

Abstract
A click chemistry-based approach was implemented to prepare peptidomimetics designed in silico and made from aromatic azides and a propargylated GIGI-mimicking platform derived from the altered Melan-A/MART-126(27L)-35 antigenic peptide ELAGIGILTV. The Cu(I)-catalyzed Huisgen cycloaddition was carried out on solid support to generate rapidly a first series of peptidomimetics, which were evaluated for their capacity to dock at the interface between the major histocompatibility complex class-I (MHC-I) human leucocyte antigen (HLA)-A2 and T-cell receptors (TCRs). Despite being a weak HLA-A2 ligand, one of those 11 first synthetic compounds bearing a p-nitrobenzyl-triazole side-chain was recognized by the receptor proteins of Melan-A/MART-1-specific T-cells. After modifications of the N- and C-termini of this agonist, which was intended to enhance HLA-A2 binding, one of the resulting 7 additional compounds triggered significant T-cell responses. Thus, these results highlight the capacity of naturally circulating human TCRs that are specific for the native Melan-A/MART-126–35 peptide to cross-react with peptidomimetics bearing organic motifs structurally different from the native central amino acids.
Keywords: immunotherapy, T-cell receptor, antigenic peptidomimetics, organic haptens, click chemistry
Entry for the Table of Contents
Click to fit. A computer-aided construction of a short series of mimetics of the tumor-derived Melan-A/MART-126(27L)-35 antigenic peptide ELAGIGILTV was achieved by clicking aromatic azido haptens on a propargylated GIGI-mimicking central platform. Out of a total of only 18 peptidomimetcis thus synthesized, one of the most heavily modified constructs harboring a p-nitrobenzyl moiety elicited significant Melan-A-specific T-cell responses.
Introduction
Melanoma is a malignant tumour arising from melanocytes and causes the majority (75%) of deaths in patients suffering of skin cancer. Over the past decade, the percentage of persons who have developed melanoma has considerably increased. According to the world health organization (WHO), this cancer disease currently affects 132,000 individuals, and globally each year incidence is increasing (AICR). The identification of melanoma-associated antigens recognized by CD8+ T-cells (MART) from melanoma patients has raised new prospects in the development of immunotherapeutic agents to treat this cancer. Melan-A/MART-1 (hereafter referred to as Melan-A) is a tissue-specific differentiation antigen expressed in melanocytes and melanoma.[1] In the late 1990s, Romero and co-workers identified the peptide segment Melan-A(26–35) (sequence: EAAGIGILTV, hereafter referred to as EAA) as being an immunodominant epitope frequently recognized by CD8+ tumor-infiltrating T- lymphocytes (TIL).[2,3] Indeed, a recent analysis of the dominant tumor-reactive clonotypes in the blood of an HLA-A2+ stage IV melanoma patient successfully treated with TIL therapy found that three out of the five most expanded clonotypes, accounting for >44% of the total response to tumor in blood following complete remission, were specific for the EAA epitope.[4] Moreover, actual therapeutic strategies aiming at blocking the down-regulation of CD8+ T-cells boost Melan-A specific T-cell immunity, which is promising for the development of melanoma immunotherapy.[5]
The EAA antigenic peptide stems from the degradation of Melan-A antigen. This agonist is then loaded onto the major histocompatibility class-I (MHC-I) HLA-A2 molecule in the endoplasmic reticulum, and finally transported to the cell surface for inspection by T-cell receptors (TCRs).[6] MHC-I proteins bind peptides adopting an extended or bulged conformation through the combination of conserved hydrogen bonds with the peptide main-chain and allele-specific pockets that accommodate some of the side-chains of peptide. For the HLA-A2 molecule, the key binding sites are two anchoring pockets that accommodate hydrophobic amino acid side-chains at P2 and C-terminal positions.[7] Because the EAA epitope presents a weak binding affinity to HLA-A2 due to the lack of an optimal residue at P2, the sequence of EAA has been optimized replacing the alanine residue at P2 by a leucine (sequence: ELAGIGILTV, hereafter referred to as ELA). The ELA peptide was found to be a high-affinity ligand capable of triggering CD8+ T cell responses stronger than those triggered by the EAA peptide.[8] However, further studies have shown that ELA and EAA prime different TCRs in vivo when used in clinical trials,[9] even though X-ray crystal structures of both EAA/HLA-A2 and ELA/HLA-A2 complexes revealed no major differences in the global architecture of the peptides with conservation of the bulged conformation.[10]
Other structural modifications of ELA were also performed in the aim of enhancing its stability in biological fluids, while maintaining high HLA-A2 binding affinity and efficacious immunogenicity.[11] These modifications consisted of replacing the N- and/or C-terminal regions, which are known to be more exposed to the action of proteases, by chemically altered or non-natural amino acids.[12] A few years ago, we opted for a less classical peptidomimetic approach for the construction of ELA analogues. Instead of iteratively modifying the terminal ELA parts bound to the HLA-A2 protein, we focused our efforts on alterations of the central TCR-contacting GIGI portion by relying on non-peptidic units, which were either tethered to this tetrapeptide or used to replace it.[13] These studies demonstrated the possibility of TCR recognition of haptens with chemical features drastically different from those of standard amino acids, while maintaining significant Melan-A specific T-cell recognition. In particular, a structure-guided rational design led us to prepare an ELA-mimetic presenting a central unit equipped with an indole acetic acid side-chain (see 1 in Scheme 1a), which triggered potent Melan-A specific T-cell responses.[13b] Our X-ray crystal structure of this ELA-mimetic 1 bound to the HLA-A2 protein[14] revealed that the central indole moiety of 1 is oriented toward the α2 helix of the HLA-A2 binding groove instead of bulging out toward the TCR loops as our in silico simulation of the A6-TCR/1/HLA-A2 ternary complex predicted.[13b,14] Nevertheless, since 1 has the ability to stimulate Melan-A specific T-cells, it likely undergoes some conformational changes when contacting TCRs, as previously observed for several HLA-A2-binding antigenic peptides.[15]
Scheme 1.

