

Abstract
The long-lived triplet excited states of transition metal photocatalysts can activate organic substrates via either energy- or electron-transfer pathways, and the rates of these processes can be influenced by rational tuning of the reaction conditions. The characteristic reactive intermediates generated, however, are distinct and can exhibit very different reactivity patterns. This mechanistic diversity available to photocatalytic reactions might thus offer an opportunity to engineer divergent reactions that give markedly different chemical outcomes under superficially similar conditions. Herein, we show that the photocatalytic reactions of benzoylformate esters with alkenes can be directed towards either Paternò–Büchi cycloadditions or allylic functionalization reactions under conditions favoring energy transfer or electron transfer, respectively. These studies provide a framework for designing other divergent photocatalytic methods that produce different sets of reaction outcomes under photoredox and triplet sensitization conditions.
Graphical Abstract
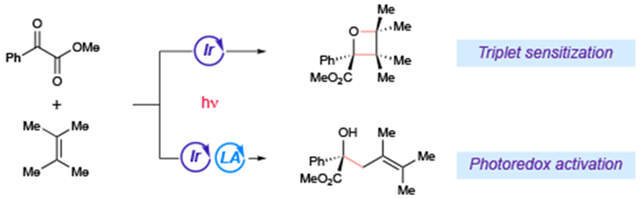
The development of new photocatalytic reactions has recently reemerged as an important theme of research in synthetic chemistry.1 The most common photocatalysts utilized for these methods belong to a family of Ru and Ir polypyridyl complexes. Several features make these inorganic complexes ideal for synthetic applications, including strong absorption in the visible range, excellent chemical stability, and predictable tunability across a wide range of excited-state redox potentials and triplet-state energies.2 Most importantly, these complexes undergo efficient intersystem crossing to produce long-lived triplet excited states that can initiate useful organic transformations via either energy-transfer (triplet sensitization) or electron-transfer (photoredox) mechanisms. Though the same photocatalytic triplet excited state is responsible for both pathways, the reactive intermediates that are generated by each are different and exhibit distinctive reactivity patterns. The optimization of photocatalytic reactions, therefore, often requires careful tuning of reaction conditions to ensure that one pathway is favored to the exclusion of the other.3 The mechanistic diversity available to photocatalytic reactions, however, might also offer an opportunity to engineer divergent reactions that give markedly different chemical outcomes under superficially similar conditions. This goal would be valuable because reactions that produce significant structural diversity using a small set of simple reactants can facilitate the discovery of compounds with new chemical and biological properties.
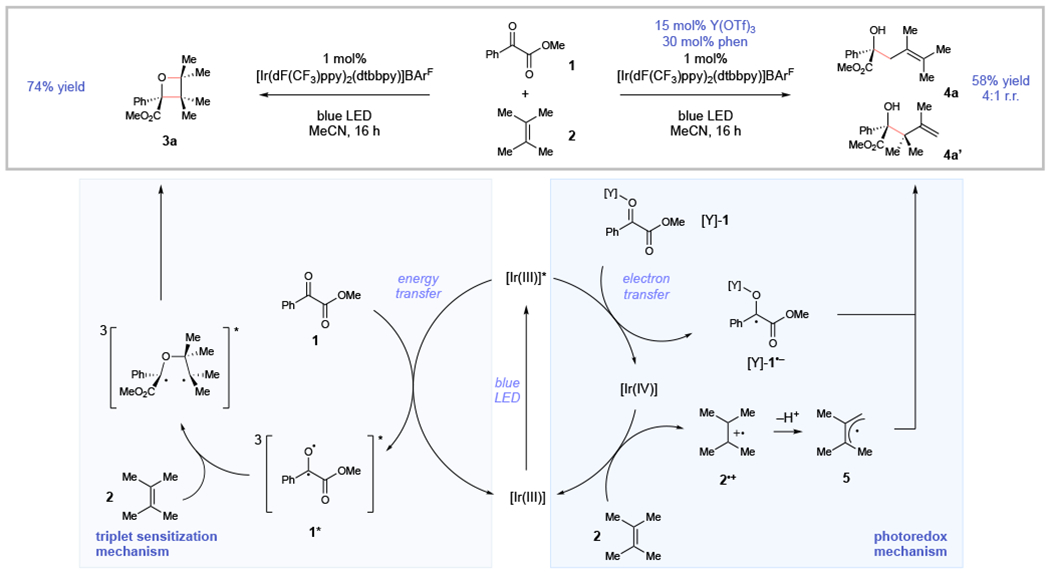
Photoreactions of carbonyl compounds have been an important topic of study throughout the history of organic photochemistry, and the mechanisms of their reactions have been extensively investigated.4 Among the oldest and best-known photocycloadditions is the Paternò–Büchi reaction,5 which classically involves the combination of excited-state aryl ketones with alkenes to afford structurally complex oxetane products.6,7 As a starting point for our investigations, we examined the photocatalytic reaction of benzoylformate esters. Neckers has explored the photochemistry of these compounds and demonstrated that they are excellent reactants for Paternò–Büchi cycloadditions.8 The triplet energies of benzoylformate esters have been reported to be ~62 kcal/mol;9 we proposed, therefore, that irradiation of methyl benzoylformate(1) and tetramethylethylene (2) in the presence of a fluorinated Ir(III) complex with a high triplet-state energy would generate long-lived triplet 1* via Dexter energy transfer (Scheme 1, left). Subsequent [2+2] cycloaddition via the standard stepwise biradical mechanism of the Paternò–Büchi reaction would then afford oxetane 3a. Indeed, upon irradiation of 1 and 2 in acetonitrile with 465 nm LEDs in the presence of 1 mol% [Ir(dF(CF3)ppy)2(dtbbpy)]BArF,10 Paternò–Büchi cycloadduct 3a was obtained in 74% yield.
