

Abstract
Alzheimer’s disease (AD) is pathologically defined by extracellular accumulation of amyloid-β (Aβ) peptides generated by the cleavage of amyloid precursor protein (APP), strings of hyperphosphorylated Tau proteins accumulating inside neurons known as neurofibrillary tangles (NFTs) and neuronal loss. The association between the two hallmarks and cognitive decline has been known since the beginning of the 20th century when the first description of the disease was carried out by Alois Alzheimer. Today, more than 40 million people worldwide are affected by AD that represents the most common cause of dementia and there is still no effective treatment available to cure the disease. In general, the aggregation of Aβ is considered an essential trigger in AD pathogenesis that gives rise to NFTs, neuronal dysfunction and dementia. During the process leading to AD, tau and Aβ first misfold and form aggregates in one brain region, from where they spread to interconnected areas of the brain thereby inducing its gradual morphological and functional deterioration. In this mini-review article, we present an overview of the current literature on the spreading mechanisms of Aβ and tau pathology in AD since a more profound understanding is necessary to design therapeutic approaches aimed at preventing or halting disease progression.
Keywords: Alzheimer’s disease, amyloid-β, tau, propagation, spreading
The Spread of Tau
The tau protein is a phosphoprotein that is codified by alternative splicing of the microtubule-associate protein tau (MAPT) gene (Goedert et al., 1988, 1989; Andreadis et al., 1992; Andreadis, 2005; Pittman et al., 2006) and is enriched in axons of mature neurons where it regulates microtubule stability to ensure proper cytoskeletal organization and trafficking (Aamodt and Williams, 1984; Aronov et al., 2001; González-Billault et al., 2002; Zhang et al., 2004; Bertrand et al., 2013). Of all different post-translational modifications that tau can undergo, the phosphorylation is of particular interest because of its involvement in a group of neurodegenerative disorders known as tauopathies (Goedert and Spillantini, 2011; Arendt et al., 2016), including Alzheimer’s disease (AD). Indeed, whereas phosphorylation is fundamental for tau function under physiological conditions, the affinity of tau for tubulin decreases under pathological conditions, and the protein starts to accumulate in the cytosol of the somatodendritic compartments where insoluble structures are built. Those neurofibrillary tangles (NFTs) disturb the microtubule network and alter the normal axoplasmatic flow, which in turn compromises the functions and viability of neurons (Brion et al., 1985; Wood and Zinsmeister, 1989; Gastard et al., 2003). In AD, the development of NFTs evolves in the brain with a predictable and hierarchical distribution pattern that starts from layer II of the entorhinal cortex, spreads through the limbic and associations areas to finally reach the hippocampus and neocortex (Braak and Braak, 1991). Pathological tau can distribute from one cell to another thereby propagating the pathology from affected to interconnected healthy areas of the brain, implicating similar mechanisms than in prion diseases (Liu et al., 2012; Jucker and Walker, 2013). A large number of pieces of evidence support a prion-like model for tau spreading, consisting of abnormal proteins with the capacity to convert normal proteins into a pathological form. The inoculation of brain extracts from mice or humans with tauopathy into the brain of wild-type animals induced tau pathology in recipient animals and its propagation from the site of injection along neuronal connections (Clavaguera et al., 2009, 2013; Lasagna-Reeves et al., 2012; Ahmed et al., 2014; Guo et al., 2016; Gibbons et al., 2017; Narasimhan et al., 2017; Smolek et al., 2019a,b). Similar results were obtained when synthetic tau fibrils were injected into young mice overexpressing mutant human tau (P301S) which resulted as well in the formation of NFT-like inclusions that propagated from the injected sites to connected brain regions in a time-dependent manner (Iba et al., 2013). Experiments of human tau viral induction in cortical neurons in young vs old mice showed age- and brain region-dependent misfolding and spreading of tau (Wegmann et al., 2019). Furthermore, in vitro studies showed, that extracellular aggregates of tau can be internalized by naïve cells promoting fibrillization of intracellular tau that can be transferred between co-cultured cells (Frost et al., 2009; Guo and Lee, 2011, 2013) also via synaptic contacts between neurons that facilitate pathology propagation (Calafate et al., 2015). Tau propagation was extensively studied and different mechanisms involved in trans-cellular diffusion were described. The prion-like propagation implies an active and regulated passage of tau in the extracellular space (secretion) and a mechanism of tau uptake by an adjacent recipient cell, although the passive release of tau from dying cells cannot be excluded as an alternative scenario.