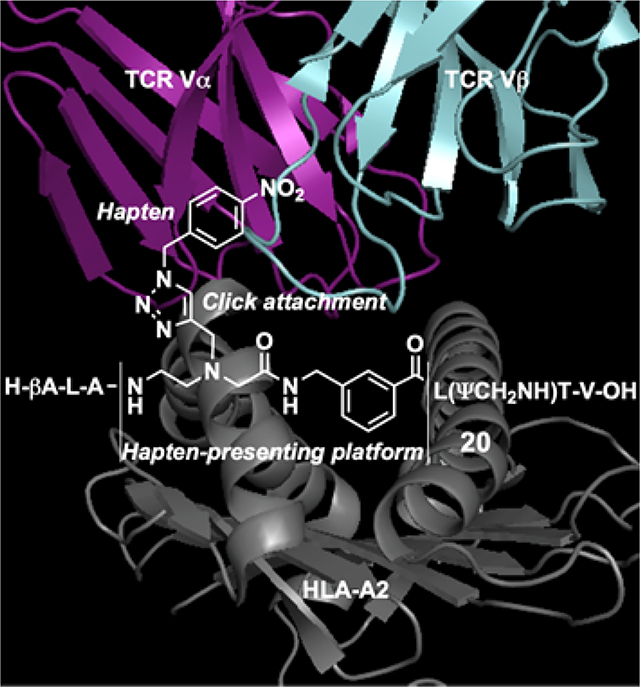
a) ELA-mimetic 1 triggers potent Melan-A specific CD8+ T-cell responses.[13b] b) Novel ELA-mimetics equipped with various clicked haptens (1R-1,2,3-triazole).
The promising results thus obtained with the ELA-mimetic 1 encouraged us to pursue this peptidomimetic approach. We used a click chemistry-based methodology to embed a selection of organic haptens in the GIGI-mimicking platform of our ELA-mimetic core structure (Scheme 1b). The copper(I)-catalyzed azide-alkyne Huisgen 1,3-dipolar cycloaddition (CuAAC) reaction was chosen for this task, since it offers a convenient and selective access to 1,4-disubstituted 1,2,3-triazoles and has found many useful applications in bioconjugate chemistry and drug discovery research.[16,17] Our selection of structures was again guided by a simulation of their docking at the TCR/MHC-I interface. The best performing ELA-mimetics were further modified to increase their HLA-A2 binding affinity and their resistance to proteases. Herein, we describe the identification of a highly heteroclitic HLA-A2 ligand for Melan-A specific CD8+ T-cells.
Results and Discussion
In silico rational design
CD8+ T-cell-mediated immune responses are initiated by molecular contacts between the TCR and peptide/MHC-I complex (pMHC-I). It is now well established that even though antigen specificity is the hallmark of the CD8+ T cell activation, a given TCR can exhibit a degree of plasticity, hence allowing it to interact with different pMHC-I ligands.[18] This cross-reactivity is, in part, the result of conformational adjustments at the TCR/pMHC-I interface that enable the formation of ternary complexes. Numerous structural and biophysical studies have emphasized the structural adaptability of TCRs and have demonstrated that significant changes between their unbound and bound states can occur upon binding to pMHC-I.[19] In this context, Baker and co-workers have explored the A6-TCR cross-reactivity by replacing the tyrosine at P5 in the viral Tax peptide sequence with a lysine, whose side-chain amino group was coupled to indole-3-butyric acid (i.e., Tax5K-IBA, depicted in orange in Figure 1).[20] The X-ray crystal structure of the A6-TCR/Tax5K-IBA/HLA-A2 complex revealed that binding proceeded through changes in the conformation of the complementarity determining region 3 (CDR3) loops of the TCR (see Figure 1), although the overall A6-TCR binding orientation was conserved.[20]
Figure 1.

Pymol superimposition of the clicked ELA-mimetic variant 11 (green) in the Tax5K-IBA (orange) binding interface of the A6-TCR/Tax5K-IBA/HLA-A2 ternary complex (PDB 2GJ6). The orientation of the anchoring residue side-chains, as well as the overall conformations, are maintained. The clicked hapten side-chain of 11 bulges out from the HLA-A2 binding groove and is well accommodated by the CDR3 loops of the A6-TCR. In the top right corner frame is depicted the phthalimide moiety with H-bonding interactions predicted by LigandFit.
These observations on the structural adaptability of TCRs lent support to our intent of equipping ELA-mimetics with sterically demanding clicked organic haptens. We explored the impact of elongating the central side-chain with a triazole-based system on the overall conformation of our ELA-mimetics within the HLA-A2 binding groove, and which orientation these central side-chains could adopt within the TCR/HLA-A2 interface. Since no ternary complex structure involving the ELA peptide or analogues thereof was reported at the time we initiated this project, we surmised that the overall conformation of ELA would be similar to those of any antigenic peptide presented by the HLA-A2 protein with a quasi-complete superimposition of the anchoring residue side-chains.[13b] We thus decided to rely on the A6/Tax5K-IBA/HLA-A2 structural data (PDB 2GJ6) to guide our docking studies. Nonetheless, Sewell and co-workers then reported the first X-ray crystal structure of ELA/HLA-A2 in complex with the MEL5-TCR (PDB 3HG1).[21] Remarkably, the conformation of the ELA peptide engaged with the MEL5-TCR is very similar to its conformation when only bound to the HLA-A2 protein with a root mean squared deviation of 0.369 Å.[10,21] Furthermore, this study emphasized that the TRAV12–2 gene, which encodes the CDR1α and CDR2α loops of the MEL5 TCR, is also expressed by the A6 TCR. Hence, the CDR1α and CDR2α loops of the A6-TCR utilize a binding mode that is identical to that observed for MEL5/ELA/HLA-A2 complex. In particular, the CDR1α loop residue Gln31 makes conserved contacts with the peptide positions P1 and P4 (Glu1 and Gly4 for ELA; Leu1 and Gly4 for Tax5K-IBA). These structural and binding mode analogies between the MEL5 and A6 TCRs validated our choice of relying on the A6/Tax5K-IBA/HLA-A2 structure as a model to guide the docking of our ELA-mimetics.
The shape-based docking engine LigandFit, from the Accelrys Cerius2 software package, was utilized to realize a virtual screening of the envisaged peptidomimetics. Hence, our initial GIGI-mimicking platform was retained (Scheme 1) and the incorporation of organic azides clicked on an alkyne installed onto its N-(2-aminoethyl)glycine unit was viewed as a convenient synthesis approach (Schemes 1 and 2), notably given the wider commercial availability of organic azides as compared to that of alkynes. Moreover, the replacement of glutamic acid at P1 by a β-alanine was maintained as this substitution improves the stability of the resulting analogues in biological fluids.[11a] Besides applying the same checkpoints as those used for designing the ELA-mimetic 1,[13b] here we also checked for the presence of potential contacts between the CDR1α residue Gln31 and the N-terminal part of our in silico engineered clicked ELA-mimetic variants, as it has been shown to play a key role in the recognition of the ELA peptide.[21]
Scheme 2.

Synthesis of clicked ELA-mimetics 7-17 via a CuAAc reaction on solid support: i) piperidine/DMF (1:4); ii) bromoacetic acid (0.4 M), N,N’-diisopropylcarbodiimide (DIC, 2 M), DMF, 5 min, rt; iii) propargylamine, DMSO; iv) Fmoc-Gly-H, NaBH3CN, 1% AcOH/DMF, 20 h, rt; v) end of SPPS; vi) R–N3, DIPEA, CuI, THF/DMF (1:1) (TBDMS = tert-butyldimethylsilyl); vii) release from resin and final deprotection using TFA/TIS/H2O (95:2.5:2.5) (see the Experimental Section and the Supporting Information for details).
