

Abstract
Peripheral T-cell lymphomas are a heterogeneous group of rare diseases with an aggressive behavior and dismal prognosis. Their classification is complex and still evolving, and several biomolecular markers now help refine the prognosis of specific disease entities, although still have limited impact in tailoring the treatment. First-line treatment strategies can cure only a minority of patients and relapsed-refractory disease still represents the major cause of failure. Frontline autologous transplantation may have an impact in the consolidation of response; however, its role is still questioned as far as complete responses obtained after induction chemotherapy are concerned. Newer drugs are now being evaluated in clinical trials, but effective salvage strategies for those who experience treatment failures are lacking. Here we review and discuss the most controversial aspects of diagnosis and treatment of peripheral T-cell lymphomas.
Introduction
Peripheral T cell lymphomas (PTCL) comprise a heterogeneous group of malignancies derived from mature T or natural killer (NK) cells, representing less than 10% of all non-Hodgkin lymphomas. With few exceptions, PTCL usually have an aggressive course. There are about 30 PTCL entities in the current World Health organization (WHO) classification of lymphomas, grouped according to their clinical presentation into predominantly leukemic, or as primarily nodal, extranodal or cutaneous diseases.1 Diagnosing and managing PTCL is always challenging: (i) it is still unclear whether certain biomarkers found within the neoplastic tissue can help us modify (ie, tailor) our approach to these diseases; (ii) responses to first-line treatment are still unsatisfactory, given that PTCL frequently affect elderly patients; (iii) we are still unsure about the impact of frontline consolidation with autologous stem cell transplantation (ASCT) in patients achieving a complete response (CR); (iv) although several new drugs are emerging, we still lack an effective salvage strategy for relapsing patients or those refractory to chemotherapy.
This work is addressed to clarify the most controversial aspects in the management of PTCL by presenting the current diagnostic and treatment standards, as well as shedding light on potential new drugs.
Biopathology
Novel data derived from high-throughput genomic studies have contributed to improve the definition of PTCL entities (Table 1) and better understand their pathogenesis.2 Mutations and copy number variations frequently target different classes of epigenetic modifiers,3,4,5,6 T-cell receptor (TCR) and co-receptors signaling pathways7,8,9,10,11 and, components or regulators of the Janus kinase (JAK) signal transducer and activator of transcription (STAT) pathway.12 Besides a few variants characteristic of certain entities,7,13 there is overlap in the mutational landscapes of different entities. Immune evasion has also emerged as another oncogenic mechanism in PTCL. Programmed death (PD) ligand 1 (PD-L1) is expressed at variable frequencies,14,15,16,17 suggesting that PD-1 blockade may represent an efficient therapy in some patients; however, since PD-1 also acts as a tumor suppressor which is inactivated in a fraction of PTCL, PD-1 checkpoint inhibition may also potentially lead to unwanted effects.18,19 Constitutive genetic background may confer susceptibility to PTCL development, as suggested by HLA associations to enteropathy-associated T-cell lymphoma. This was also demonstrated by the recent finding of germline HAVCR2 mutations altering TIM-3 in subcutaneous panniculitis-like T-cell lymphomas with hemophagocytic-lymphohistiocytic syndromes.20
Table 1.
Main nodal and extranodal mature T-cell neoplasms according to the 2017 WHO classification of lymphomas, with summary of their main characteristic (cell of origin, immunophenotype and genetic alterations) and novelties in comparison to the previous edition).
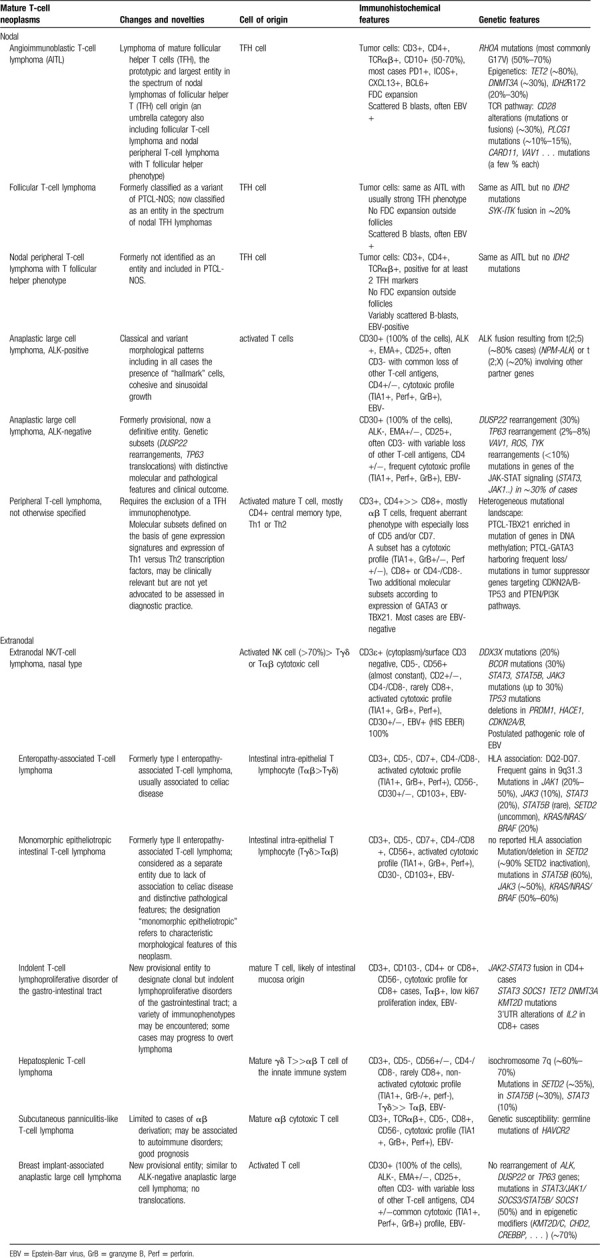
Cell of origin is a major determinant of PTCL biology. Neoplasms of innate lymphoid cells (NK, NK-like/T and γδ T-cells) are predominantly extranodal, cytotoxic and highly aggressive. Lymphomas of the adaptive immune system (derived from αβ CD4+ or CD8+ T-cells) constitute the majority of PTCL and often involve lymph nodes.
Nodal lymphomas of T follicular helper (TFH) derivation
Derivation from CD4+ TFH cells is the defining feature of the largest group of nodal lymphomas which includes angioimmunoblastic T-cell lymphoma (AITL), follicular T-cell lymphoma and nodal lymphoma with a TFH phenotype. AITL which typically manifests with systemic symptoms and biological abnormalities, consists of a polymorphous infiltrate comprising TFH neoplastic cells within an abundant polymorphous microenvironment associated with proliferation of venules and follicular dendritic cells.21 In addition to sharing a TFH immunophenotype and gene expression signature,22 these lymphomas disclose a homogeneous mutational landscape that recapitulates a multi-step oncogenic process. This typically consists of epigenetic deregulation such as TET2 ± DNMT3A mutations (in up to 80% of the cases), often occurring at early stages in hematopoietic progenitors.3,13 In addition, there are second-hit mutations including a hotspot RHOA G17V mutation (50%–80% of cases) and other gain-of-function mutations targeting the TCR signaling pathway (PLCG1, CD28, PIK3 components, CARD11).7,10 Moreover, fusions involving SYK, ITK, CD28, CTLA4 or CD28 and ICOS are detected at lower frequency.23 In addition, IDH2 R172 mutations resulting in production of an oncometabolite are found in 25-30% of AITL (Fig. 1).24 Transgenic mouse models with expression of RHOA G17V in the T-cell compartment demonstrated the role of RHOAG17V in TFH differentiation and in inducing autoimmunity.25 However, additional TET2 inactivation is required for lymphoma development, and these mouse tumors are dependent on ICOS/PIK3/MTOR signaling.26
Figure 1.