Scheme 1.
Divergent outcomes of triplet sensitization and photoredox reactions of benzoylformate esters
Our laboratory has a long-standing interest in the use of Lewis acid co-catalysts to increase the driving force for photoinduced electron transfer to carbonyl-containing substrates.11 We imagined that an appropriate Lewis acid could accelerate electron transfer to ketoester substrate 1, outcompeting the rate of energy transfer (Scheme 1, right). The resulting Ir(IV) complex could then oxidize electron-rich alkene 2 to the corresponding radical cation (2•+). Deprotonation would afford an allylic radical (5) that could generate product either by combination with the persistent Lewis acid stabilized ketyl radical ([Y]-1•−)12 or via a Lewis acid catalyzed addition to another equivalent of ketoester.13 Under otherwise identical conditions used for the Paternò–Büchi reaction, addition of 15 mol% Y(OTf)3 and 30 mol% phenanthroline led to the formation of 58% allylic functionalization products as a 4:1 mixture of regioisomers (4a and 4a’), along with 18% of the oxetane cycloadduct.
The observation that the partitioning of the reaction between Paternò–Büchi and allylic functionalization pathways could be influenced by reaction conditions was intriguing, and we elected to optimize each of these transformations independently. First, Table 1 summarizes the results of studies to improve the yield of the Paternò–Büchi reaction. As expected for a triplet sensitization process, the yield of oxetane is highest using high triplet energy photocatalysts14 (entries 1–4). Also consistent with a triplet mechanism is the relative insensitivity of the reaction towards the polarity of the solvent (entries 5–7), with the highest yields observed in toluene. The yield is increased at higher concentrations (entry 7). A control experiment shows that no product is observed in the absence of light (entry 8); however, a slow Paternò–Büchi reaction was observed in the absence of photocatalyst, which results from a tailing of the absorption of 1 into the visible region (entry 9). Nevertheless, product formation in this reaction is dominated by the photosensitized reaction pathway.
Table 1.
Optimization studies for Paternò–Büchi cycloaddition.a
 | |||
|---|---|---|---|
| entry | photocatalystb | solvent | yield |
| 1 | [Ir(dF(CF3)ppy)2(dtbbpy)]BArF (61) | MeCN | 74% |
| 2 | [Ir(Fppy)2(dtbbpy)]PF6 (53) | MeCN | 40% |
| 3 | [Ir(ppy)2(dMeObpy)]PF6 (51) | MeCN | 5% |
| 4 | [Ir(ppy)2(dtbbpy)]PF6 (51) | MeCN | 6% |
| 5 | [Ir(dF(CF3)ppy)2(dtbbpy)]BArF | CH2Cl2 | 81% |
| 6 | [Ir(dF(CF3)ppy)2(dtbbpy)]BArF | toluene | 92% |
| 7c | [Ir(dF(CF3)ppy)2(dtbbpy)]BArF | toluene | 97% |
| 8d | [Ir(dF(CF3)ppy)2(dtbbpy)]BArF | toluene | 0% |
| 9 | none | toluene | 15% |
Reactions conducted using 0.1 mmol 1, 0.5 mmol 2, 1 mol% photocatalyst, and 5 mL solvent and irradiated with a 16 W LED lamp (465 nm) for 16 h unless otherwise noted. Yields were determined by 1H NMR analysis.