Clicked ELA-mimetics Synthesis
Diverse organic azides were prepared in solution and clicked on the alkyne of the GIGI-mimicking platform, which was covalently bound to a solid support (Scheme 2). Several examples of CuAAC reactions on solid phase have been reported either to introduce 1,2,3-triazole moiety in peptoid backbones,[22] to realize cyclodimerization of peptides,[23] as well as to display various triazole-linked organic motifs on oligopeptoids.[24] Our strategy was based on a post-synthesis CuAAC reaction of the full peptoid alkyne resin 5 with various azides R–N3 (Scheme 2). Inclusion of the alkyne was thus accomplished during the course of the solid phase synthesis (SPS) via a nucleophilic substitution reaction between the α-bromoacetamide resin 3 and propargylamine. A reductive amination in the presence of N-Fmoc-glycinal was then carried out to complete the synthesis of the GIGI-mimicking moiety and was followed by the introduction of the remaining amino acids to furnish the propargylated resin 5. The CuAAC reactions were next performed using pre-synthesized azides R–N3 (see the Supporting Information for details), copper(I) iodide as catalyst and N,N-diisopropylethylamine (DIPEA) in a dimethylformamide (DMF)/tetrahydrofuran (THF) solvent mixture. No ascorbic acid was used to maintain the copper in its +1 oxidation state.[22a,25] The organic azides R–N3 selected through our preliminary virtual screening were all benzylic-type azides (see Scheme 2). After classical cleavage from the resin using trifluoroacetic acid (TFA), the resulting clicked ELA-mimetics 7-17 were purified by semi-preparative reverse-phase HPLC (see the Supporting Information).
Immunological Evaluation
Peptide antigenicity is influenced by the affinity of the peptide to the MHC-I protein, and in some cases, enhancing peptide binding has been shown to enhance T-cell recognition and immunogenicity.[26] Our clicked ELA-mimetics 7-17 were thus first evaluated by a flow cytometry-based MHC-I stabilization assay using the HLA-A2+ human mutant T2 cell line. The ELA peptide and its clicked analogues were used at a concentration of 10 μM and HLA-A2 binding was determined by the ability of those compounds to stabilize the HLA-A2 heavy chain/β2-microglobulin complex at the surface of T2 cells. Binding was quantified by mean fluorescence intensities (MFI), the values of which were then normalized relative to the value measured for the ELA peptide (Figure 2). All of our synthetically modified ELA-mimetics bound with weaker affinities to the HLA-A2 molecule as compared to that of the ELA peptide. Although the primary anchoring residues at P2 and P10 of HLA-A2 are conserved, the triazole-based central side-chains of 7-17 are both longer and bulkier than that of our original ELA-mimetic 1 (see Scheme 1). These results could be in compliance with the observations made on the crystal structure of the 1/HLA-A2 binary complex,[14] showing the central indolic side-chain of 1 oriented toward the α2 helix of the HLA-A2 protein. Thus, increasing both the length and steric demand of the central side-chain of our peptidomimetic variants could indeed affect their HLA-A2 binding affinity. Even if such a rationale is admittedly simplistic, it finds support in the fact that the binding affinity decreases when the size and hydrophobicity of the hapten units increases, the ELA-mimetics 14 to 17 exhibited the lowest HLA-A2 binding affinity. Therefore, we decided to pursue our investigations without these peptidomimetics, whose HLA-A2 binding affinities were more than 5-fold weaker than that of ELA (i.e., 14-17 with MFI below 20 in Figure 2).
Figure 2.

HLA-A2 binding affinities of the 18 clicked ELA-mimetics as measured by flow cytometry mean fluorescence intensity (MFI). Data are presented as percentage of positive control (ELA). Negative control autofluorescence has been subtracted from all values. 10 μM of compound was used for binding analysis. ELA-mimetics with binding affinities below the threshold value indicated by the dashed red line (MFI = 20) were excluded from further analyses (i.e., 14-17).
The selected ELA-mimetics 7-13 were then all evaluated for their capacity to stimulate Melan-A-specific T-cell responses in vitro. Here, an intracellular cytokine stain (ICS) was performed and T-cell activation was measured by flow cytometry. Briefly, a T-cell line was generated by stimulating HLA-A2+ peripheral blood mononuclear cells (PBMC) with the native EAA peptide. The resulting Melan-A-specific T-cell line was subsequently stimulated with our ELA-mimetics or the native EAA peptide, and T-cells examined by flow cytometry for expression of the lysosomal-associated membrane protein (LAMP) CD107a[27] and for intracellular expression of the cytokine interferon-γ (IFNγ) (Figure 3, and see the Experimental Section). Two of these ELA-mimetic variants bearing central benzyl-type haptens (i.e., 9 and 11) induced significant Melan-A specific T-cell responses, but not to the extent of the native EAA peptide ligand. Interestingly, the p-nitrobenzyl-bearing compound 11, which was the weakest HLA-A2 ligand of this selection of ELA-mimetics (see Figure 2), was capable of triggering the production of CD107a and IFNγ at notable levels (see Figure 3). The ELA-mimetic 9, which is equipped with a p-fluorobenzyl-type hapten and characterized by a much higher HLA-A2 binding affinity, was found to be a weaker agonist. In contrast to 11, the other two nitrobenzylated peptidomimetics 12 and 13 were not markedly recognized by T-cells, despite their higher HLA-A2 binding affinities (see Figure 2). These results further demonstrate that the level of binding affinity of a ligand to MHC-I proteins does not presume of its immunogenic potency. Nevertheless, it would seem that the 1-(4-nitrobenzyl)-1,2,3-triazole side-chain of 11 confers some minimum threshold for engaging TCRs, even though it is not a good ligand for the HLA-A2 molecule. At this stage of our investigations, we then wondered whether further modifying the structure of 11 by altering its N- and C-terminal portions, which are essential for the loading of any antigenic peptide onto MHC-I proteins and also highly sensitive to the action of exoproteases, would improve immunogenicity.[11a]
Figure 3.

CD107a and IFNγ expressions as measured by intracellular cellular staining (ICS) and quantified by flow cytometry. Data are presented by mean fluorescence intensity (MFI) after 10 μM stimulation with a select set of our ELA-mimetics. Negative control autofluorescence has been subtracted from all values. Positive control (native EAA peptide) is shown.