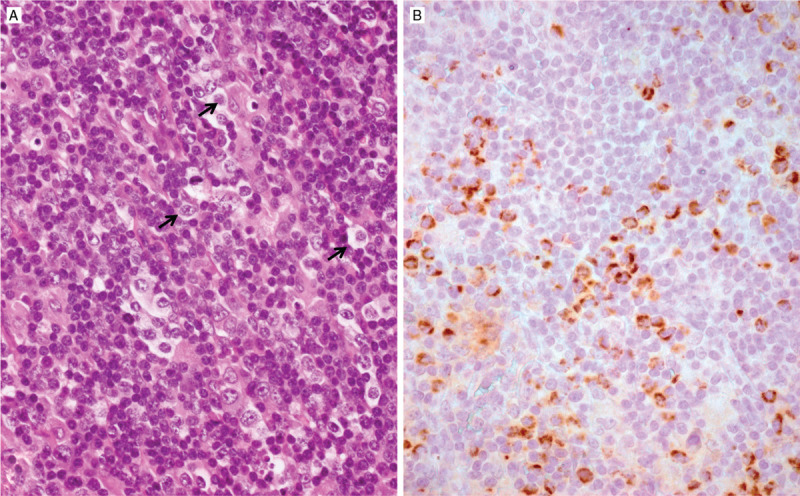
Angioimmunoblastic T-cell lymphoma (A) comprising the presence of scattered neoplastic cells with an abundant cytoplasm (arrows) which by immunohistochemistry (B) show cytoplasmic granular staining with an antibody specific for the R172K variant of IDH2, reflecting an IDH2 mutation.
Anaplastic large cell lymphomas (ALCL)
ALCL have in common several features: large-cell anaplastic morphology, strong CD30 expression and frequent phospho-STAT3 activation (Fig. 2, panels A–C). Systemic cases such as anaplastic lymphoma kinase (ALK)-positive and ALK-negative represent 15% to 20% of non-cutaneous PTCL. ALK-positive ALCL occurs mainly in children and young adults having an excellent overall prognosis. ALK fusion proteins resulting from the fusion of ALK to various gene partners, most commonly NPM1 (nucleophosmin), drive lymphomagenesis by several mechanisms, among which activation of STAT3 plays a prominent role.27 ALK-negative ALCL tend to occur in older patients and are genetically heterogeneous. Cases with rearrangement of the DUSP22 locus@6p25 (one third of the cases) have unique molecular features – lack of STAT3 activation, DNA hypomethylation and an immunogenic phenotype.17 They also frequently harbor a hotspot MSC E116K mutation in the musculin gene driving cell cycle progression that can be targeted with BET inhibitors.28 DUSP22-rearranged ALCL have a good outcome in most series.29,30,31,32 In contrast, the rare ALK-negative ALCL with TP63 rearrangements has very poor outcomes.30 A subgroup of ALK-negative ALCL have STAT3 activation resulting from rearrangements of other tyrosine kinase genes such as TYK2, ROS1, FRK or activating mutations of JAK or STAT3. 17 In addition, downregulated Wiskott-Aldrich syndrome protein (WASP) was recently identified as another oncogenic pathway in ALCL.33
Figure 2.
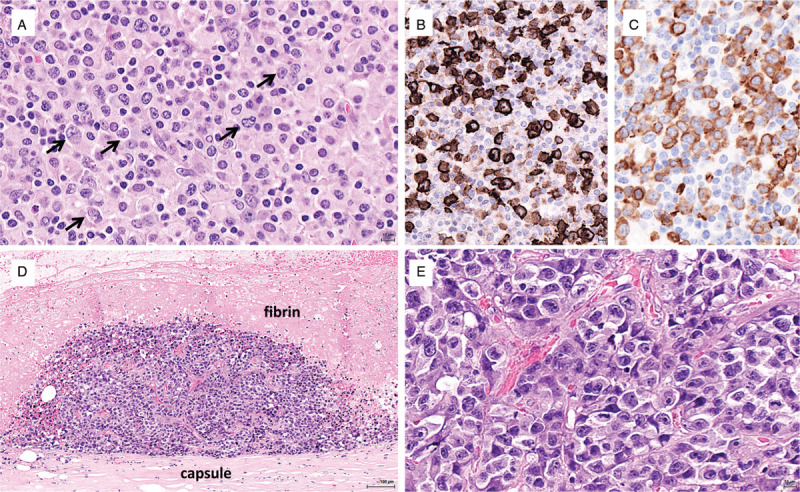
Anaplastic large cell lymphomas. (A–C) Lymphohistiocytic variant of ALK-positive ALCL comprising a polymorphous infiltrate rich in histiocytes and plasma cells with large atypical cells (arrows) which are strongly positive for CD30 (B) and show a cytoplasmic staining for ALK (C) indicative of a translocation other than NPM-ALK. (D-E) Breast implant-associated ALCL: in situ lymphoma on the inner surface of the periprosthetic capsule (D), comprising numerous large anaplastic cells (E).
Breast implant-associated (BIA) ALCL, a peculiar form of ALK-negative ALCL, is a rare complication of breast implants (Fig. 2, panels D-E).34 Most cases confined to the periprosthetic effusion and capsule (seroma or in situ lymphoma) have excellent outcomes; a minority of patients present with a tumor mass, which is an adverse prognostic factor. BIA-ALCL does not harbor recurrent translocations found in other ALK-negative ALCL; conversely, activation of STAT3 is common and recurrent mutations of STAT3, JAK1, or SOCS1 have been reported.35 Chronic inflammation elicited by the texture of the implant, silicone-derived products or bacteria adherent to the prosthesis likely play a role in triggering polyclonal lymphocyte activation and expansion through the release of cytokines like IL6 and IL10.36 Supervening genetic alterations would represent an additional step in the transformation process to a malignant monoclonal proliferation.
Peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS)
PTCL-NOS is a heterogeneous group of cases not fulfilling the criteria for more specific entities. Some cases are associated with Epstein-Barr virus infection. Gene expression profiling has unraveled molecular subgroups with differential expression of master T helper 1 (TH1) or TH2 cell-associated transcription factors (TBX21 and GATA3, respectively) and their associated target genes. GATA-3-positive PTCL-NOS and those with a cytotoxic phenotype are associated with worse outcomes compared with TBX21-positive tumors.37,38 Genetically, PTCL-GATA3 exhibit higher genomic complexity with frequent loss or mutation of tumor suppressor genes targeting the CDKN2A/B-TP53 axis and PTEN/PI3K pathway. PTCL-TBX2 have fewer copy number aberrations and are enriched in mutations of genes regulating DNA methylation.39
Frontline treatment
The standard of care for the last 30 years has been cyclophosphamide, doxorubicin, vincristine, prednisolone (CHOP) with or without additional etoposide (CHOEP). A meta-analysis of the outcome of PTCL treated with anthracycline-based regimens demonstrated a 5-year overall survival of only 36.6%.40 Attempts have been made to improve on this poor outlook by: (i) adding newer drugs to the CHOP backbone (alemtuzumab, romidepsin, brentuximab vedotin); (ii) intensifying the regimen (DA-EPOCH); (iii) developing alternative regimens (eg, gemcitabine-based combinations).
Alternative regimens to CHOP
Retrospective data collection from the German High Grade NHL study group trials, in which a small number of PTCL were included, suggested benefit for addition of etoposide (CHOEP) for younger patients, especially those with ALCL.41 A single arm phase 2 study investigated the use of DA-EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide and doxorubicin).42 This small trial (41 patients) reported after very short follow-up (median 24 months) showed favorable results with 2-year progression free survival (PFS) and overall survival (OS) of 53% and 73%, respectively. A larger study is needed to confirm these results. The European Society of Medical Oncology guidelines recommend dose dense CHOEP as initial chemotherapy for younger patients with PTCL-NOS.43
Prospective randomized trials failed to show superiority over standard CHOP for addition of alemtuzumab (ACT trial)44 or use of an alternative regimen like GEMP (gemcitabine, methylprednisolone, cisplatin) (CR rate of 62% with CHOP compared with 46% for GEMP).45 The results of CHOP + romidepsin versus CHOP alone are awaited. A randomised trial comparing CHOP with CMED (cyclophosphamide, methotrexate, etoposide, dexamethasone) in 217 patients with PTCL-NOS in a single center in South America,46 showed improved CR rate, PFS and OS. However, there were a number of difficulties with the interpretation of the data since there was no central review of pathology or imaging, the patients were young and the control arm did not receive high dose therapy.