Tau filaments can exist in the brain as a variety of distinct conformational strains associated with various tauopathy phenotypes and different rates of network propagation (Sanders et al., 2014; Guo et al., 2016; Kaufman et al., 2016; He et al., 2020). Although the distribution of tau with different conformations can be suggested as cause also of AD heterogeneity, high-resolution analysis of tau structures by using cryo-electron microscopy recently revealed no significant variation in tau filament structures within and between the brains of individuals with AD (Fitzpatrick et al., 2017; Falcon et al., 2018).
Tau can be actively secreted by neurons following three main routes (Figure 1):
Figure 1.
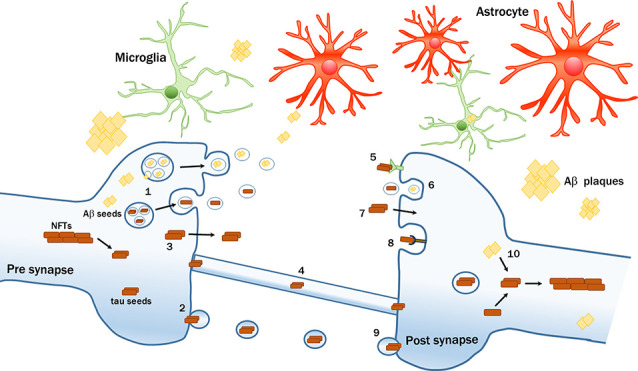
Intercellular transmission of pathological amyloid-β (Aβ) and tau proteins. Seeds of pathological proteins can be released at the presynaptic level: (1) in exosomes after the fusion of Multivesicular bodies (MVBs) with plasma membrane (PM); (2) in larger vesicles called ectosomes; (3) as naked protein freely crossing the PM; or (4) can be transferred via tunneling nanotubes. Mechanisms of up-take by recipient neurons include; (5) receptors-mediated endocytosis; (6) bulk-endocytosis; (7) fluid-phase translocation; (8) macropinocytosis mediated by heparan sulfate proteoglycans (HSPGs); and (9) fusion of large tau-containing large vescicles with PM. (10) Intraneuronal Aβ seeds can trigger or enhance the formation of tau pathological aggregates. The transmission process can be modulated by multiple factors, including glial cells.
First, at the presynaptic level, tau can be packed into microvesicles and further released by a process (Simón et al., 2012; Fontaine et al., 2016), that is modulated by neuronal electrical activity (Lachenal et al., 2011; Pooler et al., 2013; Yamada et al., 2014; Sokolow et al., 2015; Wang et al., 2017). Following this route, phosphorylated tau is internalized by cytoplasmic exosomes, so-called intraluminal vesicles formed in multivesicular bodies (MVBs) that are finally released into the extracellular space after MVBs fusion with the plasma membrane (PM; Saman et al., 2012). Alternatively, tau can also be internalized in ectosomes, larger vesicles (100–500 nm in diameter) that are formed by evaginations of the PM incorporating tau (Dujardin et al., 2014). These routes are unconventional secretion pathways since they do not involve signal peptides and exclude the endoplasmic reticulum (ER)-Golgi system. Extracellular vesicles containing phospho-tau were found in the brains of transgenic mice (Baker et al., 2016; Polanco et al., 2016) and in peripheral fluids such as blood or CSF of AD patients (Saman et al., 2012; Fiandaca et al., 2015; Winston et al., 2016).