Modification of N- and C-terminal portions
Structural and biophysical studies have highlighted the crucial role of conserved amino acids at P2 and C-terminal positions in binding to the cleft of HLA-A2 proteins.[7] Thus, to improve the HLA-A2 binding capacity of our clicked ELA-mimetic 11, as well as its resistance against proteolysis, we first envisioned to install a non-natural amino acid that could penetrate more deeply into the hydrophobic pocket occupied by the leucine at P2. To this aim, we selected the non-natural L-cyclohexylalanine (Cha, Figure 4), which was previously used as a substituent of phenylalanine to improve hydrophobic interactions.[28] Moreover, our crystal structure of the ELA-mimetic 1 bound to the HLA-A2 protein[14] revealed an unpredicted conformation of the N-terminal β-alanine disrupting key hydrogen bonds with Lys66 and Glu63 of the HLA-A2 α1 helix. In order to recover these interactions, the β-alanine could potentially be replaced by a phthalimide moiety. Molecular modeling indicated that this rigid, flat dioxoisoindolic system can promote the establishment of intermolecular contacts with the side-chains of α1 helix Lys66 and Glu63 residues (Figure 1).[29] The restoration of hydrogen bonds with these residues should stabilize binary complexes between the HLA-A2 molecule and our ELA-mimetics, and their N-terminal phthalimide unit should further protect them against the action of exoproteases.
Figure 4.

Sequences of the ELA-mimetics 18-24, analogues of the ELA-mimetic 11 designed for improving HLA-A2 binding affinity.
Finally, crystal structures involving the ELA peptide or our ELA-mimetic 1 have shown that the carbonyl oxygen of the leucine at P8 is not engaged with any HLA-A2 residue.[10,14,21] We thus introduced a reduced peptide bond (Ψ(CH2-NH)) between this leucine residue and the adjacent threonine at P9. Such a peptide bond isostere, which provides an additional degree of conformational flexibility, was viewed as a means to favor adequate positioning of the C-terminal extremity of our peptidomimetics. These three types of modifications were implemented in an iterative manner in order to facilitate the determination of their specific contribution to the improvement of the HLA-A2 binding affinity (Figure 4 and Scheme 2). Seven additional clicked ELA-mimetics 18-24, all bearing the same central 1-(4-nitrobenzyl)-1,2,3-triazole side-chain, were thus prepared by conventional solid support synthesis (see the Experimental Section and the Supporting Information).
Gratifyingly, most of the new clicked ELA-mimetics 18-24 exhibited a two- to three-fold improvement in HLA-A2 binding affinity relative to that of 11, with MFI values reaching over two-thirds of that of the ELA peptide, except for 20, being the weakest HLA-A2 binder of this series (Figure 2). In particular, the introduction of the non-natural amino acid Cha in the structure of 18 fulfilled our expectations based on the hypothesis of better occupying the hydrophobic P2 anchoring pocket. The introduction of a phthalimide moiety in place of the N-terminal β-Ala appeared to be also a suitable replacement as the corresponding mimetic 19 bound well to the HLA-A2 molecule. Of course, without any confirmation by X-ray structural analysis, it is, as yet, unclear whether this increase in binding affinity is the consequence of H-bonding interactions between the carbonyls of the phthalimide unit and the side-chains of the HLA-A2 Lys66 and Glu63. Nonetheless, a previous study reported that replacing the N-terminal glutamate by its constrained pyroglutamate lactamic analogue was deleterious for binding to HLA-A2,[11a] whereas herein the phthalimide moiety appeared to be a valuable N-terminal building block. The reduced peptide bond at P8-P9 also seems to be well tolerated, although the mimetic 20 exhibited a slightly weaker HLA-A2 binding affinity relative to those of 18 and 19 (see Figure 2). Nevertheless, when this reduced peptide bond was introduced in combination with the Cha or phthalamide residues or even both, the resulting mimetics 21, 22 and 24 exhibited very good binding affinities, given the number of structural modifications they feature relative to the structure of the native EAA antigenic peptide. All together, these results corroborate our predictions about how to restore good HLA-A2 binding affinity. Moreover, a higher protease resistance of these ELA-mimetics should be observed, since these last modifications were implemented at scissile peptide bonds.[11a]
Evaluating the immunogenicity of this second series of clicked ELA-mimetic variants 18-24 showed that the ELA-mimetic 20 was the most potent inducer of CD107a upregulation and IFN-γ secretion. Only two other peptidomimetics of the series, 18 and 23, managed to induce Melan-A-specific T-cell responses, but to a much lesser extent (Figure 3). Surprisingly, once again, the weakest HLA-A2 binder 20 of the series exhibited the strongest immunogenicity. One possible explanation could be that the lowest binding affinity is the consequence of a higher flexibility of the peptidoid backbone in the HLA-A2 binding groove, which would in turn enable more extensive conformational adjustments upon TCR engagement. The TCR could perhaps even pull the weakly binding peptidomimetic away from the HLA-A2 binding groove for establishing better contacts with both the peptidomimetic and the HLA-A2 surface.[10a,15] The more flexible ELA-mimetic 20 could thus be more capable of fitting its central 1-(4-nitrobenzyl)-1,2,3-triazole side-chain within the TCR CDR3 loops. Even though the observed T-cell responses are about one-third to one-half lower than those triggered by the native EAA antigenic peptide in terms of IFNγ and CD107a secretion, these results constitute an additional and sound experimental demonstration of the possibility of replacing natural amino acid side-chains of antigenic peptides with non-peptidic motifs still capable of eliciting T-cell responses.
Conclusions
A computer-aided design of peptidomimetics was based on the replacement of the central amino acid residues of the Melan-A/MART-1 ELA antigenic peptide by a clickable hapten-presenting platform. This approach enabled the generation of short series of ELA-mimetics equipped with a 1,4-disubstituted 1,2,3-triazole motif bearing variously substituted aromatic moieties. Even though none of our first eleven ELA-mimetics could reach the level of binding affinity to the MHC-I HLA-A2 molecule observed for the ELA peptide, the p-nitrobenzyl-bearing ELA-mimetic 11 was found to elicit Melan-A-specific T-cell responses. Further modifications of its N- and C-extremities aimed at restoring a higher level of HLA-A2 binding affinity led to seven additional ELA-mimetics. All seven compounds were expectedly observed to be much better HLA-A2 binders than 11, but it is the weakest ligand 20 of this series, simply featuring a reduced leucine-to-threonine amide bond, that triggers the most significantly enhanced T-cell stimulation. Thus, this work (i) further demonstrates that the affinity of an antigenic peptide (or mimetics thereof) to MHC-I molecules does not necessarily correlate with immunogenicity, and (ii) clearly highlights the flexibility of TCRs to cross-react with synthetic MHC-I bound peptidomimetics equipped with central organic motifs having structurally little in common with native central amino acids. A recent study demonstrated that a non-biologic T-cell ligand could protect humanized (HLA-A2+) mice against a lethal challenge with influenza virus.[30] This D-peptide had almost no sequence similarity to the wildtype L-peptide sequence and was effective even though HLA-A2 binding was far weaker than that of our agonistic ELA-mimetics.[30] A further study has shown that an altered peptide ligand could induce an improved response to the HLA-A2-restricted EAAGIGILTV epitope studied here.[31] In combination, these new developments suggest that it might be possible to build non-biologic, biostable ligands that induce improved Melan-A/MART-1 specific T-cell responses. Our study here demonstrates that the design of peptidomimetic MHC-I ligands remains a valid option for the elaboration and development of synthetic antigens as immunotherapeutic agents.
