

ABSTRACT
The mammalian target of rapamycin and the integrated stress response are central cellular hubs regulating translation upon stress. The precise proteins and pathway specificity of translation targets of these pathways remained largely unclear. We recently described a new method for quantitative translation proteomics and found that both pathways control translation of the same sets of proteins.
KEYWORDS: Translation proteomics, proteostasis, mTOR, ISR, integrated stress response, translatome, stress
Measurements of protein dynamics are a useful tool to study rapid cellular changes introduced by various sorts of stimuli. Those changes are often masked in classical proteome measurements, hiding their potential for providing direct evidence of regulatory mechanisms resulting in the studied phenotype. However, protein dynamics are rarely monitored and data for most cellular stresses and modulations remains lacking. This is largely due to the limited number of methodologies available to quantify protein dynamics on a system-wide scale. In addition, many available techniques suffer from limited resolution, depth, or technical bias. Thus, new technologies and advances in proteomics approaches are needed to drive this important area of research.
Protein homeostasis is a tightly and rapidly regulated network balancing synthesis and degradation. The two central cellular hubs are the integrated stress response (ISR), causing translation attenuation and extensive remodulation of transcription upon activation, and the mammalian target of rapamycin complex 1 (mTORC1), inactivation of which inhibits translation, induces autophagy, and alters transcription.1,2 Both pathways integrate numerous stimuli emerging from a range of stresses and cell environmental conditions and ultimately result in broad remodeling of cells. Despite their central roles, the precise set of their specific translation targets had been missing. Their study is particularly difficult as the ISR and mTORC1 integrate a range of cellular stresses and conditions and are thus highly sensitive to any perturbation caused by the experimental method.
To quantify changes in the translatome, two approaches are commonly used 1) ribosome profiling (Ribo-seq), based on sequencing of mRNA pieces protected by ribosomes, 2) proteomics approaches following labeling of newly synthesized proteins to quantify translation. Both methods pose technical challenges. When monitoring conditions with strong global translation effects, such as for the ISR or mTORC1, Ribo-seq suffers from an extensive normalization bias.3 Proteomics approaches however are challenged by the low rate of newly synthesized proteins relative to the preexisting pool of (old) proteins.4,5 To overcome this issue, translation proteomics are commonly carried out using incorporation of click-reactive reagents, such as the non-natural amino acid azido-homoalanine (AHA) or puromycin, to allow biotinylation and enrichment of newly synthesized proteins before analysis.6 However, these compounds themselves cause cell stress and affect translation: 1) AHA is a methionine analogue incorporated into nascent changes through tRNA charging. To ensure high-level incorporation, cells are typically starved for methionine before addition of AHA, causing cell starvation and resulting in translation attenuation via mTORC1 inhibition.2 In addition, tRNA charging with AHA is ~400 fold less efficient than with methionine.7 Uncharged tRNAs are sensed by the eukaryotic translation initiation factor 2 alpha kinase 4 (EIF2AK4, also known as GCN2), causing translation inhibition via ISR activation,1 thereby affecting the system studied. 2) Puromycin is a translation inhibitor prematurely terminating protein synthesis leading to the release of truncated proteins. Thus, puromycin labeling directly affects translation itself.
To allow monitoring translation without perturbing the system, we used pulsed stable isotope labeling of amino acids in cell culture (SILAC, i.e. heavy amino acids) labeling to mark new proteins.8 These heavy amino acids are biologically indistinguishable from their light counterpart and have no effect on cellular behavior. However, they cannot be enriched, which has largely prevented the use of pulse-SILAC labeling for measuring acute translation changes, as the intensity (amount) of newly synthesized versus old proteins is too low to be reasonably detectable by mass spectrometry with sufficient depth and accuracy.4,5 To overcome this problem, we developed multiplexed enhanced Protein Dynamics (mePROD) proteomics, combining pulsed-SILAC and tandem mass tag (TMT) multiplexing,9 with the addition of spike-ins specifically enhancing the signal of newly synthesized peptides to allow their determination.5 Overall, this equipped us with a method capable of quantifying translation of thousands of proteins in multiplexed experiment with up to nine samples (14 for 16-plex TMTpro), with small input requirements (e.g. <100,000 HeLa cells), and short required labeling times (i.e. minutes).
The ISR and mTORC1 both target translation of capped mRNAs. Nevertheless, the specific groups of proteins whose translation is inhibited by these pathways had been broadly assumed to be separate, despite their similar features in vivo.10 We applied mePROD translation proteomics to conditions activating the ISR or inhibiting mTORC1.5 Measured global translation rates reflected the extensive reduction observed by other unbiased methods. A large set of proteins showed significantly reduced relative translation rates. Strikingly, translation targets of the ISR and mTORC1 extensively overlapped, revealing that both pathways target the same translational processes (Figure 1). Surprisingly, our analyses also revealed that not pathway, but intrinsic features drive target specificity. Translation of a fraction of proteins is reduced under mild global translation inhibition, while others (mainly housekeeping proteins) are largely resistant to translation attenuation. This implies some features within the mRNA sequence, or specific proteins binding to these, that define how quickly or slowly translation of mRNAs is reduced upon stress. What these features are will be an important future question to address.
Figure 1.
