine Animal Studies Committee, which approved the protocols.
For crypt fission and crypt epithelial studies, postnatal mice were treated on alternate days from age 7 days to age 14 days with the following drug doses by intraperitoneal injection: (in mg/kg) 35 anionic liposomal clodronate, 0.1 dmPGE2, 1 butaprost, 1 NS-398, 0.5 tyrphostin AG-1478, and 0.5 tamoxifen.
At age 14 days, all mice were given a mixture of 5-bromo-2-deoxyuridine (BrdU, 120 mg/kg) and 5-fluoro-2-deoxyuridine (12 mg/kg) 90 min before they were euthanized to label S-phase cells. Small intestines were removed, flushed with cold Ca2+- and Mg2+-free PBS, and cut open longitudinally. Tissue strips were fixed in fresh 10% formalin (paraformaldehyde = 4%), pinned out on wax trays, and allowed to remain in fixative for 20 h. Fixed tissue strips were embedded in fresh 2% agar and then in paraffin for use in immunohistochemical studies.
Immunohistochemical analyses.
Formalin-fixed paraffin-embedded intestines from 14- day-old mice were used for all immunohistochemistry (IHC) and immunofluorescence (IF) analyses. Tissue sections were deparaffinized by warming on a 54°C slide warmer for 10 min followed by immersion in xylenes at room temperature for 20 min. Rehydration of sections was by sequential bathing for 5 min each in ethanol solutions progressing from 100%, 95%, 70%, and finally in distilled water. Sections were washed in 0.1 M Tris(hydroxymethyl)aminomethane, 0.15 M sodium chloride, and 0.05% Tween 20, pH 7.6 (TNT wash buffer). For bright-field IHC, sections were incubated in 3% hydrogen peroxide at room temperature for 10 min, whereas tissues for IF did not receive this treatment. For heat-induced epitope retrieval (HIER), sections were placed in rodent decloaker buffer (RD-913; Bio Care Medical, Concord, CA) and heated at 99°C for 18 min in a pressurized decloaking chamber (Bio Care Medical). At the end of heating, sections remained in the decloaking chamber as it depressurized for 10 min, were then moved and allowed to start cooling for 10 min on a bench, and finally placed under a stream of dH2O for 5 min to fully cool sections and displace the rodent decloaker buffer.
Hematoxylin and eosin sections from 14-day-old mice were used to determine frequency of crypt fission. Sections through fissioning crypts were identified according to Dehmer et al. (5a) and Dekaney et al. (6) as having a bifurcation creating two (and occasionally three) flask-shaped bases with a common crypt-villus junction. At least 100 crypt sections were counted in each of at least six mice per treatment group.
IHC detection of BrdU localization for assessment of intestinal crypt epithelial proliferation was carried out after HIER. Tissue sections were blocked for 30 min in Rodent Block M (RBM 961; Bio Care Medical) followed by incubation with rat anti-BrdU (1:200, catalog no. ab6326; Abcam, Cambridge, MA) overnight at 4°C. Sections were washed in TNT buffer and incubated for 15 min with rat probe, followed by 25 min in rat-on-mouse HRP polymer (RT517H; Bio Care Medical), to which was added XM Factor (XMF93; Bio Care Medical) to reduce background staining. Detection of BrdU was with 3,3-diaminobenzidine tetrahydrochloride (Millipore Sigma) and counterstaining with hematoxylin (Vector Laboratories, Burlingame, CA). BrdU-labeled crypt epithelial cells were scored on a cell-positional basis by light microscope analysis of strips of intestinal tissue sections. One hundred well-oriented half-crypt sections were counted with at least six mice per treatment group. Cell position 1 was located at the crypt base, and position numbers increased up each side of the crypt section. This generated a proliferation index for each treatment group, presented as the percentage of crypt epithelial cells at each position along the crypt that were positive for BrdU staining.
Immunofluorescence.
IF detection of TLR4, COX2, F4/80, SOX9, BrdU, and GFP (Lgr5) cellular localizations was carried out after HIER. Sections were blocked for 1 h in normal serum from the same species in which the secondary antibody was raised. After blocking was completed, sections were incubated sequentially in the first primary antibody overnight at 4°C, followed by 1.5 h in the first secondary antibody, and the procedure was repeated for the second primary and secondary antibodies. Primary antibodies were rabbit anti-TLR4 (1:200, catalog no. NB-100–56581; Novus Biologicals, Littleton, CO), mouse anti-COX2 (1:50, catalog no. 610204; BD Biosciences, San Diego, CA), rat anti-F4/80 (1:50, MCA497; Bio-Rad, Hercules, CA), rabbit anti-SOX9 (1:200, catalog no.AB5535; Millipore-Sigma), rat anti-BrdU (1:200, catalog no. ab6326; Abcam, Cambridge, MA), and chicken anti-GFP (1:100, catalog no. ab13970; Abcam). Secondary antibodies purchased from Life Technologies-Thermo Fisher (1:200; Carlsbad, CA) were AF594 and AF488 donkey anti-rabbit, donkey anti-rat, and goat anti-mouse. AF488 donkey anti-chicken was purchased from Jackson Immunoresearch (1:200, catalog no. 703–545- 155; West Grove, PA).