Experimental Section
Docking Methodology
All molecular modeling calculations were performed on a Silicon Graphics Octane workstation using Cerius2 4.7 molecular modeling softwares. First, the three-dimensional structure of the A6-TCR/Tax5K-IBA/HLA-A2 complex was taken from the PDB file 2GJ6. The Tax5K-IBA peptide ligand and all water molecules were removed. Hydrogen atoms were added using the Cerius2 templates for the protein residues. The 18 ELA-mimetic structures were constructed using Catalyst. Partial charges were assigned using the Gasteiger method[32] as implemented in Cerius2. A site model based on the Tax5K-IBA ligand, docked within the protein complex, was identified by LigandFit. Then, the docking[33] of the 18 ELA-mimetic ligands employed the following protocol: (a) a Monte Carlo conformational search for generating a candidate ligand conformation, (b) selection of a ligand position and orientation based on comparing the shape of the binding site model with that of the ligand conformation, and (c) evaluation of the goodness of docking by computing the dock energies using a grid-based energy calculation. The dock energy was expressed as the sum of the ligand internal strain energy and the interaction energy of the ligand with the protein. The position and the orientation of the ligand were optimized by minimizing the dock energy in respect to rigid body translations and rotations of the ligand using a steepest descent method. The docked conformations found were then clustered, and the first 20 were saved. A superimposition of the highest docked structure (the lowest in terms of energy) was obtained for each peptidomimetic with plausible consequence of their higher structural diversity. The parental peptide structure from the 2GJ6 PDB file enabled us to gain insight into the consequences of backbone modifications on the interactions with the protein complex.
Material and Methods
All reagents were either purchased from Aldrich, Acros, or Fluka. Amino acids, Wang resin, and N,N,N’,N’-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) were purchased from Novabiochem (Switzerland). All solution phase reactions were carried out under nitrogen (N2) atmosphere with magnetic stirring. Tetrahydrofuran (THF), diethyl ether (Et2O) and dichloromethane (CH2Cl2) were dried through alumina columns. All other organic solvents were of analytical quality and Milli-Q (Millipore) water was used for reverse phase (RP) HPLC analyses and purifications. Peptide and peptidomimetic syntheses were performed manually in a glass reactor (vide infra). RP-HPLC analyses were performed on a Thermo system using a Chromolith performance RP-18e column (4.6 × 100 mm, 5 μm) with P1000 XR pumps. The mobile phase was composed of 0.1% (v/v) TFA-H2O (Solvent A) and 0.1% TFA-CH3CN (Solvent B). A gradient elution (0–10 min: 100% to 50% A) was applied at a flow rate of 3 mL.min−1. Column effluent was monitored by UV detection at 214 and 254 nm using a Thermo UV 6000 LP diode array detector. Semi-preparative purification of peptides were performed on a Varian PrepStar system with SD-1 Dynamax® pumps, using a Microsorb C18 column (2.14 mm × 250 mm, 100 Å pore size, 5 μm). The mobile phase was similar as for the analytic system, unless otherwise notified. A gradient elution (0–40 min: 90% to 50% A) was applied at a flow rate of 20 mL.min−1. Column effluent was monitored by UV detection at 214 and 254 nm using a Varian UV-Vis Prostar 325 diode array detector. Flash column chromatography was carried out under positive pressure using 40–60 μm silica gel (Merck) and the indicated solvents. Evaporations were conducted under reduced pressure at temperatures less than 40 °C. Further drying of the residues was accomplished under high vacuum. NMR spectra of organic molecules were recorded at 300 MHz on a Bruker DPX spectrometer in the appropriated solvent. Electrospray ionization mass spectrometric low- and high-resolution data (ESIMS, HRMS) were obtained from the Mass Spectrometry Laboratory at the European Institute of Chemistry and Biology (IECB), Pessac, France.
Peptidomimetic Synthesis
Peptidomimetics were synthesized by manual solid phase peptide synthesis (SPPS) using Fmoc chemistry on Wang resin (0.65 mmol.g–1) following standard SPPS protocols. Briefly, to a solution of N-Fmoc-Val-OH (10 equiv. relative to resin loading) in dry CH2Cl2 (a few drops of DMF were required to ensure complete dissolution) under N2 was added diisopropylcarbodiimide (DIC, 5 equiv.). The reaction mixture was stirred for 20 min at 0°C. In the meantime, the resin was suspended in dry CH2Cl2 and allowed to swell for 20 min. After filtration, the N-Fmoc-Val symmetrical anhydride solution and 4-dimethylaminopyridine (DMAP, 0.1 equiv. relative to the anhydride) were added, and the resin was shaken for 4 h. After filtration, the resin was successively washed twice with DMF, CH3OH, CH2Cl2 and again DMF. The Fmoc protecting group was removed by using twice a solution of piperidine in DMF (1:4, v/v) for 3 min, then for 7 min. After filtration, the resin was successively washed as before. The SPPS was continued using N-Fmoc amino acids (3 equiv.) in the presence of HBTU (3 equiv.) and DIPEA (5 equiv.) in DMF. Each coupling reaction was performed for 45 min, after which time the resin was washed as before.
Preparation of Resin 5
To the H-AMBA-LTV peptoid construct on Wang resin (1 g, 0.45 mmol) pre-swelled in DMF (5 mL) was added a 0.4 M solution of bromoacetic acid (438 mg, 3.15 mmol) in DMF and a 2 M solution of DIC (562 μL, 3.60 mmol) in DMF (see Scheme 2). After 5 min, the resin was filtrated and washed as described above. Then, a 1 M solution of propargylamine (309 μL, 4.50 mmol) in DMSO was added to the resin and the reaction was stirred for 1 h. After washing of the resin and confirmation of the presence of a secondary amine via the chloranil test, the resin was swollen in a solution of 1% acetic acid (AcOH) in DMF, then Fmoc-Gly-H (567 mg, 2.7 mmol) followed by NaBH3CN (424 mg, 6.75 mmol) were added. After 5 h, the resin was filtered off and successively washed with CH3OH, DMF, CH2Cl2 and again DMF. After removal of the Fmoc-protecting group, the other amino acids were coupled by following standard SPPS protocols as briefly described above to furnish the resin 5 (see Scheme 2).