Values in parentheses represent the photocatalyst triplet energies in kcal/mol.
Reaction conducted in 1.5 mL toluene.
Reaction conducted in the dark.
Figure 1 summarizes an investigation of the scope of this photosensitized Paternò–Büchi cycloaddition. We first examined the scope with respect to the ketoester reaction component. Ester moieties bearing potentially reactive functional groups including alkenes (3d), alkyn es (3e), halides (3f), and azides (3g) are well-tolerated. The arene could also be easily modified with various electron-donating and -withdrawing groups (3h–3o), although ortho substituents retard the reaction somewhat (3p). Heteroaroylformate esters participate are good substrates (3q and 3r). The scope with respect to the alkene was examined next. The reaction was fastest with relatively electron-rich alkenes, consistent with the oxyl radical character of the (n,π*) triplet ketone intermediate, although disubstituted aliphatic alkenes afforded reasonable yields of the oxetane cycloadduct (3s and 3t). In all cases, the regioselectivity of the reaction could be predicted by considering the stability of the intermediate 1,4-diradical expected from the conventional stepwise mechanism of the Paternò–Büchi cycloaddition (3u–3x).
Figure 1.
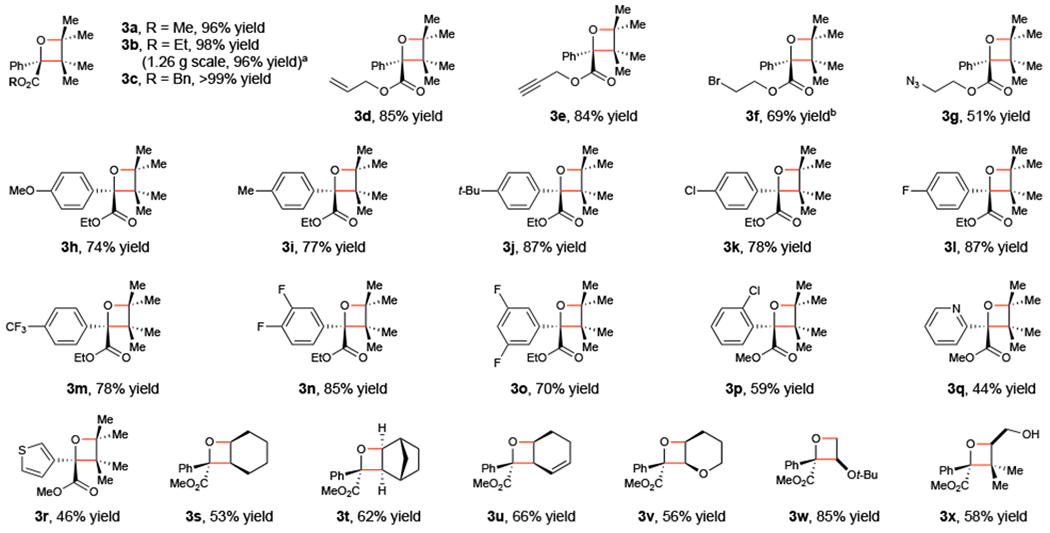
Paternò–Büchi scope studies. Reactions conducted using 0.5 mmol 1, 2.5 mmol 2, 1 mol% [Ir(dF(CF3)ppy)2(dtbbpy)]BArF, and 7.5 mL toluene and irradiated with a 16 W LED lamp (465 nm) for 16 h unless otherwise noted. a Reaction conducted using 0.5 mol% [Ir(dF(CF3)ppy)2(dtbbpy)]BArF. b Reaction conducted for 24 h.
As a further validation of the triplet mechanism of this photoreaction, we wondered if other canonical photoreactions of (n,π*) triplet ketones could be accessed using the conditions optimized for the Paternò–Büchi cycloaddition. We targeted the Norrish–Yang photocyclization15,16 of ortho-methyl benzoylformates17 to access benzofused cyclobutanes that are important structural elements of bioactive drugs and natural products.18 Indeed, a variety of ortho-methyl benzoylformates undergo smooth transformation to the benzocyclobutenol products 6a–6h (Figure 2).
Figure 2.