Small intestinal crypt epithelial stem cell proliferation in 14-day-old mice was assessed by IF. Lgr5+ intestinal crypt base columnar stem cells in formalin-fixed paraffin-embedded tissues were detected by use of an antibody against GFP. Epithelial cells labeled with GFP (Lgr5) or with BrdU, or with GFP (Lgr5) and BrdU were scored from images taken at ×200 magnification and saved as Axiovision zvi files using the Scalings program (Carl Zeiss Imaging Systems). Proliferating crypt epithelial stem cells were identified by the colocalization of GFP (Lgr5) and BrdU. The number of Lgr5+ crypt epithelial cells and the percent that were positive for both Lgr5 and BrdU were scored for 40–90 nonfissioning and for 10–40 fissioning crypts in 8–15 mice per treatment group. Census of Paneth cells was obtained by IF using rabbit anti-lysozyme (1:1,000, catalog no. ab108508; Abcam). At least 100 crypt sections were scored in each of six mice per treatment group.
Statistical analysis.
The results are shown means ± SE. Data were subjected to either a one-way analysis of variance with multiple-comparison tests using GraphPad Prism 5 or Student’s t test. F test to compare variances was used to determine if Welch’s correction was needed for Student’s t test. A P value of ≤0.05 was considered significant.
All of the authors had access to the study data and reviewed and approved the final manuscript.
RESULTS
TLR4 expressed in macrophages supports intestinal growth.
The most rapid lengthening in the mouse small intestine occurs early in life before 3 wk of age. Intestinal lengthening occurs through crypt fission, which is associated with increased proliferation of Lgr5+ epithelial stem cells (28). Administration of PEP1 to WT mice from age 7 days to age 14 days resulted in decreases in crypt fission and in Lgr5+ stem cell proliferation (29). In contrast, administration of PEP1 to TLR4−/− mice had no effect on crypt fission or Lgr5+ stem cell proliferation. These data suggest that endogenous HA promotes crypt fission and Lgr5+ stem cell proliferation in the intestine through a TLR4-mediated mechanism (29). Although that study demonstrated that crypt fission and Lgr5+ stem cell proliferation are TLR4 dependent, it did not address the cellular location of the relevant TLR4.
Pericryptal macrophages play a critical role in promoting epithelial stem cell proliferation (24). Moreover, pericryptal macrophages express TLR4 (Fig. 1A). The colonic epithelial response to injury requires MyD88 signaling in macrophages (17). We addressed the possibility that macrophage TLR4 activation mediates the effects of HA on crypt fission and Lgr5+ stem cell proliferation by two approaches. In the first approach WT mice were depleted of macrophages with anionic liposomal clodronate (hereafter referred to as clodronate; see Ref. 27). Depletion of macrophages resulted in diminished crypt fission, diminished Lgr5+ cell numbers, and diminished Lgr5+ cell proliferation in nonfissioning crypts (Fig. 1, B–D). Lgr5+ stem cell proliferation is quantified by assessing the percentage of total epithelial cells expressing both Lgr5 and BrdU in fissioning and nonfissioning crypts. As previously reported, PEP1 reduces crypt fission and Lgr5+ stem cell proliferation (29). In WT mice clodronate induced 30–45% decreases in crypt fission and in Lgr5+ stem cell proliferation in nonfissioning crypts.
Fig. 1.

Toll-like receptor 4 (TLR4) expressed in macrophages supports intestinal growth. A: photomicrographs show TLR4 expression (red) in epithelial and lamina propria cells in wild-type (WT) mice (image on left), with some F4/80-expressing macrophages (green) also expressing TLR4 (arrows in middle image). There was no TLR4 colocalization with F4/80 in macrophages in TLR4f/fLysM-Cre mice (image on right). Original magnifications were ×400 (left and right) and ×1,000 (middle). TLR4f/fLysM-Cre+/− mice have constitutive deletion of TLR4 in macrophages. Treatment of mice with anionic liposomal clodronate from age 7 days to age 14 days results in depletion of macrophage populations. Both untreated TLR4f/fLysM-Cre+/− mice and macrophage-depleted WT mice had reduced frequency of crypt fission (B), fewer Lgr5+ crypt epithelial cells (C), and reduced Lgr5+ crypt epithelial stem cell proliferation (D) compared with control mice. 16,16-Dimethyl PGE2 (dmPGE2) treatment from age 7 days to age 14 days increased epithelial proliferation in macrophage-depleted WT mice and in TLR4f/fLysM-Cre+/− mice as shown by increased frequency of crypt fission (B), increased numbers of Lgr5+ cells (C), and increased Lgr5+ crypt stem cell proliferation (D). E: positional distribution of BrdU-labeled intestinal crypt epithelial cells in 14-day-old mice. Macrophage-depleted mice had significantly reduced epithelial cell proliferation in positions 1 to 7 but slightly increased proliferation higher up in the crypt in positions 10 to 13. TLR4f/fLysM-Cre+/− control mice with deletion of TLR4 in macrophages had significantly reduced baseline crypt epithelial cell proliferation in positions 1 to 8 compared with WT controls. Values are means ± SE for 8–15 mice/treatment group. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control mice of the same genotype. xP < 0.01 compared with WT.
IF for Lgr5 and BrdU demonstrates that clodronate treatment results in fewer Lgr5+ cells per crypt and fewer cells that stain for both Lgr5 and BrdU (Fig. 1D). Because macrophage depletion resulted in a decrease in Lgr5+ stem cells, we sought to determine if there was a compensatory increase in reserve stem cells (30). Immunofluoresence for SOX9 in WT mice demonstrated some SOX9-positive cells at the base of each crypt and scattered SOX9-positive cells in the villi [Supplemental Fig. S1 (https://doi.org/10.6084/m9.figshare.11927103.v1)]. Macrophage depletion did not alter the distribution of SOX9-expressing cells.