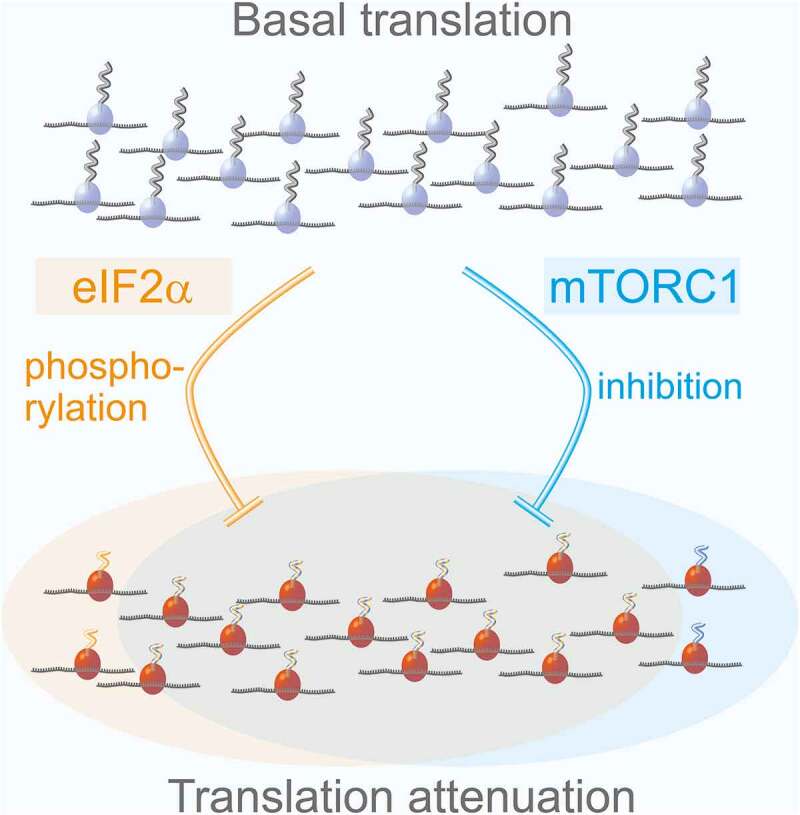
Overlapping control of translation by the ISR and mTORC1.
In summary, we developed a proteomics method to measure highly acute changes in translation by boosting the signal of interest. We employed this system to characterize target specificity of ISR or mTORC1-driven translation attenuation. Our analyses revealed the sets of proteins, for which the ISR or mTORC1 inhibit translation. Strikingly, both pathways largely regulated the same set of proteins. Instead, translation rates were dependent on global protein translation inhibition. Further work will be required to determine which elements or cellular components drive this behavior and what the functional cellular consequences are.
Funding Statement
C.M. was supported by the European Research Council (ERC StG 803565), the Emmy Noether Program (DFG, MU 4216/1-1), and the Johanna Quandt Young Academy at Goethe.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Pakos‐Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM.. 2016. The integrated stress response. EMBO Rep. 17:1–2. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Appenzeller-Herzog C, Hall MN. 2012. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 22(5):274–282. doi: 10.1016/j.tcb.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Chen K, Hu Z, Xia Z, Zhao D, Li W, Tyler JK. 2015. The Overlooked Fact: fundamental Need for Spike-In Control for Virtually All Genome-Wide Analyses. Mol. Cell. Biol. 36:662–667. doi: 10.1128/MCB.00970-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagert JD, Xie YJ, Sweredoski MJ, et al. 2014. Quantitative, Time-Resolved Proteomic Analysis by Combining Bioorthogonal Noncanonical Amino Acid Tagging and Pulsed Stable Isotope Labeling by Amino Acids in Cell Culture. Mol Cell Proteomics. 13:1352–1358. doi: 10.1074/mcp.M113.031914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klann K, Tascher G, Münch C.2020. Functional Translatome Proteomics Reveal Converging and Dose-Dependent Regulation by mTORC1 and eIF2α. Mol Cell. 77(913–925.e4):913–925.e4. doi: 10.1016/j.molcel.2019.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM. 2006. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. U. S. A. 103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiick KL, Saxon E, Tirrell DA, Bertozzi CR. 2002. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl. Acad. Sci. U. S. A. 99(1):19–24. doi: 10.1073/pnas.012583299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwanhäusser B, Gossen M, Dittmar G, Selbach M. 2009. Global analysis of cellular protein translation by pulsed SILAC. Proteomics. 9(1):205–209. doi: 10.1002/pmic.200800275. [DOI] [PubMed] [Google Scholar]
- 9.Welle KA, Zhang T, Hryhorenko JR, Shen S, Qu J, Ghaemmaghami S. 2016. Time-resolved Analysis of Proteome Dynamics by Tandem Mass Tags and Stable Isotope Labeling in Cell Culture (TMT-SILAC) Hyperplexing. Mol Cell Proteomics. 15:3551–3563. doi: 10.1074/mcp.M116.063230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wengrod JC, Gardner LB. 2015. Cellular adaptation to nutrient deprivation: crosstalk between the mTORC1 and eIF2α signaling pathways and implications for autophagy. Cell Cycle. 14(16):2571–2577. doi: 10.1080/15384101.2015.1056947. [DOI] [PMC free article] [PubMed] [Google Scholar]