Second, the majority of tau is found extracellularly as a membrane-free form. Soluble hyperphosphorylated tau can translocate directly across the PM (Plouffe et al., 2012; Pooler et al., 2012) upon interaction with phosphatidylinositol 4,5 phosphate PI(4,5)P2 cholesterol and sphingolipids. The penetration and release are facilitated by the binding with heparan sulfate proteoglycans (HSPG) on the cell surface (Katsinelos et al., 2018; Mari et al., 2018; Merezhko et al., 2018). Other studies suggested that tau is secreted via the fusion of vesicles from ER or Golgi with the PM (Ponnambalam and Baldwin, 2003). Tau was also shown to form pore-like structures in the PM that operate like channels for the passage of pathogenic proteins, a feature that can be regulated either by pathological mutations, by tau oligomerization (Lasagna-Reeves et al., 2014; Patel et al., 2015; Merezhko et al., 2018), or by specific sequences of human tau that act as binding motifs to facilitate the secretion of pathological tau (Sayas et al., 2019).
Third, another mechanism that has also been proposed involves the passage through tunneling nanotubes, filamentous actin-containing channels that connect adjacent cells, and transport proteins intercellularly (Abounit et al., 2016; Tardivel et al., 2016).
Once released, tau can be internalized by recipient cells (Figure 1). Intracranial or peripheral administration of pathological tau (Clavaguera et al., 2014; Mudher et al., 2017) together with in vitro experiments have shown that tau is mainly internalized by active endocytic processes (Frost et al., 2009; Wu et al., 2013). In particular, three kinds of endocytosis were described:
Bulk-endocytosis represents the first one, where a large portion of presynaptic PM is internalized in a dynamin-dependent manner in form of vacuoles or endosomes from which multiple synaptic vesicles can subsequently be generated (Takei et al., 1996; Wu et al., 2013).
The second, actin-dependent macropinocytosis is mediated by HSPGs on the cell surface (Holmes et al., 2013; Rauch et al., 2018; Weisová et al., 2019). Recently, the silencing of the low-density lipoprotein receptor-related protein-1 (LRP1), which works in conjunction with HSPGs, was shown to block the uptake of tau oligomers in vitro and reduced in vivo the propagation of tau between neurons (Rauch et al., 2020). Other receptors can also be involved in the uptake of pathogenic tau, such as the extracellular portion of amyloid precursor protein (APP; Takahashi et al., 2015) and muscarinic receptors (Morozova et al., 2019). Recently, the cellular prion protein was also shown to act as a receptor that facilitates the uptake of tau aggregates by cultured cells (De Cecco et al., 2020).
Finally, clathrin-mediated endocytosis was also proposed as mechanism (Evans et al., 2018), but is still under debate because the use of specific clathrin inhibitors or its silencing resulted in continued tau aggregate uptake (Calafate et al., 2016).
In general, the different mechanisms of secretion and internalization depend largely on the cell types, the size, and the different tau species involved (Dujardin et al., 2018; Evans et al., 2018). Once tau is internalized, it can escape the endosomal vesicles inducing their rupture (Calafate et al., 2016; Flavin et al., 2017) and accumulates in the cytoplasm where it becomes a potential template for the misfolding of tau (Clavaguera et al., 2009; Guo et al., 2016; Figure 1). Although the biochemical mechanisms driving the conversion of normal tau into the pathological form are still unclear, different models of tau seeding were proposed (Congdon et al., 2008; Mirbaha et al., 2018).
Glial cells such as astrocytes (Martini-Stoica et al., 2018; Perea et al., 2019), oligodendrocytes (Narasimhan et al., 2017) and in particular microglia were implicated in tau spreading (Asai et al., 2015; Maphis et al., 2015; Hopp et al., 2018). Recently, a study demonstrated that microglia isolated from AD cases and mouse models of tauopathy contain tau seeds that can be released into the medium (Hopp et al., 2018), proposing that microglia can uptake tau but not to completely digest it, thus representing a possible source for tau spreading. The ability of microglia to engulf tau aggregates was already documented by different in vitro and in vivo studies (Luo et al., 2015; Bolós et al., 2016, 2017). Moreover, microglia depletion was shown to prevent tau propagation. Microglia phagocytosed and released tau-containing exosomes whereas inhibiting exosome synthesis significantly reduced tau propagation in vitro and in vivo (Asai et al., 2015). Finally, increased microglial activation has been reported not only to accelerate tau pathology and behavioral abnormalities in the human Tau mouse model of tauopathy (Bhaskar et al., 2010; Bemiller et al., 2017; Ising et al., 2019), but also its propagation in the brain (Maphis et al., 2015). Together, these studies highlight the involvement of microglia in spreading tau pathology.