The only clearly positive study was ECHELON-2, a large randomized trial in 452 CD30+ PTCL comparing CHOP (6 courses) with CHP (i.e. with vincristine omitted) combined with brentuximab vedotin (BV).47 The results showed superior PFS for the BV-CHP combination. However, 75% of the patients in the study had ALCL and the benefit for the BV-CHP regimen was not statistically significant in the non-ALCL subtypes (72 PTCL-NOS and 54 AITL). Thus, there is insufficient evidence to recommend BV-CHP for CD30+ non-ALCL PTCL.
CNS involvement is rare (1.5% at presentation, 4%–8% at relapse) in PTCL-NOS and AITL48: CNS prophylaxis is not usually indicated, but it may be considered when >1 extranodal site is involved, given that risk of CNS relapse rises to 10% to 20%. Certain sites of disease such as paranasal sinus, skin, testis and gastro-intestinal tract are also associated with higher risk.
Extranodal NK/T-cell lymphoma merit a different approach: although a detailed description of the first line treatment in these rare disease entities is beyond the scopes of this paper, it is important to note that they seem to be quite sensitive to asparaginase-based regimens. In particular, the dexamethasone, methotrexate, ifosfamide, L-asparaginase, etoposide (SMILE) regimen has brought better outcomes than any anthracycline-containing regimen in this context.49 More recently, the dexamethasone, cisplatin, gemcitabine and pegaspargase (DDGP) regimen has demonstrated better efficacy over SMILE, now confirming its role as the standard first line approach in NK/T-cell lymphoma.50
Front line treatment: consolidation
International guidelines now recommend that consolidation with high dose therapy and ASCT in first remission should be considered for patients with chemo-sensitive PTCL-NOS, AITL and other rare subtypes, but not for ALK-positive ALCL.43 Both retrospective and prospective studies have suggested that patients may benefit from this strategy by prolonging PFS (3-year PFS of 40%–50%) and OS.51 A systematic review and meta-analysis of 27 publications covering 1368 patients reported a PFS rate of 33% (95% confidence interval 14%–56%) for the prospective studies analyzed.52 Results from the Comprehensive Oncology Measure in Peripheral T-cell Lymphoma Treatment study (COMPLETE study) have recently been reported. In this observational study that identified 119 patients with nodal PTCL in first CR, 36 underwent ASCT and 83 did not. In multivariate analyses, ASCT was independently associated with improved OS.53 However, these studies are inherently biased by the selection of better outcome patients. About one-third of patients in prospective trials failed to receive the planned ASCT either because they did not achieve a remission or due to early progression.51 Furthermore, some registry studies failed to show an advantage for ASCT, particularly if the analysis focused on patients in CR after induction. In a French study of over 500 patients, the 57% responders had the same outcome regardless of whether they received ASCT or not, with a 5-year OS and PFS of 60% and 45%, respectively, for both groups.54 Thus, there is still some controversy on the role of ASCT in first remission although it has been recognized that for some subtypes such as AITL this may be more beneficial.
Allogeneic SCT (alloSCT) is not recommended frontline other than for very rare subtypes with extremely poor outcome, such as hepatosplenic T cell lymphoma.43
Positron emission tomography (PET) in PTCL
18F-fluorodeoxyglucose (FDG) PET scan is not applied routinely for disease staging, although it has proven to be helpful in detecting FDG-avid nodal or extranodal lesions that can be missed by a CT scan evaluation.55 PET positivity found at the end of induction treatment56,57 and in patients who have received ASCT is a strong predictor of reduced survival58 and this seems particularly true for PTCL-NOS and AITL patients. On the contrary, uncertainty still exists on the role of interim PET evaluation during induction treatment. Some published experiences document that a negative interim scan (described in terms of the International Harmonization Project or applying the Deauville 5-point scale) has a favorable impact on OS59 and maybe on progression-free survival (PFS),60 possibly because PET-negative patients were more likely to receive consolidation therapy which contributed to their better survival rates. On the other hand, others show a lack of any prognostic value.56
Major controversies:
Who should get etoposide? Should non-ALCL CD30 + PTCL all get brentuximab vedotin? Who should get ASCT in first remission (in terms of disease subtype) and does level of response matter (CR vs PR)?
The treatment of elderly patients
PTCL remain a group of diseases most commonly affecting the elderly with a median age at onset of 67 years.61 Although some subtypes target younger age groups (eg, ALK-positive ALCL, median age of 41 years), the most frequent subtypes of PTCL-NOS, TFH-PTCL and AITL remain more common in patients over 60 years. However, patients in the older age group and those who have comorbidities and poor performance status are often under-represented in clinical trials. There is therefore very limited data available regarding the elements on which to base treatment decisions.
Registry data: the impact of age and performance status
Several epidemiological studies using registry data have all suggested a negative impact of age and comorbidities on patient outcomes. In the Swedish registry, two-thirds of patients were over the age of 60 years and of 694 patients 38% had significant comorbidities. A Charlson comorbidity score equal to or greater than 2 was associated with inferior PFS and OS, and even limited comorbidity had an impact on survival.62 In the recent refinement of the T-cell International Prognostic Index (IPI), performance status was the most important independent predictor of outcome.63 In a larger study (775 patients) based on data from Sweden, US and Canada, the outcome was stratified according to event-free survival at 24 months, showing dismal survival for the two-thirds of patients who progressed within 2 years (OS 11% at 5 years) as compared to those who were event-free (OS 78% at 5 years). Age over 60 had a negative impact on the good-risk group compared to younger patients. Other trials have also consistently shown age >60 to be an independent risk factor.64 Results may also be confounded by the fact that this group of less fit patients will rarely benefit from a consolidation with ASCT in first remission. In the ACT-1 trial,44 patients up to 65 years were initially consolidated with ASCT; however, due to treatment-related toxicity, the age limit for inclusion into this trial had to be reduced to 60 years.
CHOP or not?
CHOP remains the standard of care in the elderly, but it is well recognized that patients are more vulnerable to associated toxicities. Clearly, omitting anthracyclines may have advantages in reducing cardiac toxicity, which is an age-related risk. The utility of including anthracyclines has been questioned, with some reports showing no benefit. For example, the International Peripheral T-cell and Natural Killer/T-cell Lymphoma Study failed to show an advantage for the use of anthracycline-containing initial chemotherapy regimen in PTCL-NOS and AITL subtypes.65 Whilst in a series of 300 PTCL patients from the Mayo Clinic, improved PFS and OS were reported with anthracycline-containing regimens, even when the analysis was restricted to PTCL-NOS and AITL subtypes.66 In a retrospective review of German study group trials, the addition of etoposide to CHOP did not show improved results in patients over 60 years of age.41 The Swedish study reported that patients over 75 years old had no difference in outcome between those using curative multi-agent chemotherapy and low intensity regimens, with similar OS when adjusted for comorbidity index.62 A registry study from Thailand of 127 patients over the age of 60 years showed that two-thirds of patients received multi-agent chemotherapy, dropping to 34% in the over 75 year age group. With relatively short follow-up (17 months), in this study 2-year PFS and OS were 38% and 48%, respectively. Older age, poor performance status and absence of multi-agent chemotherapy were all associated with poor outcome in multivariable analysis. The choice of chemotherapy remained significant even after adjusting for age, comorbidity, performance status and IPI. The authors concluded that multi-agent chemotherapy (largely CHOP) should be given whenever possible and that a comprehensive geriatric assessment is useful.67 A Korean study in 44 patients over 70 or between 65 and 70 but with co-morbidities (median age 74) gave very similar outcome data. Patients received dose-attenuated CHOP (cyclophosphamide: 562.5 mg/m2, doxorubicin: 37.5 mg/m2, vincristine: 1.4 mg/m2, and prednisolone: 100 mg for five days; 25% reduced dose of cyclophosphamide and doxorubicin) as first-line therapy. The overall response rate was 61%, and 2-year PFS and OS were 37% and 47%, respectively. Three-quarters of patients experienced grade 3/4 neutropenia and there were 6 treatment-related deaths: although efficacy is not apparently reduced compared to full-dose CHOP, the toxicity remains significant in this age group.68
Elderly patients in clinical trials
Data from prospective randomized trials are scarce. The ACT-2 trial included 116 pts aged 61-80 years (median age 69 years) that were randomized to either CHOP or CHOP + alemtuzumab. Adding alemtuzumab to CHOP increased response rates but did not improve survival due to treatment related toxicity. However, feasibility of standard CHOP with a 3-year OS of 56% demonstrated the tolerability and efficacy of anthracyline-based treatment in the elderly.69 In the ECHELON-2 trial, patients with CD30+ PTCL up to the age of 80 were to receive either CHOP or CHP + BV: 139 patients older than 65 years were included, without experiencing undue toxicity. Therefore, anthracycline-based polychemotherapy remains the standard treatment for this group of patients.47
If anthracycline-based chemotherapy is not deemed feasible, there may be merit in exploring alternative treatment regimens in individual cases. Gemcitabine-based therapy can be effective and less toxic, as seen in diffuse large B-cell lymphoma with the use of R-GCVP.70 Monotherapy with novel agents, such as BV in ALCL, romidepsin, praletrexate and duvelisib, may have activity with less toxicity in carefully selected patients.