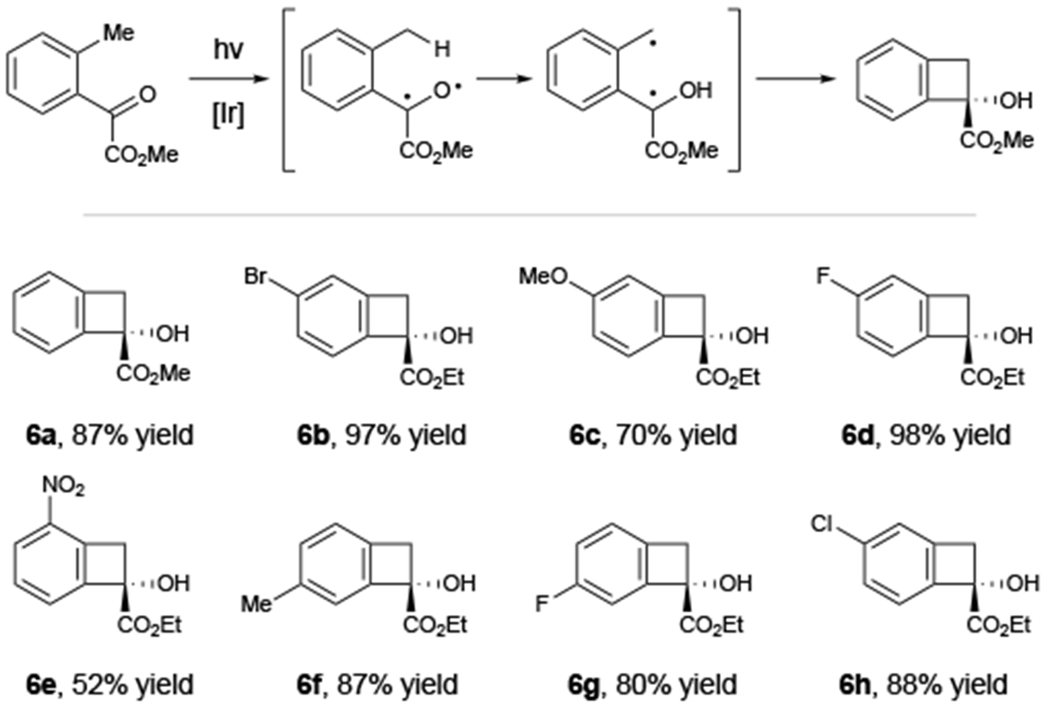
Norrish type II reactions under energy-transfer conditions. Reactions conducted using 0.5 mmol ketoester, 1 mol% [Ir(dF(CF3)ppy)2(dtbbpy)]BArF, and 7.5 mL toluene and irradiated using a 16 W LED lamp (465 nm) for 16 h.
We next undertook studies to optimize the photoredox allylic functionalization process (Table 2). We speculated that the product distribution might be diverted completely towards allylic functionalization by appropriately tuning the identity of the photocatalyst and Lewis acid co-catalyst to maximize the rate of photoredox activation and minimize the rate of energy transfer. First, a screen of alternate ligands for Y(OTf)3 revealed that a slightly more electron-deficient 5-chlorophenanthroline ligand afforded somewhat improved yields of the allylic functionalization product (entries 1–3). We next examined photocatalysts with lower triplet energy and more negative excited-state reduction potentials that might further improve the yield of this reaction (entries 4–7). Indeed, [Ir(ppy)2(dMeObpy)]PF6 gave exclusively the allylic functionalization product in 89% yield (entry 7). Control experiments indicated that the photocatalyst, Lewis acid co-catalyst, and light were all required for the formation of the allylic functionalization product (entries 8–10).
Table 2.
Optimization studies for allylic functionalization
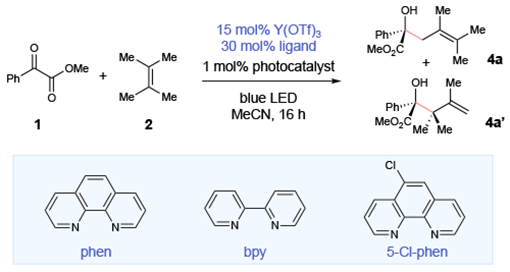 | ||||
|---|---|---|---|---|
| entry | photocatalyst | ligand | yield | r.r. |
| 1 | [Ir(dF(CF3)ppy)2(dtbbpy)]BArF | phen | 58% | 4:1 |
| 2 | [Ir(dF(CF3)ppy)2(dtbbpy)]BArF | bpy | 32% | 4:1 |
| 3 | [Ir(dF(CF3)ppy)2(dtbbpy)]BArF | 5-Cl-phen | 62% | 4:1 |
| 4 | [Ru(bpy)3](PF6)2 | 5-Cl-phen | 19% | 4:1 |
| 5 | Ir(ppy)3 | 5-Cl-phen | 78% | 4:1 |
| 6 | [Ir(Fppy)2(dtbbpy)]PF6 | 5-Cl-phen | 85% | 4:1 |
| 7 | [Ir(ppy)2(dMeObpy)]PF6 | 5-Cl-phen | 89% | 4:1 |
| 8 | none | 5-Cl-phen | 1% | -- |
| 9b | [Ir(ppy)2(dMeObpy)]PF6 | none | 2% | -- |
| 10c | [Ir(ppy)2(dMeObpy)]PF6 | 5-Cl-phen | 0% | -- |
Reactions conducted using 0.1 mmol 1, 0.5 mmol 2, 1 mol% photocatalyst, 15 mol% Y(OTf)3, 30 mol% ligand, and 1.5 mL MeCN and irradiated with a 16 W LED lamp (465 nm) for 16 h unless otherwise noted. Yields were determined by 1H NMR analysis.