The Spread of Aβ
Aβ is a proteolytic product of APP, that is highly expressed in neurons and physiologically involved in many functions such as regulation of neurite outgrowth and axonal guidance, regulation of synaptic functions and plasticity, involvement in early nervous system development and in neuroprotection (Van den Heuvel et al., 1999; Leyssen et al., 2005; Priller et al., 2006; Young-Pearse et al., 2008; Mueller et al., 2018). APP can be processed in two different ways: in the non-amyloidogenic pathway, APP is cleaved first by α- followed by γ-secretase that cuts the protein within the Aβ domain. In the amyloidogenic pathway, APP is consecutively cut by β- and γ-secretase to be finally released extracellularly as Aβ fragments of different lengths, but mainly consist of 40 (Aβ1–40) or 42 (Aβ1–42) amino acids (Haass et al., 2012). Once released, monomeric Aβ can aggregate into different assemblies giving origin to oligomers, protofibrils, and amyloid fibrils that are insoluble and can further aggregate into amyloid plaques, while monomeric and oligomeric forms of Aβ are soluble. These different states of Aβ coexist in the AD brain making it difficult to dissect the most relevant and toxic forms concerning pathogenesis. Albeit in vivo studies demonstrated that Aβ plaques lead to neuronal loss, neuronal dystrophy and alters their normal neuritic functionality (Meyer-Luehmann et al., 2008, 2009; Shah et al., 2010), different studies have identified soluble oligomeric Aβ species as the toxic drivers responsible for synaptic dysfunction, in particular in the early stage of the disease (Lambert et al., 1998; Shankar et al., 2008; Koffie et al., 2009; Forloni et al., 2016). The fact that the incidence of senile plaques increases with age even in healthy subjects and that the number of plaques often does not correlate with neuronal loss and cognitive decline (Katzman, 1988; Villemagne and Rowe, 2011) nourishes the hypothesis that compact plaques may sequester toxic Aβ oligomers until they reach a saturation point (Esparza et al., 2013; Selkoe and Hardy, 2016). Although the ultimate proof for a causal relationship between fibrillar aggregates and neurodegenerative diseases has not been delivered yet, the “amyloid cascade hypothesis” is still the most prevalent theory (Hardy and Higgins, 1992; Hardy and Selkoe, 2002) with the constrain that Aβ alone is most likely not able to cause the entire damage found in AD patient brains (Ricciarelli and Fedele, 2017). In addition to their different aggregation state, Aβ was detected in the brain of AD patients in distinct misfolded strains with specific propagation properties (Qiang et al., 2017; Condello and Stöehr, 2018), suggesting that also these structural variations may modulate the disease phenotype.
Unlike tau that spreads in a highly predictive pattern as anticipated by computational systems (Fornari et al., 2019), Aβ deposition does not always follow a stereotypic spatio-temporal pattern of progression. Nevertheless, amyloid plaques in general first appear in the neocortex from where they spread into the allocortex and the subcortical regions (Thal et al., 2002; Serrano-Pozo et al., 2011; Grothe et al., 2017). As has been described above for tau, several studies on intracerebral injections of Aβ-rich brain extracts either from AD mice or patients propose that Aβ aggregation can be initiated by prion-like seeding (Kane et al., 2000; Meyer-Luehmann et al., 2006; Eisele et al., 2009; Jucker and Walker, 2013; Ziegler-Waldkirch et al., 2018; Friesen and Meyer-Luehmann, 2019; Katzmarski et al., 2020). These misfolded protein assemblies act as seeds of aggregation to accelerate the polymerization processes of normal proteins (Harper and Lansbury, 1997) that can expand from the injection site to distant regions as well as the contro-lateral side of the brain, thus suggesting a possible spread of seeded pathology via neuronal transport along axonally interconnected brain regions (Walker et al., 2002; Rönnbäck et al., 2012; Domert et al., 2014; Ye et al., 2015).