Major controversies:
Lack of data and randomized trials to indicate clearly the most suitable first-line approach. Anthracyclines or not? Full dose or dose-attenuated regimens? Are any older patients eligible for ASCT?
The conventional approach in relapsed and refractory patients
Disease refractoriness or relapse occurs in nearly two-thirds of patients with PTCL, except ALK-positive ALCL which will not be specifically discussed in this section. Median PFS and OS are known to be extremely poor in this setting, not exceeding 4 or 7 months, respectively.71 Of main concern is the fact that results are only marginally better for patients who receive chemotherapy at relapse (3.7 months for PFS and 6.7 months for OS) questioning a pure palliative approach for patients who cannot be offered a transplant procedure.
Approved single-agents
For the last 2 decades, phase 2 clinical trials have demonstrated that overall response (OR) and CR rates were consistently limited around 25% to 40% and 10% to 15% respectively for single agent romidepsin, pralatrexate, belinostat, BV, and others.72,73,74,75,76 Likewise, median PFS and OS usually did not exceed 6 and 14 months in these studies. The Food and Drug Administration (but not the European Medicines Agency) has granted approval for romidepsin (in 2011), pralatrexate (in 2009) and belinostat (in 2014) in relapsed/refractory PTCL after at least one previous line of treatment. Overall, all 3 agents showed similar disappointing long-term results, but specific subtypes such as AITL seemed to benefit the most from epigenetic modifying agents, although any subgroup analysis needs to be interpreted cautiously. With romidepsin used as monotherapy at a dose of 14 mg/m2 at day 1, 8, 15 of 28-day cycles, OR and CR rates reached 33% and 22% in AITL compared to 25% and 15% in PTCL in general. With belinostat used as monotherapy at a dose of 1000 mg/m2 from day 1 to 5 of 21-day cycles, OR reached 45% in AITL compared to 25% in PTCL-NOS. Although not approved in Europe, romidepsin and belinostat can be used in some countries based on a compassionate use program. Pralatrexate, administered intravenously at the dose of 30 mg/m2/week for 6 weeks, followed by 1 week of rest, until progressive disease or unacceptable toxicity, has showed significant activity in PTCL-NOS and ALCL patients in the pivotal PROPEL trial, with an OR rate of 32% and 35%, respectively.73 For this reason, it is considered the first salvage treatment in this context of patients, if available. Mucositis is the most relevant adverse event with pralatrexate: vitamin B12 and folate supplementation, along with dose reduction to 20 mg/m2 are required in patients experiencing drug-related mucosal toxicity.
Cyclosporine A is a therapeutic option in second or subsequent line especially for AITL patients who are not candidate to ASCT. A recently published review gathering 26 patients has reported an outstanding OR rate of 86% in heavily pretreated AITL cases, including patients who had failed a previous ASCT.77
Investigational drugs will be detailed in the last paragraph of this paper.
Conventional chemotherapy salvage regimen
If ASCT has not been performed frontline as a consolidation procedure, it is usually considered the standard of care for fit patients in the relapsed/refractory setting although no study has formally demonstrated its superiority over conventional therapy. The optimal salvage regimen before high dose therapy and transplant is still a matter of debate since no large randomized trial or compelling evidence has demonstrated a clear advantage for a peculiar regimen over another. Puig et al presented retrospectively collected data from 40 patients with refractory disease or in first relapse aiming for ASCT. Most patients were treated with DHAP (cisplatin, cytosine arabinoside, dexamethasone) or ESHAP (etoposide, methylprednisolone, cytarabine, cisplatin). OR rate was 63% with those regimens.78 The study from Mikesch et al retrospectively assessed the outcome of patients receiving either DexaBEAM (dexamethasone, carmustine, etoposide, cytarabine and melphalan; n = 16) vs ICE (ifosfamide, carboplatin and etoposide; n = 15) as first salvage regimen followed by ASCT. The OR and CR rates were higher with DexaBEAM than ICE (69% vs 20%, p = 0.01; and 38% vs 7%, p = not significant) although toxicity was also more common after Dexa-BEAM.79 The study from Yao et al reported a new salvage regimen for patients unsuitable for high dose therapy and ASCT combining gemcitabine, oxaliplatin and dexamethasone (GemOD) for up to 6 courses every 21 days.80 GemOD provided a 38% OR rate, which might appear disappointing, but comparison with other regimen for patients fit for high-dose therapy as in the aforementioned study from Mikesch et al is not possible. The Royal Marsden Hospital reported an OR rate of 69% with a regimen combining gemcitabine, cisplatin and methylprednisolone (GEMP-P) for 16 patients among who 15 were pretreated.81 To date, DHAP or DHAOx or DHAC (DHAP in which platine is replaced by oxaliplatin or carboplatin), ESHAP, GDP (gemcitabine, dexamethasone, cisplatin), GemOx (gemcitabine, oxaliplatin) or ICE (ifosfamide, carboplatin, etoposide) are the most frequently used regimen at relapse or in refractory disease worldwide.
Role of ASCT and allogeneic stem cell transplant (alloSCT)
Mostly retrospective studies or prospective non-randomized trials have been reported concerning the role of ASCT and alloSCT in PTCL. Extensive bibliography from previous reviews is reported elsewhere and will not be part of this chapter.82,83,84,85,86,87,88
Except for ALK-positive ALCL and for unfit patients, the National Comprehensive Cancer Network guidelines recommend ASCT, observation or clinical trial enrollment in first-line complete metabolic response in PTCL. The ESMO43 and the American Society for Blood and Marrow Transplantation89 guidelines recommend ASCT for partial or complete responders. However, recently published data from the Lymphoma Study Association (LYSA) suggest that in routine practice, roughly half of patients are not planned for ASCT in intention-to-treat, due to low consensus among the community on the real benefit associated with ASCT in first remission in the absence of randomized trial.54 Consequently, a substantial fraction of patients with relapsed/refractory PTCL does not undergo ASCT in first line and might be considered for the procedure in case of disease sensitive to conventional chemotherapy, approved single agents or investigational drugs. ASCT is therefore often considered the standard of care for eligible relapsed/refractory patients if they did not receive consolidation strategy in first line. However, as in newly diagnosed patients, no convincing data from randomized trials exist to support this strategy compared to no further consolidation after conventional chemotherapy or approved single agent like pralatrexate, romidepsin or belinostat. Indeed, most studies on ASCT in relapsed/refractory patients suffer from the following drawbacks: retrospective selection bias, mixed histologies, various salvage regimen, heterogeneous patient characteristics and response status before ASCT.
Concerning the role of alloSCT, recent data from the AATT trial (EudraCT Number: 2007-001052-39) in first line confirmed that a better control of disease over ASCT is obtained at the expense of greater toxicity, resulting in similar OS. In the relapsed/refractory setting, data from the literature are scarce and suffer from identical biases as for ASCT studies.
Major controversies:
Which is the best treatment to adopt for patients with PTCL (except ALK-positive ALCL) with a relapsed/refractory disease? Can we think about a disease-specific approach with any of the new agents (e.g. pralatrexate in PTCL-NOS, romidepsin versus belinostat in AITL)? Is there a role for ASCT if patients are considered fit enough and if not performed in first line? Can alloSCT be superior to ASCT in relapsed/refractory patients?