Reaction conducted without Y(OTf)3.
Reaction conducted in the dark.
Scope studies exploring the generality of this allylic functionalization are summarized in Figure 3. All of the benzoylformates examined in the Paternò–Büchi reaction performed well in this transformation but afforded the allylic functionalization products (4a–4p) to the exclusion of the previously observed oxetanes. In general, we obtained relatively modest regioselectivity for the formation of the tetrasubstituted olefin product over the 1,1 -disubstituted alkene isomer. However, those benzoylformates bearing bulky ortho substituents gave better selectivity (4p–4v). Importantly, in these cases, we observed no trace of the Norrish–Yang cyclization products.
Figure 3.
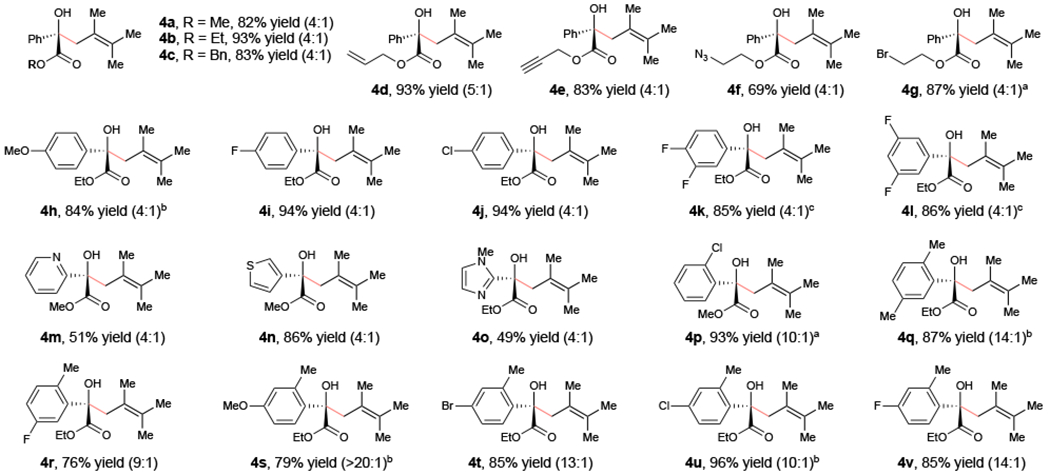
Allylic functionalization scope studies. Reactions conducted using 0.5 mmol 1, 2.5 mmol 2, 1 mol% [Ir(ppy)2(dMeObpy)]PF6, 15 mol% Y(OTf)3, 30 mol% 5-Cl-phen, and 7.5 mL toluene and irradiated with a 16 W LED lamp (465 nm) for 16 h unless otherwise noted. Values in parentheses represent the ratio of regioisomers. a Reaction conducted for 32 h. b Reaction conducted for 24 h. c Reaction conducted using 1 mol% [Ir(Fppy)2(dtbbpy)]PF6.
Several lines of evidence support our proposal that the allylic functionalization reaction involves a photoredox process. First, cyclic voltammetry performed with benzoylformate 1 showed a quasi-reversible reduction wave at −1.28 V vs SCE. The addition of Y(OTf)3 and phen results in a positive shift to −0.70 V, which is easily within the range accessible by the excited states of both [Ir*(dF(CF3)ppy)2(dtbbpy)]+ (−0.89 V)10 and the more strongly reducing [Ir*(ppy)2(dMeObpy)]+ (−1.26 V). Second, this mechanism predicts that the oxidation of tetramethylethylene (+1.45 V) by the oxidized [Ir(ppy)2(dMeObpy)]2+ catalyst (+1.21 V) would be slightly endergonic by 5.5 kcal/mol. Consistent with this expectation, less-electron-rich trisubstituted alkenes such as 7 (+1.98 V)19 fail to give any C–H functionalization products (Scheme 2A).20 On the other hand, electron-rich methallylsilane 8 (+1.24 V)21 participates readily in this reaction to afford Sakurai addition product 9 in 90% yield (Scheme 2B).