Aβ peptides are collected in intraluminal vesicles within MVBs and, upon fusion with the PM, the intraluminal vesicles are released into the extracellular space as exosomes (Rajendran et al., 2006; Sharples et al., 2008; Hu et al., 2009; Figure 1). Furthermore, a recent study reported that Aβ-rich exosomes isolated from AD patients can act as vehicles for cell-to-cell transfer of such toxic species in recipient cultured neurons (Sinha et al., 2018). However, even though the cell-to-cell passage of Aβ represents a plausible hypothesis, substantiated by the fact that the protein is found inside neurons (Wertkin et al., 1993; Turner et al., 1996; Gouras et al., 2000; LaFerla et al., 2007), there is no conclusive evidence for active transport of Aβ along neurons. Transplantations of WT neurons into brains of pre-depositing AD mice revealed that Aβ from the transgenic host tissue can enter and deposit within WT grafts (Meyer-Luehmann et al., 2003; Bachhuber et al., 2015; Espuny-Camacho et al., 2017), thus suggesting a possible passive extracellular diffusion of Aβ from the outside to the inside of the grafts. Although over the last few years many different groups have described glial cells as an alternative source of Aβ (Siman et al., 1989; Joshi et al., 2014) or their involvement in the formation of amyloid plaque deposition (Wisniewski et al., 1990; Venegas et al., 2017; Spangenberg et al., 2019), there are to date no studies that implicate a direct involvement of glial cells in Aβ trafficking across different areas of the brain. Since Aβ40 and Aβ42 circulate in body fluids such as plasma and CSF (Mehta et al., 2000), another potential route of Aβ diffusion from the periphery to the brain may be constituted by the vascular system. Indeed, intraperitoneal or intravenous administration of Aβ-rich extracts in pre-depositing APP23 mice promoted cerebral amyloid angiopathy (CAA; Eisele et al., 2010; Burwinkel et al., 2018) pointing again to a vascular component of circulating immune cell involvement in the spread of Aβ seeds (Cintron et al., 2015).
Protein Cross-Seeding
It is well known that different neuropathological lesions such as Aβ, NFTs, or Lewy bodies can co-exist in the brains of AD patients (Braak and Braak, 1997; Hamilton, 2000), predicting cross protein interactions. Indeed, several studies have shown that the interaction between Aβ and tau can exaggerate AD pathology (Ribé et al., 2005; Bennett et al., 2017; He et al., 2018; Vergara et al., 2019) and that amyloid deposition, preceding the NFT formation, can actively influence tau spreading to neocortical regions (Braak and Braak, 1997; Hardy and Selkoe, 2002; Jacobs et al., 2018; Vogel et al., 2020). Furthermore, oligomeric forms of Aβ were found to be abundant in synapses of AD patients early in the disease before the appearance of phospo-tau at later stages, suggesting that soluble Aβ oligomers in synaptic terminals are associated with dementia onset and may initiate a cascade that drives phosphorylated tau accumulation and its synaptic spread (Bilousova et al., 2016).
Nevertheless, the finding that Aβ and tau deposition starts in different brain areas and follows distinct temporal sequences, argues against the idea that tau pathology may be driven exclusively by the presence of amyloid and rather speaks for an Aβ independent pathway (Raj et al., 2015; Jack et al., 2019; van der Kant et al., 2020). Tau aggregation assays with tau isolated from patients containing both lesions showed an enhanced ability to induce tau aggregates when compared to tau isolated from human cases without plaques (Bennett et al., 2017). Similar results have been obtained in double-transgenic mice overexpressing both, mutated APP and tau (Lewis et al., 2001; Hurtado et al., 2010). Furthermore concurrent cortical amyloid deposition in double transgenic mice strongly accelerated interneuronal transfer of tau and boosted its spreading to distal brain regions with an increase in neuronal loss (Hurtado et al., 2010; Pooler et al., 2015).