New experimental agents
Targeting epigenetic modifiers
PTCL harbor a multitude of genetic alterations affecting epigenetic regulators. While the functional consequences of many of these alterations remain poorly understood, the clinical activity of drugs like histone deacetylase inhibitors is still intriguing: durable responses with a median duration of 13 and up to 17 months can be achieved with belinostat and romidepsin, respectively, leading to FDA approval.90 Unfortunately, both belinostat and romidepsin showed overall response rates of only roughly 25% in relapsed/refractory PTCL and predictive biomarkers of response do not exist. However, taking advantage of their potential activity, multiple combination strategies including HDAC inhibitors are currently being tested in clinical trials, including a front-line phase 3 trial of romidepsin + CHOP (NCT01796002).91 Furthermore, novel derivatives of HDAC inhibitors like the alkylating deacetylase inhibitor molecule tinostamustine (EDO-S101) were developed recently and have now entered clinical trials including PTCL (NCT02576496).92
The high incidence of mutations affecting the TET family of DNA methylcytosine hydroxylases with subsequently increased DNA methylation, provides a rationale for hypomethylating agents (HMA) like 5-azacytidine. Indeed, treatment with 5-azacytidine induced sustained responses in AITL.93 Further clinical data will be needed, though, to assess the predictive role of mutations affecting TET2, IDH1/2 or DNMT3A for HMA, as reported for MDS and AML.94,95,96 Furthermore, novel combinations are on their way: in a recent phase 1 combination trial of 5-azacytidine and romidepsin, O’Connor et al reported OR and CR rates of 73% and 55%, respectively.97
Taken together, the frequency of genetic alterations affecting epigenetic modifiers and the clinical activity of HDAC inhibitors and hypomethylating agents is intriguing. However, as these drugs have pleiotropic effects, patient stratification and development of predictive biomarkers of response remain a major obstacle and clinical data on the safety and efficacy of combination strategies need to be awaited.
Emerging immunotherapeutic strategies
PTCL are commonly infiltrated by a variety of non-neoplastic immune cells, but sufficiently evade anti-tumor immune responses. From a therapeutic perspective, novel strategies targeting distinct mechanisms of immune-evasion are currently emerging.
The development of checkpoint inhibitors, targeting the interaction of PD-1 and PD-L1/PD-L2 with subsequent repression of T cell activation has revolutionized therapeutic options in multiple malignancies.98 In PTCL, increased expression of PD-L1 has been described in both malignant and stromal cells across various subtypes, suggesting therapeutic checkpoint-inhibition. Indeed, partial responses in PTCL were described in a phase 1b study with nivolumab.99 However, PD-1 was also recently described as a potential haploinsufficient tumor-suppressor and cases of rapid disease progression after PD-1 inhibitor therapy have been described, particularly in ATLL.19, 100 Thus, as robust clinical data are scarce, results of clinical studies including PD-1 inhibition are urgently needed to clarify the role of PD-1 checkpoint-inhibitors in T cell lymphomas.
Further targetable mechanisms of immune evasion recently emerged in TCL: CD47, a glycoprotein of the immunoglobulin superfamily providing a “don’t eat me” signal by binding to signal regulatory protein α (SIRPα) on macrophages has been found to be overexpressed in PTCL-NOS and cutaneous T cell lymphomas.101 Therapeutic antibodies have been designed to interrupt the CD47-SIRPα interaction. Of these, the CD47 decoy receptor signal regulatory protein αFc (SIRPαFc; TTI-621) recently showed activity in patients with Sézary syndrome.102 Similarly, the killer cell immunoglobulin-like receptor KIR3DL2 was found to be expressed in CTCL and IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody recruiting and activating NK cells showed activity in patients with relapsed or refractory CTCL.103 Notably, high expression levels were also found in subtypes of PTCL and the efficacy of IPH4102 in PTCL is currently being tested in a multi-cohort, phase 2 trial (TELLOMAK).
Ultimately, concepts for chimeric antigen receptor (CAR) T cells are being developed for PTCL. However, as pan T cell aplasia causes severe immunodeficiency, these concepts are challenged by the lack of specific antigens discriminating malignant and normal T cells. The mutually exclusive expression pattern of the T cell receptor β-chain constant domains 1 and 2 (TRBC1 and TRBC2), though, might be a promising approach to target neoplastic T cells harboring either TRBC1 or TRBC2 while preserving a substantial proportion of normal T cells.104
Targeting SYK and phosphatidyl-inositol 3-kinase (PI3K) pathways
The SYK signaling pathway plays a role in oncogenic transformation: experimental data suggest that SYK is an oncogenic driver in subsets of T cell lymphomas, being highly expressed (> 90% of cases of systemic and cutaneous ALCL, AITL, PTCL-NOS, mycosis fungoides) in its native form or uncommonly as a fusion protein with ITK.105
Cerdulatinib is an orally available, ATP-competitive inhibitor of SYK, JAK1, JAK3 and Tyk2, whose efficacy has been demonstrated in T cell lines of ATLL.106 Its role in relapsed/refractory PTCL and cutaneous T cell lymphomas (CTCL) has been evaluated in a phase 2a study in which it was administered orally at the dose of 30 mg bid in 28-day cycles until progression or intolerance. In 41 PTCL patients, the treatment produced an overall response rate of 34% according to the Lugano criteria (CR in 27% and PR in 7% of cases), whereas in 27 evaluable patients with CTCL the overall response rate was 26%, with a CR in 7% and a PR in 19% of cases. Overall responses were seen across multiple histologies, mainly in ATLL (33%), TFH/AITL (57%, with 50% of CR), ALCL (33%).107
PI3K inhibition blocks mitogen and prosurvival signals from tumor and microenvironment cells and concomitantly activates an anti-lymphoma immune response.
Duvelisib, an oral dual inhibitor of the isoforms PI3Kγ and PI3Kδ, was applied as single agent in a phase 1 trial involving 16 PTCL and 19 CTCL patients with relapsed/refractory disease. The drug was administered on a dose-escalation basis, starting from 25 mg bid up to 100 mg bid on a 28-day cycle and the maximum tolerated dose was 75 mg bid. In the PTCL population, the overall response rate was 50%, with 3 CR and 5 PR, all occurring within the first 2 to 4 months of treatment. Responses were seen across multiple histologies, including PTCL-NOS, and AITL. Median OS and PFS were 8.4 and 8.3 months, respectively. In the CTCL population, 6 patients obtained a PR, all within 4 months since treatment inception. Median PFS was 4.5 months, with 1-year PFS and OS being 26.5% and 78.9%, respectively. Transaminase elevation was the most relevant adverse event (57% of cases), with grade 3–4 toxicity described in 40% of patients.108
Based on these results and on an in vitro documented synergism, a phase 1 trial of duvelisib+romidepsin (arm A) and duvelisib+bortezomib (arm B) was initiated in relapsed/refractory PTCL and CTCL patients. Duvelisib was given at 25 mg bid, 50 mg bid or 75 mg bid, romidepsin at 10 mg/m2 on days 1, 8, 15 and bortezomib at 1 mg/m2 on days 1, 4, 8 and 11, on 28-day cycles. In expansion cohorts, duvelisib was given with romidepsin at 75 mg bid and at 25 mg bid with bortezomib. Thirty-eight patients in arm A and 31 in arm B were evaluable for response: the combination duvelisib+romidepsin produced an overall response rate of 55% (59% in PTCL and 45% in CTCL patients), with a CR rate of 24% (33% in PTCL and 0 in CTCL patients). AITL and PTCL-NOS appeared as the most sensitive entities, with 75% and 64% of OR rate, and 63% and 36% of CR rate, respectively. The combination duvelisib + bortezomib yielded an OR rate of 35% (44% in PTCL and 27% in CTCL patients), with a CR rate of 13% (25% in PTCL and 0 in CTCL patients). Incidence of transaminase elevation was interestingly much lower than with single agent duvelisib.109
Major controversies:
Which single agent is likely to be more promising? Can we think about combining experimental agents with approved target therapies?