The second approach to addressing the possibility of a role for macrophage TLR4 activation in mediating the effects of HA on crypt fission and Lgr5+ stem cell proliferation was the use of TLR4f/fLysM-CRE+/− mice, which are deficient in TLR4 in myeloid cells. The deletion of TLR4 in myeloid cells was validated by IF for TLR4 and F4/80, a macrophage marker. In WT mice there are pericryptal cells that express both TLR4 and F4/80. In mice deficient in myeloid cell TLR4 there are cells that express TLR4 and cells that express F4/80 but no cells that express bothTLR4 and F4/80 (Fig. 1A). At baseline, depletion of TLR4 in myeloid cells resulted in 30% decreases in crypt fission, in the number of Lgr5+ stem cells in both fissioning and nonfissioning crypts, and in Lgr5+ proliferation in both fissioning and nonfissioning crypts [Fig. 1, B–D, and Supplemental Fig. S2 (https://doi.org/10.6084/m9.figshare.11927103.v1)].
We also assessed the percentage of epithelial cells that are BrdU positive at each position along the crypt villus axis. The cells at the base of the crypt are assigned position 1; cells higher up the crypt are assigned progressively higher numbers. In positional BrdU incorporation experiments, treatment of WT mice with clodronate resulted in diminished BrdU incorporation at positions 1 through 7 (Figs. 1E and 3B). Similarly mice deficient in myeloid cell TLR4 had diminished BrdU incorporation at positions 1 through 8 compared with WT mice.
COX2 expressed in macrophages supports intestinal growth.
Having established that crypt fission and Lgr5+ stem cell proliferation are dependent on TLR4 activation in macrophages, we next sought to define the downstream events. In the DSS colitis model, epithelial proliferation is mediated by TLR4 activation and prostaglandin production through COX2 (3). Pericryptal macrophages express COX2 (Fig. 2A). We used two approaches to determine if crypt fission and Lgr5+ stem cell proliferation in the postnatal mouse are COX2 dependent. In the first approach we used the COX2 inhibitor NS-398. In NS-398-treated mice there was a 26% decrease in crypt fission and decreased Lgr5+ stem cell numbers and decreased Lgr5+ stem cell proliferation in fissioning crypts (Fig. 2, B–D). Our second approach was the use of COX2−/− mice. In COX2−/− mice there was a 40% decrease in crypt fission, with similar decreases in Lgr5+ stem cell number and Lgr5+ stem cell proliferation compared with WT mice [Fig. 2, B–D, and Supplementary Fig. S2, (https://doi.org/10.6084/m9.figshare.11927103.v1)]. Treatment of WT mice with NS-398 results in diminished BrdU incorporation at positions 1 through 6 (Fig. 2E). Similarly, COX2−/− mice incorporate BrdU at a lower level than WT controls in positions 1 through 7 (Figs. 2E and 3B).
Fig. 2.

Cyclooxygenase 2 (COX2) expressed in macrophages supports intestinal growth. A: photomicrograph of 14-day-old control wild-type (WT) mouse small intestine illustrates expression of COX2 (red) in F4/80- expressing macrophages (green) and in scattered other cells in the lamina propria. COX2−/− mice had reduced frequency of crypt fission (B), fewer Lgr5+ crypt epithelial cells (C), and reduced Lgr5+ crypt epithelial stem cell proliferation (D) compared with WT control mice. 16,16-Dimethyl PGE2 (dmPGE2) treatment increased epithelial proliferation in COX2−/− mice as shown by increased frequency of crypt fission (B), increased numbers of Lgr5+ cells (C), and increased Lgr5+ crypt stem cell proliferation (D). B: WT mice treated with the EP2 receptor agonist butaprost had increased frequency of crypt fission similar to dmPGE2 treated mice. WT mice treated with NS-398 had decreased crypt epithelial proliferation as shown by reduced crypt fission (B), decreased numbers of Lgr5+ crypt epithelial cells (C), and decreased Lgr5+ crypt epithelial stem cell proliferation (D) compared with WT controls. E: positional distribution of BrdU-labeled intestinal crypt epithelial cells in 14-day-old mice. COX2−/− mice had significantly reduced crypt epithelial cell proliferation in positions 1 to 7 compared with controls. The COX2 specific inhibitor NS-398 reduced crypt epithelial cell proliferation in positions 1 to 6 in WT mice. Values are means ± SE for 8–15 mice/treatment group. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control mice of the same genotype. xxP < 0.01 and xxxP < 0.001 compared with WT control.
Fig. 3.

Exogenous PGE2 rescues positional crypt epithelial cell proliferation in mice having reduced proliferation due to macrophage depletion, deletion of Toll-like receptor 4 (TLR4) in macrophage, or deletion of cyclooxygenase 2 (COX2). A: 5-bromo-2-deoxyuridine (BrdU) was administered to mice 90 min before being euthanized. The percentage of epithelial cells that were BrdU labeled was assessed for each epithelial position. 16,16-Dimethyl PGE2 (dmPGE2) treatment of wild-type (WT) mice from age 7 days to age 14 days significantly increased epithelial proliferation in positions 1 to 3. In WT mice depleted of macrophages due to treatment with anionic liposomal clodronate, in TLR4f/fLysM-Cre+/− mice having constitutive deletion of TLR4 in macrophage, and in COX2−/− mice there is significantly reduced epithelial cell proliferation in the lower one-half of the intestinal crypt. Simultaneous treatment with dmPGE2 significantly increased crypt epithelial cell proliferation in all three conditions; positions 1 to 5 in TLR4f/fLysM-Cre+/− and macrophage-depleted WT mice and positions 1 to 7 in COX2−/− mice. Furthermore, in the crypt base, dmPGE2 increased proliferation at or above that found in WT control mice. Values are means ± SE for 8–15 mice/treatment group. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control mice of the same genotype. ++P < 0.01 and +++P < 0.001 compared with WT control mice. B: representative histological sections showing BrdU staining.