Intracerebral infusion of Aβ-rich extracts into tau-transgenic mice resulted in significantly more NFT formation compared to tau-rich or WT extracts (Götz et al., 2001; Bolmont et al., 2007; Vasconcelos et al., 2016) indicating that the presence of Aβ triggers the formation of tau pathology and proposing synergistic toxicity on the neuronal network. Inoculation of human AD-tau extracts into the brains of APP transgenic mice that normally do not form NFTs resulted in rapid fibrillization of endogenous tau (Bennett et al., 2017; He et al., 2018). Moreover, ipsi- and contralateral tau propagation was enhanced in tau-injected 5xFAD mice compared to tau-injected WT mice (Vergara et al., 2019).
Transgenic APPPS1+Tau mice, that express WT human tau within a mouse tau-deficient background showed that Aβ and tau can synergistically cooperate to cause a hyperactivity behavioral phenotype and resulted in a downregulation of genes involved in synaptic function (Pickett et al., 2019). Treatment strategies that aim at reducing tau levels in mice that co-express Aβ and human tau prevented neuronal loss (DeVos et al., 2018) as well as excitotoxin-induced neuronal dysfunctions (Roberson et al., 2007) and ameliorated the behavioral and gene expression phenotypes (Pickett et al., 2019). Together, these results demonstrate the therapeutic benefit of tau reduction with a positive impact on the Aβ-induced cytotoxic effects.
Despite all these observational evidence, the mechanism of Aβ and tau interplay remains largely unknown. Further studies are necessary to unravel whether it is a direct interaction between the two pathogenic proteins or instead mediated by other factors.
Conclusions and Future Prospective
Two of the most remarkable features of AD are: (i) the stereotypic pattern of Aβ and tangle spreading through interconnected areas of the brain (Braak and Braak, 1991) that is closely related to cognitive decline years before the onset of clinical symptoms; and (ii) the ability of pathogenic misfolded Aβ and tau to serve as templates to convert their innocuous counterparts into toxic forms in a prion-like manner (Clavaguera et al., 2009; Jucker and Walker, 2011, 2013). These two aspects, together with the fact that the two hallmarks often coexist in the brain of AD patients and amplify each other’s toxic effects downstream (Ittner and Götz, 2011), make the development of an effective therapy challenging. Currently, clinical trials targeting Aβ have reported limited success, implying the notion that the timing of intervention is too late and that directing to only one target might be not sufficient to halt the spreading of both pathogenic proteins and to avoid their synergistic impact on neuronal networks. Furthermore, abundant pieces of evidence have recently highlighted the role that different strains of tau and Aβ may play in modulating the clinical picture of the disease, turning away the possibility to develop therapies against different AD subtypes. For this purpose, a better understanding of the conformational heterogeneity of tau and Aβ is necessary to better design the best intervention methods.
Although in the last years many molecular and cellular mechanisms involved in the formation, aggregation, deposition, and propagation of Aβ and tau were uncovered, many questions remain still open. Which mechanisms guide Aβ diffusion? What is the exact sequence of events leading to clinical symptoms? Which role might other cellular systems or molecular pathways play in promoting the pathological bond of Aβ and tau? These are only some of the questions that need to be clarified to design the best strategies to finally arrest disease progression.
Author Contributions
Pd’E and MM-L wrote the manuscript. Pd’E and MM-L read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) ME 3542/2-1 (MM-L). The article processing charge was funded by the German Research Foundation (DFG) and the University of Freiburg in the funding programme Open Access Publishing.