Preparation of Resins 6 by Click Conjugation
To a suspension of resin 5 in a dry THF-DMF mixture (1:1) was added copper iodide (CuI, 1 mg, 5% M), a solution of each azides R–N3 (10 equiv., see Scheme 2) in dry THF-DMF (1:1) and DIPEA (5 equiv.). After stirring overnight, the resins were washed twice with DMF and CH2Cl2.
Release of ELA-mimetics 7–18 from Resin
Final deprotection and cleavage from the resins were carried out using a TFA-H2O-triisopropylsilane (TIS) mixture (95:2.5:2.5, v/v/v) for 4 h. The peptidomimetics were precipitated using cold Et2O and then filtered. The filtrates were washed with cold Et2O, and the peptidomimetics were extracted from the residues with H2O-CH3CN mixtures containing 0.1% TFA. The resulted solutions were frozen and lyophilized to afford the ELA-mimetics 7-18 as white solids. These crude products were checked for purity by RP-HPLC and purified by semi-preparative RP-HPLC.
Preparation of Resin-Bound ELA-mimetics 20–22 and 24
To the H-TV dipeptide construct on Wang resin (270 mg, 0.13 mmol) pre-swelled in a solution of 1% AcOH in DMF was added Fmoc-Leu-H[34] (0.40 mmol) and NaBH3CN (59 mg, 0.94 mmol). The resin was shaken for 2 h after which time it was filtered off and successively washed with MeOH, DMF, CH2Cl2 and again DMF. Coupling with the AMBA residue was then performed, and the preparation of these resin-bound ELA-mimetics was continued according to the general procedures described for the preparation of resins 5 and 6.
Incorporation of the Phthalimide Moiety on Resin-Bound ELA-mimetics 19 and 22–24.[35]
To the corresponding resin-bound peptidomimetic intermediates without the terminal β-Ala residue and suspended in DMF was added phthalimide anhydride (5 equiv.) and DIPEA (5 equiv.). After 1 h, each resulting resin was filtered off and washed according to our standard procedure (vide infra). Each resin was again swollen in DMF, and HBTU (1.5 equiv.), hydroxybenzotriazole (HOBt, 1.5 equiv.) and DIPEA (3 equiv.) were successively added. After stirring overnight, the resins were filtered off and washed according to our standard procedure.
The release from resin and final deprotection of ELA-mimetics 19-24 were carried out as described for the ELA-mimetics 7-18 (vide infra).
Peptidomimetic-MHC binding assay
The mutant LCL T-lymphoblastoid hybrid cell line, 174xCEM.T2 (referred to as T2 cells) is an antigen-presenting mutant line. The cells express stable HLA-A2 molecules on their surface upon addition of an exogenous HLA-A2-binding peptide. T2 cells were incubated with the ELA-mimetics 7–24 at 10 μM in AIM-V serum-free medium (Invitrogen Co., Carlsbad, California) at 26 °C for 16 h, followed by incubation at 37 °C for 2 h. Quantification of stable surface HLA was analyzed by surface staining using the anti-HLA-A2-FITC BB7.2 antibody (from the ATCC hybridoma HB-82), and measured as mean fluorescent intensity (MFI) on a FACSCanto II flow cytometer (BD Biosciences).
T-cell Stimulation
2×106 HLA-A2+ PBMC were cultured for 3 days in in the presence of 10 μM of the native EAA peptide in RPMI media in a 24-well plate. At day 3, the culture was supplemented with T-cell media (10% human serum in RPM, R10). At day 10, the T-cell culture resulted in approximately 15% of the total CD8+ population measured by Melan-A-HLA-A2 APC tetramer by flow cytometry. For intracellular staining, T cells were rested overnight at 1×106 per mL in R2 (as for R10 with 2% FCS) and added to peptide-pulsed targets (i.e., HLA A*0201+-C1R pulsed with 10 μM of the native EAA peptide or our ELA-mimetics) at an effector:target ratio of 1:2 (i.e., T-cell culture effectors:HLA A*0201+-C1R targets) in the presence of 5 μg/mL brefeldin A (Sigma-Aldrich), 0.35 μl/mL monensin and 5 μl/mL αCD107a-FITC (BD Biosciences). After 5 h at 37 °C, the cells were washed and stained with LIVE/DEAD Fixable Aqua (Life Technologies) followed by anti-CD3-PacificBlue, anti-CD8-APCH7 (BioLegend). The cells were then fixed/permeabilized using a Cytofix/Cytoperm Kit (BD Biosciences) and stained intracellularly with anti-IFNγ-PECy7 on ice for 30 min. Data were acquired using a FACSCanto II flow cytometer (BD Biosciences) and analyzed with FlowJo software (Tree Star Inc.). Cell population gates were set using fluorescence minus one staining control. ICS MFIs are shown.
