
Figure 2.
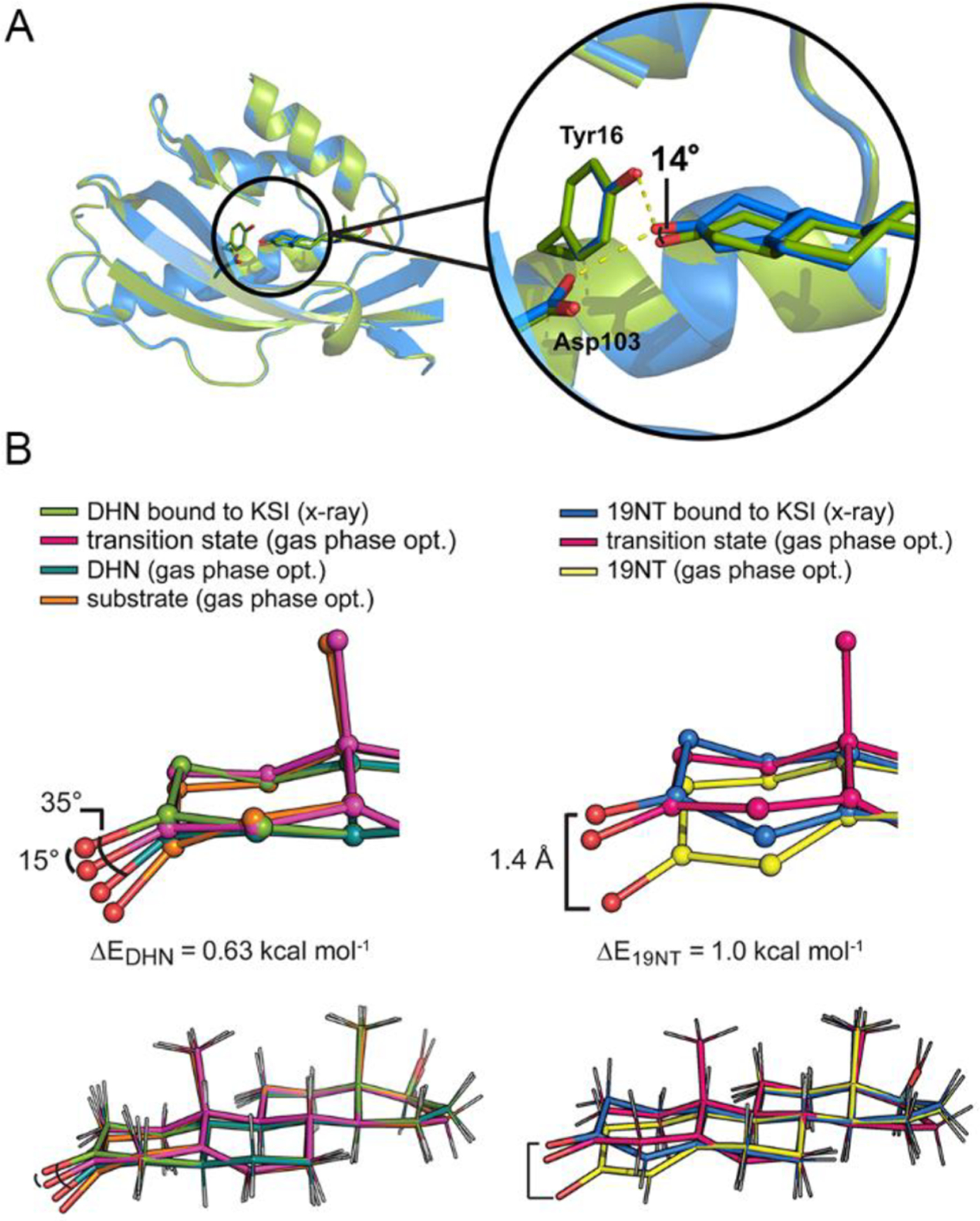
Crystal structures of wild type KSI bound with DHN (6UFS) and with 19NT (5KP4) show perturbed geometry of the ligands. (A) The structure of KSI bound to DHN (green, 1.5 Å) is globally aligned with that of KSI bound to 19NT (blue, 1.7 Å), giving an RMS deviation of 0.177 Å. Within this alignment frame, the angle shift between the carbonyl of DHN and the carbonyl of 19NT is about 14°, smaller than the predicted difference of 31° from the optimized gas-phase geometries (Figure 1B). (B) The C=O bonds of DHN and 19NT get distorted from their gas-phase optimal geometries to more closely resemble the TS structure when bound to KSI. A small energy penalty (ΔE) is calculated from the energy difference between the gas-phase optimal structure of the ligand and the gas-phase structure “constrained” to the crystal coordinates (Figure 3 and Figure S3).