Endogenous PGE2 supports intestinal growth.
PGE2 produced through COX2 promotes epithelial proliferation in the DSS colitis model (3). To determine if PGE2 is the COX2 product that mediates crypt fission and Lgr5+ stem cell proliferation we treated mice with dmPGE2, which binds to EP receptors 1–4, and with butaprost, a specific EP2 receptor agonist. In WT mice treatment with either dmPGE2 or butaprost resulted in 44–50% increases in crypt fission (Fig. 2B). Treatment with dmPGE2 resulted in similar increases in Lgr5+ stem cell number and proliferation (Fig. 2, C and D). dmPGE2 also rescued the decreases in crypt fission and Lgr5+ stem cell proliferation in WT mice with clodronate-induced depletion of macrophages, in mice deficient in TLR4 in myeloid cells, and in COX2−/− mice (Figs. 1, B–D, and 2, B–D). Studies of positional BrdU incorporation in dmPGE2-treated mice demonstrated increased incorporation in positions 1–3 (Fig. 3). Treatment of clodronate-treated WT mice, mice deficient in myeloid cell TLR4, and COX2−/− mice with dmPGE2 resulted in increased BrdU incorporation in the crypt (Fig. 3).
PGE2 promotes intestinal growth through EGFR activation.
Lgr5+ stem cell proliferation is mediated through both β-catenin and EGFR activation (7, 5, 13, 32). PGE2 can act through both β-catenin and EGFR activation (1, 4, 11, 23). PGE2 binding to the EP2 receptor transactivates EGFR (42). We used two approaches to assess the contribution of EGFR activation to the effects of PGE2 on crypt fission and Lgr5+ stem cell proliferation. The first approach was the administration of tyrphostin AG-1478 (hereafter referred to as tyrphostin), an inhibitor of EGFR phosphorylation. In WT animals administration of tyrphostin resulted in a 37% decrease in crypt fission, and similar decreases in Lgr5+ cell number and in Lgr5+ stem cell proliferation in both fissioning and nonfissioning crypts (Fig. 4, A–C). Coadministration of dmPGE2 with tyrphostin did not rescue the effects of tyrphostin on crypt fission or Lgr5+ stem cell proliferation. Tyrphostin also reduced BrdU incorporation at positions 1–8, an effect that was not rescued by coadministration of dmPGE2 (Fig. 4D).
Fig. 4.

Blocking transactivation of the epidermal growth factor receptor (EGFR) by endogenous PGE2 reduces intestinal crypt epithelial proliferation. Tyrphostin AG-1478 is a selective tyrosine kinase inhibitor that blocks phosphorylation of the EGFR. WT mice treated with tyrphostin from age 7 days to age 14 days had reduced frequency of crypt fission (A), fewer Lgr5+ crypt epithelial cells (B), and reduced Lgr5+ crypt epithelial stem cell proliferation (C) compared with wild-type (WT) control mice. Exogenous 16,16-dimethyl PGE2 (dmPGE2) failed to rescue these measures of crypt epithelial proliferation in tyrphostin-treated mice. D: positional distribution of 5-bromo-2-deoxyuridine (BrdU)-labeled intestinal crypt epithelial cells in 14-day-old WT mice. The highest rate of epithelial cell proliferation in control mice was in the range of position 4 to position 8. Tyrphostin treatment resulted in significantly decreased proliferation in positions 1 to 8 compared with controls. dmPGE2 treatment of mice given tyrphostin failed to rescue BrdU+ epithelial cell proliferation in positions 1 to 6 but restored proliferation to control levels higher up in the crypt. Values are means ± SE for 8–15 mice/treatment group. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with WT control mice.
The second approach to assessing the contribution of EGFR activation to the effects of PGE2 on crypt fission and Lgr5+ stem cell proliferation was the use of EGFRf/fVil-Cre+/−/ERT2 mice in which administration of tamoxifen resulted in the deletion of EGFR in intestinal epithelial cells. Administration of tamoxifen resulted in a 33% decrease in crypt fission, and similar decreases in Lgr5+ cell number and Lgr5+ stem cell proliferation in both fissioning and nonfissioning crypts [Fig. 5, A–C, and Supplemental Fig. S2 (https://doi.org/10.6084/m9.figshare.11927103.v1)]. Administration of dmPGE2 did not increase crypt fission or Lgr5+ stem cell proliferation in mice deficient in epithelial cell EGFR. BrdU incorporation was diminished at positions 1–7 in mice deficient in epithelial cell EGFR (Figs. 5D and 3B). Administration of dmPGE2 did not increase BrdU incorporation in these mice.
Fig. 5.

The effects of PGE2 are mediated through transactivation of the epidermal growth factor receptor (EGFR). Tamoxifen treatment of EGFRf/fVil-Cre/ERT2 mice from age 7 days to age 14 days resulted in depletion of the EGFR in intestinal epithelial cells. These mice had reduced frequency of crypt fission (A), fewer numbers of Lgr5+ crypt epithelial cells (B), and reduced Lgr5+ crypt epithelial stem cell proliferation (C) compared with control mice. D: positional distribution of 5-bromo-2-deoxyuridine (BrdU)-labeled intestinal crypt epithelial cells in 14-day-old EGFRf/fVil-Cre/ERT2 mice. The highest rate of epithelial cell proliferation in control mice was in the range of position 4 to position 8. Tamoxifen treatment resulted in significantly decreased positional epithelial proliferation in the lower one-half of the crypt compared with controls. 16,16-Dimethyl PGE2 (dmPGE2) treatment of mice having tamoxifen-induced depletion of EGFR failed to rescue crypt epithelial proliferation. Tamoxifen treatment had no effect on crypt epithelial proliferation in EGFRf/f mice, which are functionally the same as wild-type (WT) mice and responded to dmPGE2 treatment with increased crypt epithelial proliferation. Values are means ± SE for 8–15 mice/treatment group. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control mice.