References
- Aamodt E. J., Williams R. C., Jr. (1984). Microtubule-associated proteins connect microtubules and neurofilaments in vitro. Biochemistry 23, 6023–6031. 10.1021/bi00320a019 [DOI] [PubMed] [Google Scholar]
- Abounit S., Wu J. W., Duff K., Victoria G. S., Zurzolo C. (2016). Tunneling nanotubes: a possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 10, 344–351. 10.1080/19336896.2016.1223003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z., Cooper J., Murray T. K., Garn K., McNaughton E., Clarke H., et al. (2014). A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 127, 667–683. 10.1007/s00401-014-1254-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadis A. (2005). Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim. Biophys. Acta 1739, 91–103. 10.1016/j.bbadis.2004.08.010 [DOI] [PubMed] [Google Scholar]
- Andreadis A., Brown W. M., Kosik K. S. (1992). Structure and novel exons of the human tau gene. Biochemistry 31, 10626–10633. 10.1021/bi00158a027 [DOI] [PubMed] [Google Scholar]
- Arendt T., Stieler J. T., Holzer M. (2016). Tau and tauopathies. Brain Res. Bull. 126, 238–292. 10.1016/j.brainresbull.2016.08.018 [DOI] [PubMed] [Google Scholar]
- Aronov S., Aranda G., Behar L., Ginzburg I. (2001). Axonal tau mRNA localization coincides with tau protein in living neuronal cells and depends on axonal targeting signal. J. Neurosci. 21, 6577–6587. 10.1523/JNEUROSCI.21-17-06577.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai H., Ikezu S., Tsunoda S., Medalla M., Luebke J., Haydar T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. 10.1038/nn.4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachhuber T., Katzmarski N., McCarter J. F., Loreth D., Tahirovic S., Kamp F., et al. (2015). Inhibition of amyloid-β plaque formation by α-synuclein. Nat. Med. 21, 802–807. 10.1038/nm.3885 [DOI] [PubMed] [Google Scholar]
- Baker S., Polanco J. C., Götz J. (2016). Extracellular vesicles containing P301L mutant tau accelerate pathological tau phosphorylation and oligomer formation but do not seed mature neurofibrillary tangles in ALZ17 mice. J. Alzheimers Dis. 54, 1207–1217. 10.3233/jad-160371 [DOI] [PubMed] [Google Scholar]
- Bemiller S. M., McCray T. J., Allan K., Formica S. V., Xu G., Wilson G., et al. (2017). TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 12:74. 10.1186/s13024-017-0216-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett R. E., DeVos S. L., Dujardin S., Corjuc B., Gor R., Gonzalez J., et al. (2017). Enhanced tau aggregation in the presence of amyloid β. Am. J. Pathol. 187, 1601–1612. 10.1016/j.ajpath.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand A., Khan U., Hoang D. M., Novikov D. S., Krishnamurthy P., Rajamohamed Sait H. B., et al. (2013). Non-invasive, in vivo monitoring of neuronal transport impairment in a mouse model of tauopathy using MEMRI. NeuroImage 64, 693–702. 10.1016/j.neuroimage.2012.08.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar K., Konerth M., Kokiko-Cochran O. N., Cardona A., Ransohoff R. M., Lamb B. T. (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. 10.1016/j.neuron.2010.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilousova T., Miller C. A., Poon W. W., Vinters H. V., Corrada M., Kawas C., et al. (2016). Synaptic amyloid-β oligomers precede p-tau and differentiate high pathology control cases. Am. J. Pathol. 186, 185–198. 10.1016/j.ajpath.2015.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T., Clavaguera F., Meyer-Luehmann M., Herzig M. C., Radde R., Staufenbiel M., et al. (2007). Induction of tau pathology by intracerebral infusion of amyloid-β -containing brain extract and by amyloid-β deposition in APP x Tau transgenic mice. Am. J. Pathol. 171, 2012–2020. 10.2353/ajpath.2007.070403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolós M., Llorens-Martín M., Jurado-Arjona J., Hernández F., Rábano A., Avila J. (2016). Direct evidence of internalization of tau by microglia in vitro and in vivo. J. Alzheimers Dis. 50, 77–87. 10.3233/jad-150704 [DOI] [PubMed] [Google Scholar]