Scheme 2.
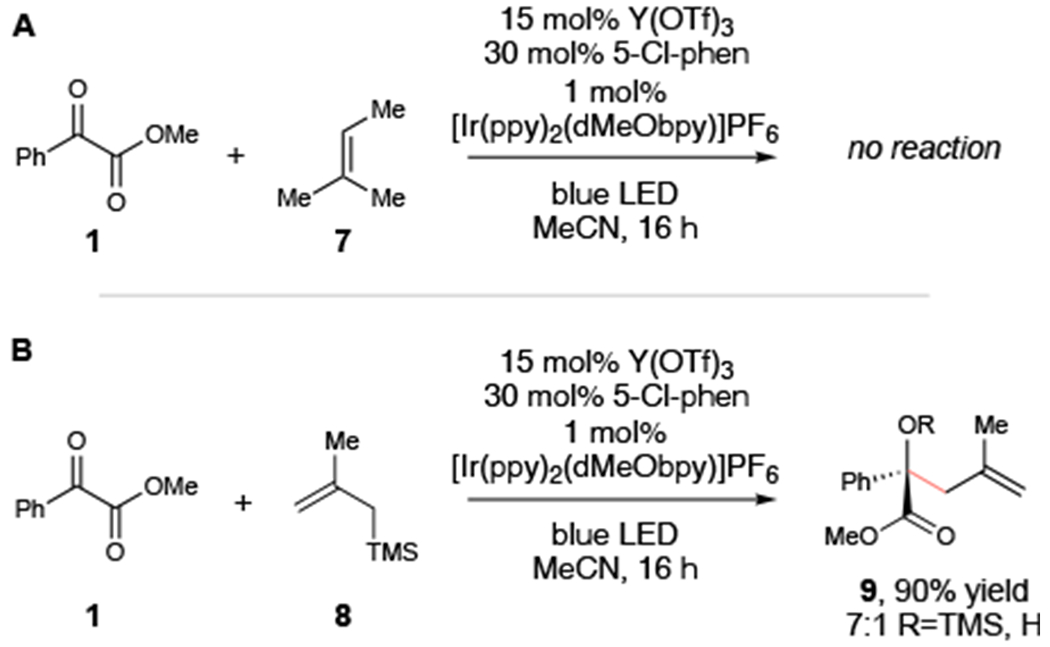
Support for a photoredox mechanism.
In summary, we have shown that divergent reactivity is accessible in the photocatalytic reaction of benzoylformates with alkenes. Under conditions that favor energy transfer, the triplet-state benzoylformate undergoes smooth Paternò–Büchi cycloaddition to afford complex oxetane scaffolds. Under photoredox conditions, on the other hand, the same starting materials undergo selective transformation to allylic functionalization products. These results provide a valuable demonstration that the ability to rationally favor either energy- or electron-transfer mechanisms offers a means not only to impact the rate of useful photocatalytic transformations but also to alter their chemical outcomes.
Supplementary Material
ACKNOWLEDGMENT
We dedicate this paper to Prof. Ilhyong Ryu (Osaka Prefecture University) on the occasion of his 70th birthday. We are grateful to Katie Rykaczewski and Prof. Corinna Schindler (University of Michigan) for sharing unpublished data. Funding for this project was provided by the NIH (GM095666). NMR and MS facilities at UW–Madison are funded by the NIH (1S10 OD020022-1) and a generous gift from the Paul J. and Margaret M. Bender Fund.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Detailed experimental procedures, full spectroscopic data for all new copounds (PDF).
FAIR Data is available as Supporting Information for Publication and includes the primary NMR FID files for compounds 3a–x, 4a–v, 6a–h, 9, and S1–S10.