PGE2 promotes Paneth cell numbers in postnatal mice.
Paneth cells are an important component of the epithelial stem cell niche and are derived from epithelial stem cells (31). We sought to determine if Paneth cell number was affected by myeloid TLR4 signaling, PG production, or the presence of macrophages. Mice deficient in COX-2, or in myeloid TLR4, had a 50% reduction in Paneth cell number (Fig. 6). Similarly, depletion of macrophages with clodronate resulted in a 60% reduction in Paneth cells. Mice deficient in epithelial cell EGFR had a 50% reduction in Paneth cells. We next sought to determine if PGE2, which increases Lgr5+ epithelial stem cell proliferation, would also increase Paneth cell number. Treatment with dmPGE2 increased Paneth cell number by 40% in WT mice. dmPGE2 induced lesser increases in Paneth cell number in mice deficient in COX-2, in mice deficient in myeloid TLR4, or in macrophage-depleted mice. However, dmPGE2 did not increase Paneth cell number in mice deficient in epithelial cell EGFR.
Fig. 6.

PGE2 increased Paneth cell numbers in postnatal wild-type (WT), Toll-like receptor 4 (TLR4)f/fLysM-Cre, and cyclooxygenase 2 (COX2)−/− mice, but not in postnatal epidermal growth factor receptor (EGFR)f/fVil-Cre/ERT2 mice. A: photomicrographs show lysozyme-expressing Paneth cells in small intestines of control and 16,16-dimethyl PGE2 (dmPGE2)-treated 14-dayold mice. B: compared with WT mice, the number of Paneth cells/crypt section was reduced in WT mice depleted of macrophages due to clodronate treatment, in TLR4f/fLysM-Cre mice having constitutive deletion of TLR4 in macrophages, in COX2−/− mice, and in EGFRf/fVil-Cre/ERT2 mice with tamoxifen-induced deletion of EGFR in intestinal epithelial cells. Treatment with dmPGE2 increased Paneth cell numbers in WT mice, in mice deficient in myeloid TLR4, and in mice deficient in COX2 but not in mice deficient in epithelial cell EGFR. Values are means ± SE for 6 mice/treatment group. ***P < 0.001 compared with similar mice not treated with dmPGE2. +++P < 0.001 compared with WT control mice.
DISCUSSION
These studies demonstrate that normal intestinal lengthening through crypt fission is promoted by endogenous HA binding to macrophage TLR4. These macrophages produce PGE2 through COX2 (36, 46). The positioning of COX2-expressing macrophages immediately adjacent to crypt Lgr5+ stem cells is of particular importance in that PGE2 has a very short half-life in tissues (9, 20). As a consequence, PGE2 only acts on cells adjacent to the cell that produce it. PGE2 transactivation of EGFR results in Lgr5+ stem cell proliferation and crypt fission. Blocking this pathway, either pharmacologically or by genetic manipulation, at any step (HA binding to TLR4, TLR4 activation, PGE2 production, or EGFR activation) results in 30% reductions in Lgr5+ proliferation and crypt fission. This is consistent with the earlier demonstration that treatment of mice with PEP1 from 3 to 8 wk of age results in a 30% reduction in intestinal length and the demonstration that PEP1 reduced crypt fission and Lgr5+ stem cell proliferation by 30% in postnatal mice (28, 29). Thus, the HA/TLR4/PGE2/EGFR pathway accounts for ~30% of Lgr5+ stem cell proliferation and intestinal elongation. dmPGE2, which activates receptors EP1–4, and the selective EP2 agonist butaprost promote crypt fission equally, suggesting that the effects of PGE2 are mediated through the EP2 receptor. This is consistent with the demonstration that in HCT-116 cells PGE2 binding to EP2 transactivates EGFR (42).
Both the baseline epithelial proliferation seen in normal growth and the enhanced epithelial proliferation seen in the repair phase of DSS colitis are dependent on TLR4 activation and PGE2 production (3). The distinction between epithelial proliferation in normal growth and epithelial proliferation in wound repair relates to the cellular source of the PGE2. In normal growth PGE2 is produced by pericryptal macrophages in response to TLR4 activation. In the repair phase of DSS colitis the intestinal injury promotes the TLR4-dependent migration of COX2-expressing mesenchymal stem cells to an area near the crypt epithelial stem cells. These COX2-expressing mesenchymal stem cells produce large amounts of PGE2 (43). In growth experiments presented here administration of exogenous dmPGE2 results in a significant increase in Lgr5+ stem cell proliferation and crypt fission. This suggests that, under homeostatic conditions, endogenous PGE2 is present at levels sufficient to promote Lgr5+ cell proliferation but not sufficient to achieve maximal rates of proliferation. It may be that the COX2-expressing mesenchymal stem cells recruited to the crypt base during injury produce enough PGE2 to maximize epithelial cell proliferation. The earlier studies did not address the mechanism by which PGE2 enhanced epithelial proliferation in the DSS model. However, based on the current studies, it is possible that PGE2 enhances epithelial cell proliferation in the DSS model through transactivation of EGFR. The decreases in Lgr5+ stem cell proliferation and crypt fission seen in mice treated with tyrphostin were equal to those seen in mice treated with PEP1 (29) or NS-398. This suggests that transactivation of EGFR by PGE2 is the major pathway for EGFR activation in Lgr5+ stem cells. It also suggests that other mechanisms for EGFR activation, such as activation by EGF made by Paneth cells, are less important in promoting Lgr5+ stem cell proliferation under homeostatic conditions. The reduction in Lgr5+ stem cell proliferation and crypt fission seen in PEP1-treated WT mice, COX2 knockout mice, and mice lacking epithelial cell EGFR are similar. Moreover, dmPGE2 fails to rescue the decreases in Lgr5+ stem cell proliferation and crypt fission in tyrphostin-treated mice and in mice deficient in epithelial cell EGFR. This suggests that all of the crypt fission driven by the HA/TLR4/PGE2 pathway is mediated through EGFR activation. Positional BrdU studies demonstrate that the HA/TLR4/PGE2/EGFR pathway is important in regulating proliferation at the crypt base but not in the rapid proliferation seen in the transit-amplifying cells higher up in the crypt. This is consistent with localization of HA in the intestine to an area surrounding the crypt base (46).