Supplementary Material
Acknowledgements
The authors thank the Servier Laboratories and the Société Française de Chimie Thérapeutique for financial support and M.T.’s research assistantship. This work was also supported by the Biotechnology and Biological Sciences Research Council (grant BB/H001085/1 to A.K.S.), and by the National Institute of General Medical Sciences, NIH (grant R35GM118166 to B.M.B). J.J.M was supported by an NHMRC CDF2 APP1131732 fellowship, and E.S.J.E by a studentship from the MRC and Cardiff University’s I3 Interdisciplinary Research Group. Dr Céline Douat is gratefully acknowledged for her contributions to the design and synthesis of the peptidomimetics and for her co-supervisorship on M.T.’s doctoral research studies, Mathew Clement and Kristin Ladell for their contributions to the production of T-cell lines and FACS analysis, Prof Linda Wooldridge (University of Bristol, UK) for her supervision and advice concerning the T-cell cytokine production experiments, and Dr Nathalie Marchand-Geneste for her advices in the realization of the docking modeling procedure.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Hofbauer GF, Kamarashev J, Geertsen R, Boni R, Dummer R, Melanoma Res. 1998, 8, 337–343. [DOI] [PubMed] [Google Scholar]
- [2].Romero P, Gervois N, Schneider J, Escobar P, Valmori D, Pannetier C, Steinle A, Wolfel T, Lienard D, Brichard V, van Pel A, Jotereau F, Cerottini JC, J. Immunol 1997, 159, 2366–2374. [PubMed] [Google Scholar]
- [3].Saint-Jean M, Knol A-C, Volteau C, Quéreux G, Peuvrel L, Brocard A, Pandolfino M-C, Saiagh S, Nguyen J-M, Bedane C, Basset-Seguin N, Khammari A, Dréno B, J. Immunol. Res 2018, 3530148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rius C, Attaf M, Tungatt K, Bianchi V, Legut M, Bovay A, Donia M, thor Straten P, Peakman M, Svane IM, Ott S, Connor T, Szomolay B, Dolton G, Sewell AK, J. Immunol 2018, 200, 2263–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Retseck J, Nasr A, Lin Y, Lin H, Mendiratta P, Butterfield LH, Tarhini AA, J. Transl. Med 2018, 16, 184–194; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Simon S, Vignard V, Varey E, Parrot T, Knol A-C, Khammari A, Gervois N, Lang F, Dreno B, Labarriere N, Cancer Res. 2017, 77, 7083–7093. [DOI] [PubMed] [Google Scholar]
- [6].Chapatte L, Servis C, Valmori D, Burlet-Schiltz O, Dayer J, Monsarrat B, Romero P, Lévy F, J. Immunol 2004, 173, 6033–6040. [DOI] [PubMed] [Google Scholar]
- [7].Rudolph MG, Stanfield RL and Wilson IA, Annu. Rev. Immunol 2006, 24, 419–466. [DOI] [PubMed] [Google Scholar]
- [8].Valmori D, Fonteneau JF, Maranon Lizana C, Gervois N, Lienard D, Rimoldi D, Jongeneel CV, Jotereau F, Cerottini JC, Romero P, J. Immunol 1998, 160, 1750–1758. [PubMed] [Google Scholar]
- [9].a) Wieckowski S, Baumgaertner P, Corthesy P, Voelter V, Romero P, Speiser DE, Rufer N, J. Immunol 2009, 183, 5397–5306; [DOI] [PubMed] [Google Scholar]; b) Speiser DE, Baumgaertner P, Voelter V, Devevre E, Barbey C, Rufer N, Romero P, Proc. Natl. Acad. Sci. USA 2008, 105, 3849–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Madura F, Rizkallah PJ, Holland CJ, Fuller A, Bulek A, Godkin AJ, Schauenburg AJ, Cole DK, Sewell AK, Eur. J. Immunol 2015, 45, 584–591; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cole DK, Edwards ESJ, Wynn KK, Clement M, Miles JJ, Ladell K, Ekeruche J, Gostick E, Adams KJ, Skowera A, Peakman M, Wooldridge L, Price DA, Sewell AK, J. Immunol 2010, 185, 2600–2610; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Borbulevych OY, Insaidoo FK, Baxter TK, Jr Powell DJ, Johnson LA, Restifo NP, Baker BM, J. Mol. Biol 2007, 372, 1126–1136; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sliz P, Michielin O, Cerottini JC, Luescher I, Romero P, Karplus M and Wiley DC, J. Immunol 2001, 167, 3276–3284. [DOI] [PubMed] [Google Scholar]
- [11].a) Blanchet JS, Valmori D, Dufau I, Ayyoub M, Nguyen C, Guillaume P, Monsarrat B, Cerottini JC, Romero P, Gairin JE, J. Immunol 2001, 167, 5852–5861; [DOI] [PubMed] [Google Scholar]; b) Beck A, Bussat M-C, Klinguer-Hamour C, Goetsch L, Aubry J-P, Champion T, Julien E, Haeuw J-F, Bonnefoy J-Y, Corvaia N, J. Peptide Res 2001, 57, 528–538. [DOI] [PubMed] [Google Scholar]
- [12].Croft NP, Purcell AW, Expert Rev. Vaccines 2011, 10, 211–226. [DOI] [PubMed] [Google Scholar]
- [13].a) Douat-Casassus C, Marchand-Geneste N, Diez E, Aznar C, Picard P, Geoffre S, Huet A, Bourguet-Kondracki M-L, Gervois N, Jotereau F, Quideau S, Mol. BioSyst 2006, 2, 240–249; [DOI] [PubMed] [Google Scholar]; b) Douat-Casassus C, Marchand-Geneste N, Diez E, Gervois N, Jotereau F, Quideau S, J. Med. Chem 2007, 50, 1598–1609; [DOI] [PubMed] [Google Scholar]; c) Tarbe M, Azcune I, Balentova E, Miles JJ, Edwards EE, Miles KM, Do P, Baker BM, Sewell AK, Aizpurua JM, Douat-Casassus C, Quideau S, Org. Biomol. Chem 2010, 8, 5345–5353. [DOI] [PubMed] [Google Scholar]
- [14].Douat-Casassus C, Borbulevych O, Tarbe M, Gervois N, Jotereau F, Baker BM, Quideau S, J. Med. Chem 2010, 53, 7061–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Madura F, Riskallah PJ, Legut M, Holland CJ, Fuller A, Bulek A, Schauenburg AJ, Trimby A, Hopkins JR, Wells SA, Godkin A, Miles JJ, Sami M, Li Y, Liddy N, Jacobsen BK, Loveridge EJ, Cole DK, Sewell AK, Eur. J. Immunol 2019, 49, 1052–1066; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ayres CM, Corcelli SA, Baker BM, Front. Immunol 2017, 8, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Huisgen R, 1,3-Dipolar Cycloaddition Chemistry, Wiley, New York, 1984; [Google Scholar]; b) Tornøe CW, Christensen C, Meldal M, J. Org. Chem 2002, 67, 3057–3064; [DOI] [PubMed] [Google Scholar]; c) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB, Angew. Chem. Int. Ed 2002, 41, 2596–2599; [DOI] [PubMed] [Google Scholar]; d) Kolb HC, Finn MG, Sharpless KB, Angew. Chem. Int. Ed 2001, 40, 2004–2021; [DOI] [PubMed] [Google Scholar]; e) Kolb HC, Sharpless KB, Drug Discovery Today 2003, 3, 1128–1137; [DOI] [PubMed] [Google Scholar]; f) Meldal M, Tornøe CW, Chem. Rev 2008, 108, 2952–3015. [DOI] [PubMed] [Google Scholar]