Conclusions
PTCL are rare and heterogeneous diseases: their classification is still evolving, given the significant amount of knowledge in terms of biomolecular mechanisms gained in the last few years. First-line antracycline containing regimens are currently the uniformly accepted standard of care, albeit results are heterogeneous. ASCT is part of the frontline approach to PTCL, even though its consolidative role may be weakened in complete responders after induction. On the other hand, it is important to note that the majority of PTCL are diagnosed in elderly patients, for whom a recognized standard of care is still lacking. Newly approved drugs are capable of inducing relevant OR and CR rates in relapsed and refractory disease, although response durations are limited over time. This is why new molecules are eagerly needed. More specifically, multiple novel concepts targeting immune-escape mechanisms and key pro-survival pathways are emerging for T cell lymphomas, needing to be tested in clinical trials. Alongside, thorough correlative studies are warranted to understand mechanisms of response and resistance and to incorporate these approaches into rational combination strategies.
Disclosures
Emmanuel Bachy: Employment/consultation for Gilead, Novartis, Amgen, Roche, Janssen, Sanofi. Alessandro Broccoli: Nothing to disclose. Claire Dearden: Nothing to disclose. Laurence de Leval: Nothing to disclose. Philippe Gaulard: Employment/consultation for Takeda, Gilead. Grants/pending grants from Innate Pharma, Takeda. Raphael Koch: Employment with University Medical Center of Goettingen (academic). Grants/pending grants from German Cancer Aid Foundation. Franck Morschhauser: Advisory boards for Celgene, Roche, Epizyme, Giliead, AbbVie. Honoraria from scientific lectures for Roche, Celgene, Janssen. Lorenz Trümper: Employment/consultation with University of Göttingen. Grants/pending grants from EU Grant Transcan Program TCLymph. Pier Luigi Zinzani: Advisory boards for Verastem, Takeda, Gilead, Celltrion, Roche, Eusapharma, Kirin Kyowa, MEI Pharma, Janssen, Servier.
Footnotes
Citation: Bachy E, Broccoli A, Dearden C, de Leval L, Gaulard P, Koch R, Morschhauser F, Trümper L, Zinzani PL. Controversies in the Treatment of Peripheral T-cell Lymphoma. HemaSphere, 2020;4:4:(e461). http://dx.doi.org/10.1097/HS9.0000000000000461
References
- 1. WHO classification of tumours of the haematopoietic and lymphoid tissues. (Revised 4th, edition) Lyon: IARC; 2017. [Google Scholar]
- 2. Lemonnier F, Gaulard P, de Leval L. New insights in the pathogenesis of T-cell lymphomas. Curr Opin Oncol. 2018;30:277–284. [DOI] [PubMed] [Google Scholar]
- 3. Lemonnier F, Couronne L, Parrens M, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120:1466–1469. [DOI] [PubMed] [Google Scholar]
- 4. Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012;366:95–96. [DOI] [PubMed] [Google Scholar]
- 5. Roberti A, Dobay M, Bisig B, et al. Type II Enteropathy-Associated T-Cell Lymphoma Features a Unique Genomic Profile with Highly Recurrent SETD2 Alterations. Nat Commun. 2016;7:12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McKinney M, Moffitt AB, Gaulard P, et al. The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov. 2017;7:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Palomero T, Couronne L, Khiabanian H, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46:166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kataoka K, Nagata Y, Kitanaka A, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47:1304–1315. [DOI] [PubMed] [Google Scholar]
- 9. Rohr J, Guo S, Huo J, et al. Recurrent activating mutations of CD28 in peripheral T-cell lymphomas. Leukemia. 2016;30:1062–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vallois D, Dobay MP, Morin RD, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood. 2016;128:1490–1502. [DOI] [PubMed] [Google Scholar]
- 11. Abate F, da Silva-Almeida AC, Zairis S, et al. Activating mutations and translocations in the guanine exchange factor VAV1 in peripheral T-cell lymphomas. Proc Natl Acad Sci U S A. 2017;114:764–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kucuk C, Jiang B, Hu X, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat Commun. 2015;6:6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sakata-Yanagimoto M, Enami T, Yoshida K, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:171–175. [DOI] [PubMed] [Google Scholar]
- 14. Muhamad H, Suksawai N, Assanasen T, et al. Programmed Cell Death 1 and programmed cell death ligands in extranodal natural Killer/T Cell lymphoma: expression pattern and potential prognostic relevance. Acta Haematol. 2020;143:78–88. [DOI] [PubMed] [Google Scholar]
- 15. Song TL, Nairismagi ML, Laurensia Y, et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood. 2018;132:1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Asano N, Miyoshi H, Kato T, et al. Expression pattern of immunosurveillance-related antigen in adult T cell leukaemia/lymphoma. Histopathology. 2018;72:945–954. [DOI] [PubMed] [Google Scholar]
- 17. Luchtel RA, Dasari S, Oishi N, et al. Molecular profiling reveals immunogenic cues in anaplastic large cell lymphomas with DUSP22 rearrangements. Blood. 2018;132:1386–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wartewig T, Ruland J. PD-1 tumor suppressor signaling in T Cell lymphomas. Trends Immunol. 2019;40:403–414. [DOI] [PubMed] [Google Scholar]
- 19. Ratner L, Waldmann TA, Janakiram M, et al. Rapid progression of adult T-Cell leukemia-lymphoma after PD-1 inhibitor therapy. N Engl J Med. 2018;378:1947–1948. [DOI] [PubMed] [Google Scholar]
- 20. Gayden T, Sepulveda FE, Khuong-Quang DA, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nat Genet. 2018;50:1650–1657. [DOI] [PubMed] [Google Scholar]
- 21. de Leval L, Gisselbrecht C, Gaulard P. Advances in the understanding and management of angioimmunoblastic T-cell lymphoma. Br J Haematol. 2010;148:673–689. [DOI] [PubMed] [Google Scholar]
- 22. Dobay MP, Lemonnier F, Missiaglia E, et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica. 2017;102:e148–e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vallois D, Dupuy A, Lemonnier F, et al. RNA fusions involving CD28 are rare in peripheral T-Cell lymphomas and concentrate mainly in those derived from follicular helper T cells. Haematologica. 2018;103:e360–e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lemonnier F, Cairns RA, Inoue S, et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc Natl Acad Sci U S A. 2016;113:15084–15089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ng SY, Brown L, Stevenson K, et al. RhoA G17 V is sufficient to induce autoimmunity and promotes T cell lymphomagenesis in mice. Blood. 2018;132:935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cortes JR, Ambesi-Impiombato A, Couronne L, et al. RHOA G17 V Induces T follicular helper cell specification and promotes lymphomagenesis. Cancer Cell. 2018;33:259–273. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Werner MT, Zhao C, Zhang Q, et al. Nucleophosmin-anaplastic lymphoma kinase: the ultimate oncogene and therapeutic target. Blood. 2017;129:823–831. [DOI] [PubMed] [Google Scholar]
- 28. Luchtel RA, Zimmermann MT, Hu G, et al. Recurrent MSC (E116K) mutations in ALK-negative anaplastic large cell lymphoma. Blood. 2019;133:2776–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parilla Castellar ER, Jaffe ES, Said JW, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014;124:1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pedersen MB, Hamilton-Dutoit SJ, Bendix K, et al. DUSP22 and TP63 rearrangements predict outcome of ALK-negative anaplastic large cell lymphoma: a Danish cohort study. Blood. 2017;130:554–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hapgood G, Ben-Neriah S, Mottok A, et al. Identification of high-risk DUSP22-rearranged ALK-negative anaplastic large cell lymphoma. Br J Haematol. 2019;186:e28–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Drieux F, Ruminy P, Abdel-Sater A, et al. Defining the signatures of peripheral T-cell lymphoma, with a targeted 20-markers gene expression profiling assay (RT-MLPA). Haematologica. 2020;105:1582–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Menotti M, Ambrogio C, Cheong TC, et al. Wiskott-Aldrich syndrome protein (WASP) is a tumor suppressor in T cell lymphoma. Nat Med. 2019;25:130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Leval L. Breast implant-associated anaplastic large cell lymphoma and other rare T-cell lymphomas. Hematol Oncol. 2019;37 (Suppl 1):24–29. [DOI] [PubMed] [Google Scholar]