The wnt/β-catenin pathway is important in promoting Lgr5+ stem cell proliferation (7). The data presented here demonstrate that blocking EGFR results in a 30% reduction in Lgr5+ stem cell proliferation. This may reflect a direct effect of EGFR activation on Lgr5+ stem cell proliferation or it may reflect an indirect effect of EGFR activation acting through the wnt/β-catenin pathway. AKT activation is downstream of EGFR activation, and β-catenin signaling is regulated by AKT activation (14). Thus it is possible that the effects of PGE2 on epithelial stem cell proliferation are mediated by the EGFR/AKT/β-catenin pathway.
TLR4 is expressed on a number of cell types in the intestine, including epithelial cells (45). In these experiments, TLR4 deficiency in myeloid cells and global TLR4 deficiency (29) had the same effects on Lgr5+ stem cell proliferation and crypt fission, suggesting that, under homeostatic conditions, the TLR4 activation that promotes crypt fission and Lgr5+ stem cell proliferation is in myeloid cells. Similarly, TLR4 deficiency in myeloid cells had the same effect on Lgr5+ stem cell proliferation and crypt fission as did clodronate depletion of macrophages, suggesting that all of the effects of macrophages on Lgr5+ stem cell proliferation and crypt fission are mediated through TLR4 activation and PGE2 production.
In neonatal mice depleted of macrophages with clodronate there was decreased crypt fission, decreased Lgr5+ stem cell proliferation, and decreased Paneth cell numbers. In an earlier study, Sehgal et al. depleted adult mice of macrophages expressing the colony-stimulating factor-1 receptor (CSF1R; see Ref. 33). Macrophage ablation following CSF1R blockade affected Paneth cell differentiation and reduced Lgr5+ stem cell proliferation and Lgr5+ stem cell number. These mice also demonstrated an increase in epithelial proliferation in the upper crypt consistent with the proliferation of SOX9+ BMI1+ reserve stem cells. We did not see evidence for proliferation of reserve stem cells in neonatal mice depleted of macrophages with clodronate. This may reflect a distinction between neonatal and adult mice.
Paneth cells are produced through Lgr5+ stem cell proliferation (7, 32). All of the genetic and pharmacologic manipulations of the HA/TLR4/PGE2/EGFR pathway had similar effects on Lgr5+ stem cell proliferation and Paneth cell number. Depletion of COX2, myeloid TLR4, or epithelial cell EGFR decreased Lgr5+ stem cell proliferation and Paneth cell number, whereas exogenous dmPGE2 increased Lgr5+ stem cell proliferation and Paneth cell number. These findings are consistent with PGE2 acting directly on Lgr5+ stem cells to increase their proliferation. In that case the increase in Paneth cell number would be the result of increased Lgr5+ stem cell proliferation. Support for a direct action of PGE2 on Lgr5+ stem cells comes from the demonstration that PGE2 supports the growth of chicken embryo intestinal organoids and the demonstration that PGE2 promotes Lgr5+ stem cell proliferation in mouse colon organoids where there are no Paneth cells (8). However, it is also possible that PGE2 acts primarily on Paneth cells or subepithelial myofibroblasts, resulting in the production of EGFR agonists such as amphiregulin and epiregulin (34).
The role of COX2 and PGE2 in colon cancer tumorigenesis has been much more extensively studied than their role in epithelial proliferation in normal growth (12, 23). PGE2 transactivation of EGFR promotes Lgr5+ stem cell proliferation under homeostatic conditions. This is consistent with PGE2 transactivation of EGFR promoting proliferation in colon cancer cell lines (23). Inactivation of EGFR kinase with selective inhibitors reduced PGE2-induced proliferation in colon cancer cell lines just as tyrphostin reduced the proliferation of Lgr5+ stem cells (23).
Here we demonstrate that HA/TLR4/PGE2/EGFR, a novel pathway, plays an important role in promoting Lgr5+ stem cell proliferation, crypt fission, and normal intestinal growth. This raises the possibility that drugs such as nonsteroidal anti-inflammatory drugs that inhibit this pathway may impair normal intestinal growth. It also raises the possibility that agents that activate this pathway such as exogenous HA or TLR4 agonists or prostanoids may enhance intestinal growth in short bowel syndrome and other conditions in which enhanced growth may be desirable.
GRANTS
This work supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants R37-DK-33165 (W. F. Stenson), DK-100737 (M. A. Ciorba), and DK-109384 (M. A. Ciorba), Crohn’s and Colitis Senior Fellowship Award No. 370863 (D. M. Alvarado), Digestive Diseases Research Core Centers NIDDK Grant P30-DK-052574, and philanthropic support from the Givin’ It All For Guts Foundation (https://givinitallforguts.org/, M. A. Ciorba) and the Lawrence C. Pakula MD IBD Research, Innovation, and Education Fund (M. A. Ciorba and D. M. Alvarado).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.E.R., D.A., and X.E. performed experiments; T.E.R., D.A., X.E., M.A.C., and W.F.S. analyzed data; T.E.R., D.A., M.A.C., and W.F.S. interpreted results of experiments; T.E.R. and D.A. prepared figures; T.E.R., D.A., and W.F.S. drafted manuscript; T.E.R., M.A.C., and W.F.S. edited and revised manuscript; T.E.R., D.A., X.E., M.A.C., and W.F.S. approved final version of manuscript; M.A.C. and W.F.S. conceived and designed research.