- [17].a) Angell YL, Burgess K, Chem. Soc. Rev 2007, 36, 1674–1689; [DOI] [PubMed] [Google Scholar]; b) Gopi HN, Tirupula KC, Baxter S, Ajith S, Chaiken IM, ChemMedChem, 2006, 1, 54–57, and references cited therein; [DOI] [PubMed] [Google Scholar]; c) J Pieters R, Rijkers DTS, Liskamp RMJ, QSAR Comb. Sci 2007, 26,1181–1190; [Google Scholar]; d) Fournier D, Hoogenboom R, Schubert US, Chem. Soc. Rev 2007, 36, 1369–1380; [DOI] [PubMed] [Google Scholar]; e) Gopi H, Umashankara M, Pirrone V, LaLonde J, Madani N, Tuzer F, Baxter S, Zentner I, Cocklin S, Jawanda N, Miller SR, Schön A, Klein JC, Freire E, Krebs FC, Smith AB, Sodroski J, Chaiken I, J. Med. Chem 2008, 51, 2638– 2647; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Sokolova NV, Nenajdenko VG, RSC Advances 2013, 3, 16212–16242; [Google Scholar]; g) Li H, Aneja R, Chaiken I, Molecules 2013, 18, 9797–9817; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Romanelli A, Affinito A, Avitabile C, Catuogno S, Ceriotti P, Iaboni M, Modica J, Condorelli G, Catalucci D, PlosONE 2018, 13, e0193392; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Bundle DR, Paszkiewicz E, Elsaidi HRH, Mandal SS, Sarkar S, Molecules 2018, 23, 1961–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sewell AK, Nat. Rev. Immunol 2012, 12, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Garcia KC, Degano M, Pease LR, Huang M, Peterson PA, Teyton L, Wilson IA, Science, 1998, 279, 1166–1172; [DOI] [PubMed] [Google Scholar]; b) Reiser JB, Grégoire C, Darnault C, Mosser T, Guimezanes A, Schmitt-Verhulst AM, Fontecilla-Camps JC, Mazza G, Malissen B, Housset D, Immunity, 2002, 16, 345–354; [DOI] [PubMed] [Google Scholar]; c) Tynan FE, Reid HH, Kjer-Nielsen L, Miles JJ, Wilce MC, Kostenko L, Borg NA, Williamson NA, Beddoe T, Purcell AW, Burrows SR, McCluskey J, Rossjohn J, Nat. Immunol 2007, 8, 268–276; [DOI] [PubMed] [Google Scholar]; d) Baker BM, Scott DR, Blevins SJ, Hawse WF, Immunol. Rev 2012, 250, 10–31; [DOI] [PubMed] [Google Scholar]; e) Holland CJ, MacLachlan BJ, Bianchi V, Hesketh SJ, Morgan R, Vickery O, Bulek AM, Fuller A, Godkin A, Sewell AK, Rizkallah PJ, Wells S, Cole DK, Front. Immunol 2018, 9, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gagnon SJ, Borbulovych OY, Davis-Harrison RL, Turner RV, Damirjian M, Wojnarowicz A, Biddison WE, Baker BM, J. Mol. Biol 2006, 363, 228–243. [DOI] [PubMed] [Google Scholar]
- [21].Cole DK, Yuan F, Rizkallah PJ, Miles JJ, Rizkallah, Gostick E, Price DA, Gao GF, Jakobsen BK, Sewell AK, J. Biol. Chem 2009, 284, 27281–27289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Tornøe CW, Sanderson SJ, Mottram JC, Coombs GH, Meldal M, J. Comb. Chem 2004, 6, 312–324; [DOI] [PubMed] [Google Scholar]; b) Zhang ZS, Fan E, Tetrahedron Lett. 2006, 47, 665–669. [Google Scholar]
- [23].a) Jagasia R, Holub JM, Bollinger M, Kirshenbaum K, Finn MG J. Org. Chem 2009, 74, 2964–2974; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gopi HN, Tirupula KC, Baxter S, Ajith S, Chaiken IM, ChemMedChem, 2006, 1, 54–57. [DOI] [PubMed] [Google Scholar]
- [24].Jang H, Fafarman A, Holub JM, Kirshenbaum K, Org. Lett 2005, 7, 1951–1954. [DOI] [PubMed] [Google Scholar]
- [25].Zhang ZS, Fan E, Tetrahedron Lett. 2006, 47, 665–669. [Google Scholar]
- [26].Tan MP, Gery AB, Brewer JE, Melchiori L, Bridgeman JS, Bennett AD, Pumphrey NJ, Jacobsen BK, Price DA, Ladell K, Sewell AK, Clin. Exp. Immunol 2014, 180, 255–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA, J. Immunol. Methods 2003, 281, 65–78; [DOI] [PubMed] [Google Scholar]; b) Rubio V, Stuge TB, Singh N, Betts MR, Weber JS, Roederer M, Lee PP, Nat. Med 2003, 9, 1377–1382. [DOI] [PubMed] [Google Scholar]
- [28].a) Bolin DR, Swain AL, Sarabu R, Berthel SJ, Gillespie P, Huby NJ, Makofske R, Orzechowski L, Perrotta A, Toth K, Cooper JP, Jiang N, Falcioni F, Campbell R, Cox D, Gaizband D, Belunis CJ, Vidovic D, Ito K, Crowther R, Kammlott U, Zhang X, Palermo R, Weber D, Guenot J, Nagy Z, Olson GL J. Med. Chem 2000, 43, 2135–2148; [DOI] [PubMed] [Google Scholar]; b) Falcioni F, Ito K, Vidovic D, Belunis C, Campbell R, Berthel SJ, Bolin DR, Gillespie PB, Huby N, Olson GL, Sarabu R, Guenot J, Madison V, Hammer J, Sinigaglia F, Steinmetz M, Nagy ZA, Nat. Biotechnol 1999, 17, 562–567. [DOI] [PubMed] [Google Scholar]
- [29].Beck A, Bussat MC, Klinguer-Hamour C, Goetsch L, Aubry JP, Champion T, Julien E, Haeuw JF, Bonnefoy JY, Corvaia N, J. Pept. Res 2001, 57, 528–538. [DOI] [PubMed] [Google Scholar]
- [30].Miles JJ, Tan MP, Dolton G, Edwards ESJ, Galloway SAE, Laugel B, Clement M, Makinde J, Ladell K, Matthews KK, Watkins TS, Tungatt K, Wong Y, Siean Lee H, Clark RJ, Pentier JM, Attaf M, Lissina A, Ager A, Gallimore A, Rizkallah PJ, Gras S, Rossjohn J, Burrows SR, Cole DK, Price DA, Sewell AK, J. Clin. Invest 2018, 128, 1569–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Galloway SAE, Dolton G, Attaf M, Wall A, Fuller A, Rius C, Bianchi V, Theaker S, Lloyd A, Caillaud ME, Svane IM, Donia M, Cole DK, Szomolay B, Rizkallah P, Sewell AK, Front. Immunol 2019, 10, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gasteiger J, Marsili M, Tetrahedron Lett. 1978, 19, 3181–3184. [Google Scholar]
- [33].Nagy A, Armatis P, Z Cai R, Szepeshazi K, Halmos G, Vieth M, Hirst JD, Dominy BN, Daigler H, Brooks CL, J. Comput. Chem 1998, 19, 1623–1631. [Google Scholar]
- [34].Douat C, Heitz A, Martinez J, Fehrentz JA, Tetrahedron Lett. 2000, 41, 37–40. [Google Scholar]
- [35].Eugenio Vázquez M, Rothman DM, Imperiali B, Org. Biomol. Chem, 2004, 2, 1965–1966. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.