- 35. Letourneau A, Maerevoet M, Milowich D, et al. Dual JAK1 and STAT3 mutations in a breast implant-associated anaplastic large cell lymphoma. Virchows Arch. 2018;473:505–511. [DOI] [PubMed] [Google Scholar]
- 36. Laurent C, Haioun C, Brousset P, et al. New insights into breast implant-associated anaplastic large cell lymphoma. Curr Opin Oncol. 2018;30:292–300. [DOI] [PubMed] [Google Scholar]
- 37. Iqbal J, Wright G, Wang C, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123:2915–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Amador C, Greiner TC, Heavican TB, et al. Reproducing the molecular subclassification of peripheral T-cell Lymphoma-NOS by immunohistochemistry. Blood. 2019;134:2159–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heavican TB, Bouska A, Yu J, et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood. 2019;133:1664–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abouyabis AN, Shenoy PJ, Sinha R, et al. A systematic review and meta-analysis of front-line anthracycline-based chemotherapy regimens for peripheral T-Cell Lymphoma. ISRN Hematol. 2011;2011:623924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schmitz N, Trümper L, Ziepert M, et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood. 2010;116:3418–3425. [DOI] [PubMed] [Google Scholar]
- 42. Maeda Y, Nishimori H, Yoshida I, et al. Dose-adjusted EPOCH chemotherapy for untreated peripheral T-Cell Lymphomas: a multicenter phase II trial of west-JHOG PTCL0707. Haematologica. 2017;102:2097–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. d’Amore F, Gaulard P, Trümper L, et al. ESMO Guidelines Committee Peripheral T-cell lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26 (Suppl 5):v108–v115. [DOI] [PubMed] [Google Scholar]
- 44. d’Amore F, Leppä S, Gomes da Silva M, et al. First interim efficacy and safety analysis of an international phase III randomized trial in newly diagnosed systemic peripheral T-cell lymphoma treated with or without alemtuzumab and consolidated with high dose therapy. Blood. 2012;120:57. [Google Scholar]
- 45. Gleeson M, Peckitt C, To YM, et al. CHOP versus GEM-P in previously untreated patients with peripheral T-cell lymphoma (CHEMO-T): a phase 2, multicentre, randomised, open-label trial. Lancet Haematol. 2018;5:e190–e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Avilés A, Castañeda C, Neri N, et al. Results of a phase III clinical trial: CHOP versus CMED in peripheral T-cell lymphoma unspecified. Med Oncol. 2008;25:360–364. [DOI] [PubMed] [Google Scholar]
- 47. Horwitz S, O’Connor OA, Pro B, et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet. 2019;393:229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chukwueke UN, Nayak L. Central nervous system lymphoma. Hematol Oncol Clin North Am. 2019;33:597–611. [DOI] [PubMed] [Google Scholar]
- 49. Yamaguchi M, Kwong YL, Kim WS, et al. Phase II study of SMILE chemotherapy for newly diagnosed stage IV, relapsed, or refractory extranodal natural killer (NK)/T-cell lymphoma, nasal type: the NK-cell tumor study group study. J Clin Oncol. 2011;29:4410–4416. [DOI] [PubMed] [Google Scholar]
- 50. Li X, Cui Y, Sun Z, et al. DDGP versus SMILE in newly diagnosed advanced natural killer/T-cell lymphoma: a randomized controlled, multicenter open-label study in China. Clin Cancer Res. 2016;22:5223–5228. [DOI] [PubMed] [Google Scholar]
- 51. d’Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30:3093–3099. [DOI] [PubMed] [Google Scholar]
- 52. El-Asmar J, Reljic T, Ayala E, et al. Efficacy of high-dose therapy and autologous hematopoietic cell transplantation in peripheral T cell lymphomas as front-line consolidation or in the relapsed/refractory setting: a systematic review/meta-analysis. Biol Blood Marrow Transplant. 2016;22:802–814. [DOI] [PubMed] [Google Scholar]
- 53. Park SI, Horwitz SM, Foss FM, et al. The role of autologous stem cell transplantation in patients with nodal peripheral T-cell lymphomas in first complete remission: Report from COMPLETE, a prospective, multicenter cohort study. Cancer. 2019;125:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fossard G, Broussais F, Coelho I, et al. Role of up-front autologous stem-cell transplantation in peripheral T-cell lymphoma for patients in response after induction: an analysis of patients from LYSA centers. Ann Oncol. 2018;29:715–723. [DOI] [PubMed] [Google Scholar]
- 55. Casulo C, Schöder H, Feeney J, et al. 18F-fluorodeoxyglucose positron emission tomography in the staging and prognosis of T cell lymphoma. Leuk Lymphoma. 2013;54:2163–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. El-Galaly TC, Pedersen MB, Hutchings M, et al. Utility of interim and end-of-treatment PET/CT in peripheral T-cell lymphomas: a review of 124 patients. Am J Hematol. 2015;90:975–980. [DOI] [PubMed] [Google Scholar]
- 57. Tomita N, Hattori Y, Fujisawa S, et al. Post-therapy 18F-fluorodeoxyglucose positron emission tomography for predicting outcome in patients with peripheral T cell lymphoma. Ann Hematol. 2015;94:431–436. [DOI] [PubMed] [Google Scholar]
- 58. Sohn BS, Yoon DH, Kim KP, et al. The role of 18F-fluorodeoxyglucose positron emission tomography at response assessment after autologous stem cell transplantation in T-cell non-Hodgkin's lymphoma patients. Ann Hematol. 2013;92:1369–1377. [DOI] [PubMed] [Google Scholar]
- 59. Ham JS, Kim SJ, Choi JY, et al. The prognostic value of interim and end-of-treatment PET/CT in patients with newly diagnosed peripheral T-cell lymphoma. Blood Cancer J. 2016;6:e395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pellegrini C, Argnani L, Broccoli A, et al. Prognostic value of interim positron emission tomography in patients with peripheral T-cell lymphoma. Oncologist. 2014;19:746–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ellin F, Landström J, Jerkeman M, et al. Real-world data on prognostic factors and treatment in peripheral T-cell lymphomas: a study from the Swedish Lymphoma Registry. Blood. 2014;124:1570–1577. [DOI] [PubMed] [Google Scholar]
- 62. Ellin F, Jerkeman M, Törnqvist J, et al. Impact of comorbidity on survival in peripheral T-cell lymphomas: A Swedish lymphoma registry study. Hematol Oncol. 2018;36:159–165. [DOI] [PubMed] [Google Scholar]
- 63. Federico M, Bellei M, Marcheselli L, et al. Peripheral T cell lymphoma, not otherwise specified (PTCL-NOS). A new prognostic model developed by the International T cell Project Network. Br J Haematol. 2018;181:760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gleeson M, Peckitt C, Cunningham D, et al. Outcomes following front-line chemotherapy in peripheral T-cell lymphoma: 10-year experience at The Royal Marsden and The Christie Hospital. Leuk Lymphoma. 2018;59:1586–1595. [DOI] [PubMed] [Google Scholar]
- 65. Vose J, Armitage J, Weisenburger D, et al. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26:4124–4130. [DOI] [PubMed] [Google Scholar]
- 66. Briski R, Feldman AL, Bailey NG, et al. The role of front-line anthracycline-containing chemotherapy regimens in peripheral T-cell lymphomas. Blood Cancer J. 2014;4:e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wudhikarn K, Bunworasate U, Julamanee J, et al. Characteristics, treatment patterns, prognostic determinants and outcome of peripheral T cell lymphoma and natural killer/T cell non-Hodgkin Lymphoma in older patients: The result of the nationwide multi-institutional registry Thai Lymphoma Study Group. J Geriatr Oncol. 2020;11:62–68. [DOI] [PubMed] [Google Scholar]
- 68. Choi EJ, Hong JY, Yoon DH, et al. Treatment outcomes of dose-attenuated CHOP chemotherapy in elderly patients with peripheral T cell lymphoma. Blood Res. 2017;52:270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wulf GG, Altmann B, Ziepert M, et al. Alemtuzumab plus CHOP versus CHOP in elderly patients with peripheral T-cell lymphoma: the DSHNHL2006-1B/ACT-2 trial. Leukemia. 2020 May 7. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 70. Fields PA, Townsend W, Webb A, et al. De novo treatment of diffuse large B-cell lymphoma with rituximab, cyclophosphamide, vincristine, gemcitabine, and prednisolone in patients with cardiac comorbidity: a United Kingdom National Cancer Research Institute trial. J Clin Oncol. 2014;32:282–287. [DOI] [PubMed] [Google Scholar]