REFERENCES
- 1.Ansari KM, Rundhaug JE, Fischer SM. Multiple signaling pathways are responsible for prostaglandin E2-induced murine keratinocyte proliferation. Mol Cancer Res 6: 1003–1016, 2008. doi: 10.1158/1541-7786.MCR-07-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449: 1003–1007, 2007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 3.Brown SL, Riehl TE, Walker MR, Geske MJ, Doherty JM, Stenson WF, Stappenbeck TS. Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury. J Clin Invest 117: 258–269, 2007. doi: 10.1172/JCI29159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem 278: 35451–35457, 2003. doi: 10.1074/jbc.M302474200. [DOI] [PubMed] [Google Scholar]
- 5.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell 149: 1192–1205, 2012. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 5a.Dehmer JJ, Garrison AP, Speck KE, Dekaney CM, Van Landeghem L, Sun X, Henning SJ, Helmrath MA. Expansion of intestinal epithelial stem cells during murine development. PLoS One 6: e27070, 2011. doi: 10.1371/journal.pone.0027070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dekaney CM, Gulati AS, Garrison AP, Helmrath MA, Henning SJ. Regeneration of intestinal stem/progenitor cells following doxorubicin treatment of mice. Am J Physiol Gastrointest Liver Physiol 297: G461–G470, 2009. doi: 10.1152/ajpgi.90446.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Lau W, Barker N, Low TY, Koo BK, Li VSW, Teunissen H, Kujala P, Haegebarth A, Peters PJ, van de Wetering M, Stange DE, van Es JE, Guardavaccaro D, Schasfoort RBM, Mohri Y, Nishimori K, Mohammed S, Heck AJR, Clevers H. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476: 293–297, 2011. doi: 10.1038/nature10337. [DOI] [PubMed] [Google Scholar]
- 8.Fan YY, Davidson LA, Callaway ES, Goldsby JS, Chapkin RS. Differential effects of 2- and 3-series E-prostaglandins on in vitro expansion of Lgr5+ colonic stem cells. Carcinogenesis 35: 606–612, 2014. doi: 10.1093/carcin/bgt412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forstermann U, Neufang B. Elimination from circulation of cats of 6-keto-PGE1compared with PGE2 and PGI2. J Pharm Pharmacol 35: 724–728, 1983. doi: 10.1111/j.2042-7158.1983.tb02878.x. [DOI] [PubMed] [Google Scholar]
- 10.Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, Xu R, Inoue H, Arditi M, Dannenberg AJ, Abreu MT. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology 131: 862–877, 2006. doi: 10.1053/j.gastro.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, Zon LI. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 136: 1136–1147, 2009. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 30: 377–386, 2009. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]
- 13.Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev 19: 877–890, 2005. doi: 10.1101/gad.1295405. [DOI] [PubMed] [Google Scholar]
- 14.He XC, Yin T, Grindley JC, Tian Q, Sato T, Tao WA, Dirisina R, Porter-Westpfahl KS, Hembree M, Johnson T, Wiedemann LM, Barrett TA, Hood L, Wu H, Li L. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet 39: 189–198, 2007. doi: 10.1038/ng1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia L, Vianna CR, Fukuda M, Berglund ED, Liu C, Tao C, Sun K, Liu T, Harper MJ, Lee CE, Lee S, Scherer PE, Elmquist JK. Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun 5: 3878, 2014. doi: 10.1038/ncomms4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 11: 1173–1179, 2005. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 17.Malvin NP, Seno H, Stappenbeck TS. Colonic epithelial response to injury requires Myd88 signaling in myeloid cells. Mucosal Immunol 5: 194–206, 2012. doi: 10.1038/mi.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miguel JC, Maxwell AA, Hsieh JJ, Harnisch LC, Al Alam D, Polk DB, Lien CL, Watson AJ, Frey MR. Epidermal growth factor suppresses intestinal epithelial cell shedding through a MAPK-dependent pathway. J Cell Sci 130: 90–96, 2017. doi: 10.1242/jcs.182584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mummert ME, Mohamadzadeh M, Mummert DI, Mizumoto N, Takashima A. Development of a peptide inhibitor of hyaluronan-mediated leukocyte trafficking. J Exp Med 192: 769–779, 2000. doi: 10.1084/jem.192.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nomura T, Chang HY, Lu R, Hankin J, Murphy RC, Schuster VL. Prostaglandin signaling in the renal collecting duct: release, reuptake, and oxidation in the same cell. J Biol Chem 280: 28424–28429, 2005. doi: 10.1074/jbc.M408286200. [DOI] [PubMed] [Google Scholar]
- 22.O’Neill LAJ. A feed-forward loop involving hyaluronic acid and toll-like receptor-4 as a treatment for colitis? Gastroenterology 137: 1889–1891, 2009. doi: 10.1053/j.gastro.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 23.Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med 8: 289–293, 2002. doi: 10.1038/nm0302-289. [DOI] [PubMed] [Google Scholar]
- 24.Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci USA 102: 99–104, 2005. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118: 229–241, 2004. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Reinhart WH, Müller O, Halter F. Influence of long-term 16,16-dimethyl prostaglandin E2 treatment on the rat gastrointestinal mucosa. Gastroenterology 85: 1003–1010, 1983. doi: 10.1016/S0016-5085(83)80064-X. [DOI] [PubMed] [Google Scholar]