An early preprint of this research appeared on ChemRxiv.22
REFERENCES
- 1.For reviews, see:; a) Narayanam JMR; Stephenson CRJ “Visible Light Photoredox Catalysis: Applications in Organic Synthesis,” Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; b) Prier CK; Rankic DA; MacMillan DWC “Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis,” Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC “The Merger of Transition Metal and Photocatalysis,” Nature Rev. Chem. 2017, 1, 0052. [Google Scholar]
- 2.a) Juris A; Balzani V; Barigelletti F; Campagna S; Belser P; von Zelewsky A “Ru(II) Polypyridine Complexes: Photophysics, Photochemistry, Electrochemistry, and Chemiluminescence,” Coord. Chem. Rev 1988, 84, 85–277. [Google Scholar]; b) Huckaba AJ; Nazeeruddin MK “Strategies for Tuning Emission Energy in Phosphorescent Ir(III) Complexes.” Comments Inorg. Chem. 2017, 37, 117–145. [Google Scholar]
- 3.a) Lin S; Ischay MA; Fry CG; Yoon TP “Radical Cation Diels-Alder Cycloadditions by Visible Light Photocatalysis.” J. Am. Chem. Soc 2011, 133, 19350–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ischay MA; Ament MS; Yoon TP “Crossed Intermolecular [2 + 2] Cycloaddition of Styrenes by Visible Light Photocatalysis.” Chem. Sci 2012, 3, 2807–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nguyen JD; D’Amato EM; Narayanam JMR; Stephenson CRJ “Engaging Unactivated Alkyl, Alkenyl and Aryl Iodides in Visible-Light-Mediated Free Radical Reactions.” Nat. Chem 2012, 4, 854–859. [DOI] [PubMed] [Google Scholar]; d) Daub ME; Jung H; Lee BJ; Won J; Baik M-H; Yoon TP “Enantioselective [2+2] Cycloadditions of Cinnamate Esters: Generalizing Lewis Acid Catalysis of Triplet Energy Transfer.” J. Am. Chem. Soc 2019, 141, 9543–9547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Dinda B, “Photochemistry of Carbonyl Compounds” In Essentials of Pericycli cand Photochemical Reactions, vol. 93, Springer, Switzerland, 2017, pp. 241–275. [Google Scholar]; b) Dantas JA; Correia JTM; Paixão MW; Corrêa AG Photochemistry of Carbonyl Compounds: Application in Metal-Free Reactions. ChemPhotoChem 2019, 3, 506–520. [Google Scholar]
- 5.a) Paternò E; Chieffi G “Sintesi in chimica organica per mezzo della luce. Nota II. Composti degli idrocarburi non saturi con aldeidi e chetoni.” Gazz. Chim. Ital 1909, 39, 341–361. [Google Scholar]; b) Büchi G; Inman CG; Lipinsky ES “Light-Catalyzed Organic Reactions. I. The Reaction of Carbonyl Compounds with 2-Methyl-2-Butene in the Presence of Ultraviolet Light.” J. Am. Chem. Soc 1954, 76, 4327–4331. [Google Scholar]
- 6.For reviews, see:; a) Fréneau M; Hoffmann N “The Paternò-Büchi Reaction — Mechanisms and Application to Organic Synthesis.” J. Photochem. Photobiol. C Photochem. Rev 2017, 33, 83–108. [Google Scholar]; b) D’Auria M “The Paternò-Büchi Reaction — A Comprehensive Review.” Photochem. Photobiol. Sci 2019, 18, 2297–2362. [DOI] [PubMed] [Google Scholar]
- 7.For recent contributions to Paternò–Büchi methodology, see:; a) Kumarasamy E; Raghunathan R; Kandappa SK; Sreenithya A; Jockusch S; Sunoj RB; Sivaguru J “Transposed Paternò–Büchi Reaction.” J. Am. Chem. Soc 2017, 139, 655–662. [DOI] [PubMed] [Google Scholar]; b) Flores DM; Schmidt VA “Intermolecular 2 + 2 Carbonyl–Olefin Photocycloadditions Enabled by Cu(I)–Norbornene MLCT.” J. Am. Chem. Soc 2019, 141, 8741–8745. [DOI] [PubMed] [Google Scholar]; c) Mateos J; Vega-Peñaloza A; Franceschi P; Rigodanza F; Andreetta P; Companyó X; Pelosi G; Bonchio M; Dell’Amico L “A Visible-Light Paternò–Büchi Dearomatisation Process towards the Construction of Oxeto-Indolinic Polycycles.” Chem. Sci 2020, 11, 6532–6538. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rykaczewski KA; Schindler CS “Visible Light-Enabled Paternò–Büchi Reaction via Triplet Energy Transfer for the Synthesis of Oxetanes.” ChemRxiv 2020, Preprint. 10.26434/chemrxiv.12606377.v1. [DOI] [PubMed] [Google Scholar]