- 71. Mak V, Hamm J, Chhanabhai M, et al. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: spectrum of disease and rare long-term survivors. J Clin Oncol. 2013;31:1970–1976. [DOI] [PubMed] [Google Scholar]
- 72. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30:631–636. [DOI] [PubMed] [Google Scholar]
- 73. O’Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29:1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. O’Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-Cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) Study. J Clin Oncol. 2015;33:2492–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Horwitz SM, Advani RH, Bartlett NL, et al. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood. 2014;123:3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Damaj G, Gressin R, Bouabdallah K, et al. Results from a prospective, open-label, phase II trial of bendamustine in refractory or relapsed T-cell lymphomas: the BENTLY trial. J Clin Oncol. 2013;31:104–110. [DOI] [PubMed] [Google Scholar]
- 77. Ohmoto A, Fuji S. Cyclosporine for angioimmunoblastic T-cell lymphoma: a literature review. Expert Rev Hematol. 2019;12:975–981. [DOI] [PubMed] [Google Scholar]
- 78. Puig N, Wang L, Seshadri T, et al. Treatment response and overall outcome of patients with relapsed and refractory peripheral T-cell lymphoma compared to diffuse large B-cell lymphoma. Leuk Lymphoma. 2013;54:507–513. [DOI] [PubMed] [Google Scholar]
- 79. Mikesch JH, Kuhlmann M, Demant A, et al. DexaBEAM versus ICE salvage regimen prior to autologous transplantation for relapsed or refractory aggressive peripheral T cell lymphoma: a retrospective evaluation of parallel patient cohorts of one center. Ann Hematol. 2013;92:1041–1048. [DOI] [PubMed] [Google Scholar]
- 80. Yao YY, Tang Y, Zhu Q, et al. Gemcitabine, oxaliplatin and dexamethasone as salvage treatment for elderly patients with refractory and relapsed peripheral T-cell lymphoma. Leuk Lymphoma. 2013;54:1194–1200. [DOI] [PubMed] [Google Scholar]
- 81. Arkenau HT, Chong G, Cunningham D, et al. Gemcitabine, cisplatin and methylprednisolone for the treatment of patients with peripheral T-cell lymphoma: the Royal Marsden Hospital experience. Haematologica. 2007;92:271–272. [DOI] [PubMed] [Google Scholar]
- 82. Shustov A. Controversies in autologous and allogeneic hematopoietic cell transplantation in peripheral T/NK-cell lymphomas. Best Pract Res Clin Haematol. 2013;26:89–99. [DOI] [PubMed] [Google Scholar]
- 83. Jethwa KD, Bishton MJ, Fox CP. The role of high-dose chemotherapy and autologous stem cell transplant for treatment-naïve patients with peripheral T-cell lymphoma: a systematic review of the literature. Br J Haematol. 2017;178:476–479. [DOI] [PubMed] [Google Scholar]
- 84. Casulo C, Horwitz S. Should eligible patients with T-cell lymphoma receive high-dose therapy and autologous stem cell transplant in the upfront setting? Curr Oncol Rep. 2010;12:374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Friedrichs B, Stelljes M, Schmitz N. The role of allogeneic stem cell transplantation in T-cell lymphoma. Curr Opin Oncol. 2018;30:301–307. [DOI] [PubMed] [Google Scholar]
- 86. Schmitz N, Lenz G, Stelljes M. Allogeneic hematopoietic stem cell transplantation for T-cell lymphomas. Blood. 2018;132:245–253. [DOI] [PubMed] [Google Scholar]
- 87. Wei J, Xu J, Cao Y, et al. Allogeneic stem-cell transplantation for peripheral T-cell lymphoma: a systemic review and meta-analysis. Acta Haematol. 2015;133:136–144. [DOI] [PubMed] [Google Scholar]
- 88. Perrone G, Corradini P. Autologous stem cell transplantation for T-cell lymphomas. Semin Hematol. 2014;51:59–66. [DOI] [PubMed] [Google Scholar]
- 89. Kharfan-Dabaja MA, Kumar A, Ayala E, et al. Clinical practice recommendations on indication and timing of hematopoietic cell transplantation in mature T Cell and NK/T Cell lymphomas: an international collaborative effort on behalf of the guidelines committee of the American society for blood and marrow transplantation. Biol Blood Marrow Transplant. 2017;23:1826–1838. [DOI] [PubMed] [Google Scholar]
- 90. Coiffier B, Pro B, Prince HM, et al. Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma: pivotal study update demonstrates durable responses. J Hematol Oncol. 2014;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dupuis J, Morschhauser F, Ghesquieres H, et al. Combination of romidepsin with cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated patients with peripheral T-cell lymphoma: a non-randomised, phase 1b/2 study. Lancet Haematol. 2015;2:e160–e165. [DOI] [PubMed] [Google Scholar]
- 92. Mehrling T, Chen Y. The Alkylating-HDAC inhibition fusion principle: taking chemotherapy to the next level with the first in class molecule EDO-S101. Anticancer Agents Med Chem. 2016;16:20–28. [DOI] [PubMed] [Google Scholar]
- 93. Lemonnier F, Dupuis J, Sujobert P, et al. Treatment with 5-azacytidine induces a sustained response in patients with angioimmunoblastic T-cell lymphoma. Blood. 2018;132:2305–2309. [DOI] [PubMed] [Google Scholar]
- 94. Bejar R, Lord A, Stevenson K, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124:2705–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Emadi A, Faramand R, Carter-Cooper B, et al. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am J Hematol. 2015;90:E77–E79. [DOI] [PubMed] [Google Scholar]
- 96. Metzeler KH, Walker A, Geyer S, et al. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26:1106–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. O’Connor OA, Falchi L, Lue JK, et al. Oral 5-azacytidine and romidepsin exhibit marked activity in patients with Ptcl: a multicenter phase I study. Blood. 2019;134:1395–1405. [DOI] [PubMed] [Google Scholar]
- 98. Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. 2007;19:813–824. [DOI] [PubMed] [Google Scholar]
- 99. Lesokhin AM, Ansell SM, Armand P, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib Study. J Clin Oncol. 2016;34:2698–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wartewig T, Kurgyis Z, Keppler S, et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature. 2017;552:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. de Leval L, Rickman DS, Thielen C, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109:4952–4963. [DOI] [PubMed] [Google Scholar]
- 102. Johnson LDS, Banerjee S, Kruglov O, et al. Targeting CD47 in Sezary syndrome with SIRPalphaFc. Blood Adv. 2019;3:1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Bagot M, Porcu P, Marie-Cardine A, et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: an international, first-in-human, open-label, phase 1 trial. Lancet Oncol. 2019;20:1160–1170. [DOI] [PubMed] [Google Scholar]
- 104. Maciocia PM, Wawrzyniecka PA, Philip B, et al. Targeting the T cell receptor beta-chain constant region for immunotherapy of T cell malignancies. Nat Med. 2017;23:1416–1423. [DOI] [PubMed] [Google Scholar]
- 105. Feldman AL, Sun DX, Law ME, et al. Overexpression of Syk tyrosine kinase in peripheral T-cell lymphomas. Leukemia. 2008;22:1139–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ishikawa C, Senba M, Mori N. Anti-adult T-cell leukemia/lymphoma activity of cerdulatinib, a dual SYK/JAK kinase inhibitor. Int J Oncol. 2018;53:1681–1690. [DOI] [PubMed] [Google Scholar]
- 107. Horwitz SM, Feldman TA, Hess BT, et al. The novel SYK/JAK inhibitor cerdulatinib demonstrates good tolerability and clinical response in a phase 2a study in relapsed and refractory peripheral T-cell lymphoma and cutaneous T-cell lymphoma. Blood. 2018;132:1001a. [Google Scholar]
- 108. Horwitz SM, Koch R, Porcu P, et al. Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood. 2018;131:888–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Horwitz SM, Moskowitz AJ, Jacobsen ED, et al. The combination of duvelisib, a PI3K- δ,γ inhibitor, and romidepsin is highly active in relapsed/refractory peripheral T-cell lymphoma with low rates of transaminits: results of parallel multicentre, phase 1 combination studies with expansion cohorts. Blood. 2018;132:683a. [Google Scholar]