- 27.Riehl TE, Alvarado D, Ee X, Zuckerman A, Foster L, Kapoor V, Thotala D, Ciorba MA, Stenson WF. Lactobacillus rhamnosus GG protects the intestinal epithelium from radiation injury through release of lipoteichoic acid, macrophage activation and the migration of mesenchymal stem cells. Gut 68: 1003–1013, 2019. doi: 10.1136/gutjnl-2018-316226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riehl TE, Ee X, Stenson WF. Hyaluronic acid regulates normal intestinal and colonic growth in mice. Am J Physiol Gastrointest Liver Physiol 303: G377–G388, 2012. doi: 10.1152/ajpgi.00034.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riehl TE, Santhanam S, Foster L, Ciorba M, Stenson WF. CD44 and TLR4 mediate hyaluronic acid regulation of Lgr5+ stem cell proliferation, crypt fission, and intestinal growth in postnatal and adult mice. Am J Physiol Gastrointest Liver Physiol 309: G874–G887, 2015. doi: 10.1152/ajpgi.00123.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roche KC, Gracz AD, Liu XF, Newton V, Akiyama H, Magness ST. SOX9 maintains reserve stem cells and preserves radioresistance in mouse small intestine. Gastroenterology 149: 1553–1563.e10, 2015. doi: 10.1053/j.gastro.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469: 415–418, 2011. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459: 262–265, 2009. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 33.Sehgal A, Donaldson DS, Pridans C, Sauter KA, Hume DA, Mabbott NA. The role of CSF1R-dependent macrophages in control of the intestinal stem-cell niche. Nat Commun 9: 1272, 2018. doi: 10.1038/s41467-018-03638-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shao J, Sheng H. Amphiregulin promotes intestinal epithelial regeneration: roles of intestinal subepithelial myofibroblasts. Endocrinology 151: 3728–3737, 2010. doi: 10.150/en.2010-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherman L, Sleeman J, Herrlich P, Ponta H. Hyaluronate receptors: key players in growth, differentiation, migration and tumor progression. Curr Opin Cell Biol 6: 726–733, 1994. doi: 10.1016/0955-0674(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 36.Sokolowska M, Chen LY, Eberlein M, Martinez-Anton A, Liu Y, Alsaaty S, Qi HY, Logun C, Horton M, Shelhamer JH. Low molecular weight hyaluronan activates cytosolic phospholipase A2α and eicosanoid production in monocytes and macrophages. J Biol Chem 289: 4470–4488, 2014. doi: 10.1074/jbc.M113.515106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stenson WF. Toll-like receptors and intestinal epithelial repair. Curr Opin Gastroenterol 24: 103–107, 2008. doi: 10.1097/MOG.0b013e3282f44a2a. [DOI] [PubMed] [Google Scholar]
- 38.Sun RC, Diaz-Miron JL, Choi PM, Sommovilla J, Guo J, Erwin CR, Warner BW. Both epidermal growth factor and insulin-like growth factor receptors are dispensable for structural intestinal adaptation. J Pediatr Surg 50: 943–947, 2015. doi: 10.1016/j.jpedsurg.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki A, Sekiya S, Gunshima E, Fujii S, Taniguchi H. EGF signaling activates proliferation and blocks apoptosis of mouse and human intestinal stem/progenitor cells in long-term monolayer cell culture. Lab Invest 90: 1425–1436, 2010. doi: 10.1038/labinvest.2010.150. [DOI] [PubMed] [Google Scholar]
- 40.Taylor KR, Yamasaki K, Radek KA, Di Nardo A, Goodarzi H, Golenbock D, Beutler B, Gallo RL. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll-like receptor 4, CD44, and MD-2. J Biol Chem 282: 18265–18275, 2007. doi: 10.1074/jbc.M606352200. [DOI] [PubMed] [Google Scholar]
- 41.Tessner TG, Cohn SM, Schloemann S, Stenson WF. Prostaglandins prevent decreased epithelial cell proliferation associated with dextran sodium sulfate injury in mice. Gastroenterology 115: 874–882, 1998. doi: 10.1016/S0016-5085(98)70259-8. [DOI] [PubMed] [Google Scholar]
- 42.Tessner TG, Muhale F, Riehl TE, Anant S, Stenson WF. Prostaglandin E2 reduces radiation-induced epithelial apoptosis through a mechanism involving AKT activation and bax translocation. J Clin Invest 114: 1676–1685, 2004. doi: 10.1172/JCI2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker MR, Brown SL, Riehl TE, Stenson WF, Stappenbeck TS. Growth factor regulation of prostaglandin-endoperoxide synthase 2 (Ptgs2) expression in colonic mesenchymal stem cells. J Biol Chem 285: 5026–5039, 2010. doi: 10.1074/jbc.M109.032672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamaoka T, Yan F, Cao H, Hobbs SS, Dise RS, Tong W, Polk DB. Transactivation of EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc Natl Acad Sci USA 105: 11772–11777, 2008. doi: 10.1073/pnas.0801463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu S, Gao N. Compartmentalizing intestinal epithelial cell toll-like receptors for immune surveillance. Cell Mol Life Sci 72: 3343–3353, 2015. doi: 10.1007/s00018-015-1931-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng L, Riehl TE, Stenson WF. Regulation of colonic epithelial repair in mice by Toll-like receptors and hyaluronic acid. Gastroenterology 137: 2041–2051, 2009. doi: 10.1053/j.gastro.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]