- 8.Hu S; Neckers DC “Rapid Regio- and Diastereoselective Paternò–Büchi Reaction of Alkyl Phenylglyoxylates.” J. Org. Chem 1997, 62, 564–567. [DOI] [PubMed] [Google Scholar]
- 9.Herkstroeter WG; Lamola AA; Hammond GS “Mechanisms of Photochemical Reactions in Solution. XXVIII. 1 Values of Triplet Excitation Energies of Selected Sensitizers.” J. Am. Chem. Soc 1964, 86, 4537–4540. [Google Scholar]
- 10.Lowry MS; Goldsmith JI; Slinker JD; Rohl R; Pascal RA Jr.; Malliaras GG; Bernhard S “Single-layer electroluminescent devices and photoinduced hydrogen production from an ionic iridium(III) complex.” Chem. Mater 2005, 17, 5712–5719. [Google Scholar]
- 11.Yoon TP “Photochemical Stereocontrol Using Tandem Photoredox-Chiral Lewis Acid Catalysis.” Acc. Chem. Res 2016, 49, 2307–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Russell GA; Strom ET; Talaty ER; Weiner SA “Semidiones. I. Acyclic Semidione Radical Anions and Cations Containing a Single Aryl Substituent.” J. Am. Chem. Soc 1966, 88, 1998–2004. [Google Scholar]; b) Spaccini R; Pastori N; Clerici A; Punta C; Porta O “Key Role of Ti(IV) in the Selective Radical–Radical Cross-Coupling Mediated by the Ingold-Fischer Effect.” J. Am. Chem. Soc 2008, 130, 18018–18024. [DOI] [PubMed] [Google Scholar]
- 13.Reckenthäler M; Neudörfl J-M; Zorlu E; Griesbeck AG “Combined Photoredox and Lewis Acid Catalyzed α-Hydroxyalkylation of Cyclic Ethers with Aromatic Ketones.” J. Org. Chem 2016, 81, 7211–7216. [DOI] [PubMed] [Google Scholar]
- 14.Photocatalyst triplet energy values taken from Reference 3d.
- 15.a) Yang NC; Yang D-DH “Photochemical Reactions of Ketones in Solution.” J. Am. Chem. Soc 1958, 80, 2913–2914. [Google Scholar]; b) Chen C “The Past, Present, and Future of the Yang Reaction.” Org. Biomol. Chem 2016, 14, 8641–8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For recent applications of Norrish–Yang photocyclizations, see:; a) Roque JB; Kuroda Y; Jurczyk J; Xu L-P; Ham JS; Göttemann LT; Roberts CA; Adpressa D; Saurí J; Joyce LA; Musaev DG; Yeung CS; Sarpong R “C–C Cleavage Approach to C–H Functionalization of Saturated Aza-Cycles.” ACS Catal. 2020, 10, 2929–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ham JS; Park B; Son M; Roque JB; Jurczyk J; Yeung CS; Baik M-H; Sarpong R “C–H/C–C Functionalization Approach to N-Fused Heterocycles from Saturated Azacycles.” J. Am. Chem. Soc 2020, jacs.0c04278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu S; Neckers DC “Photocycloaddition and ortho-Hydrogen Abstraction Reactions of Methyl Arylglyoxylates: Structure Dependent Reactivities.” J. Chem. Soc. Perkin Trans. 2 1999, 1771–1778. [Google Scholar]
- 18.a) Peglion J-L; Vian J; Vilaine J-P; Villeneuve N; Janiak P; Bidouard J-P “3-Benzazépin-zones substituées par un groupe benzocyclobutyl- ou indanyl-alkyl-amino-alkyle, utiles dans le traitement des affections cardiovasculaires,” European Patent Office EP0534859A1, March 31, 1993.; b) Hu X-R; Chou G-X; Zhang C-G “Flavonoids, Alkaloids from the Seeds of Crotalaria Pallida and Their Cytotoxicity and Anti-Inflammatory Activities.” Phytochemistry 2017, 143, 64–71. [DOI] [PubMed] [Google Scholar]
- 19.Roth HG; Romero NA; Nicewicz DA “Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry.” Synlett 2016, 27, 714–723. [Google Scholar]
- 20.NMR analysis of the unpurified reaction mixture shows 92% of unreacted benzoylformate 1 remaining.
- 21.Mizuno K; Ikeda M; Otsuji Y “Dual Regioselectivity in the Photoallylation of Electron-Deficient Alkenes by Allylic Silanes.” Chem. Lett 1988, 17, 1507–1510. [Google Scholar]
- 22.Zheng J; Dong X; Yoon T “Divergent Photocatalytic Reactions of α-Ketoesters Under Triplet Sensitization and Photoredox Conditions,” ChemRxiv 2020. 10.26434/chemrxiv.12611459.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.