- Bolós M., Llorens-Martín M., Perea J. R., Jurado-Arjona J., Rábano A., Hernández F., et al. (2017). Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 12:59. 10.1186/s13024-017-0200-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H., Braak E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E. (1997). Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 18, 351–357. 10.1016/s0197-4580(97)00056-0 [DOI] [PubMed] [Google Scholar]
- Brion J. P., Couck A. M., Passareiro E., Flament-Durand J. (1985). Neurofibrillary tangles of Alzheimer’s disease: an immunohistochemical study. J. Submicrosc. Cytol. 17, 89–96. [PubMed] [Google Scholar]
- Burwinkel M., Lutzenberger M., Heppner F. L., Schulz-Schaeffer W., Baier M. (2018). Intravenous injection of β-amyloid seeds promotes cerebral amyloid angiopathy (CAA). Acta Neuropathol. Commun. 6:23. 10.1186/s40478-018-0511-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafate S., Buist A., Miskiewicz K., Vijayan V., Daneels G., de Strooper B., et al. (2015). Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep. 11, 1176–1183. 10.1016/j.celrep.2015.04.043 [DOI] [PubMed] [Google Scholar]
- Calafate S., Flavin W., Verstreken P., Moechars D. (2016). Loss of Bin1 promotes the propagation of tau pathology. Cell Rep. 17, 931–940. 10.1016/j.celrep.2016.09.063 [DOI] [PubMed] [Google Scholar]
- Cintron A. F., Dalal N. V., Dooyema J., Betarbet R., Walker L. C. (2015). Transport of cargo from periphery to brain by circulating monocytes. Brain Res. 1622, 328–338. 10.1016/j.brainres.2015.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F., Akatsu H., Fraser G., Crowther R. A., Frank S., Hench J., et al. (2013). Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. U S A 110, 9535–9540. 10.1073/pnas.1301175110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F., Bolmont T., Crowther R. A., Abramowski D., Frank S., Probst A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913. 10.1038/ncb1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F., Hench J., Lavenir I., Schweighauser G., Frank S., Goedert M., et al. (2014). Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 127, 299–301. 10.1007/s00401-013-1231-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello C., Stöehr J. (2018). Aβ propagation and strains: implications for the phenotypic diversity in Alzheimer’s disease. Neurobiol. Dis. 109, 191–200. 10.1016/j.nbd.2017.03.014 [DOI] [PubMed] [Google Scholar]
- Congdon E. E., Kim S., Bonchak J., Songrug T., Matzavinos A., Kuret J. (2008). Nucleation-dependent tau filament formation: the importance of dimerization and an estimation of elementary rate constants. J. Biol. Chem. 283, 13806–13816. 10.1074/jbc.m800247200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco E., Celauro L., Vanni S., Grandolfo M., Bistaffa E., Moda F., et al. (2020). The uptake of tau amyloid fibrils is facilitated by the cellular prion protein and hampers prion propagation in cultured cells. J. Neurochem. [Epub ahead of print]. 10.1111/jnc.15040 [DOI] [PubMed] [Google Scholar]
- DeVos S. L., Corjuc B. T., Commins C., Dujardin S., Bannon R. N., Corjuc D., et al. (2018). Tau reduction in the presence of amyloid-β prevents tau pathology and neuronal death in vivo. Brain J. Neurol. 141, 2194–2212. 10.1093/brain/awy117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domert J., Rao S. B., Agholme L., Brorsson A.-C., Marcusson J., Hallbeck M., et al. (2014). Spreading of amyloid-β peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 65, 82–92. 10.1016/j.nbd.2013.12.019 [DOI] [PubMed] [Google Scholar]
- Dujardin S., Bégard S., Caillierez R., Lachaud C., Carrier S., Lieger S., et al. (2018). Different tau species lead to heterogeneous tau pathology propagation and misfolding. Acta Neuropathol. Commun. 6:132. 10.1186/s40478-018-0637-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin S., Bégard S., Caillierez R., Lachaud C., Delattre L., Carrier S., et al. (2014). Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One 9:e100760. 10.1371/journal.pone.0100760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele Y. S., Bolmont T., Heikenwalder M., Langer F., Jacobson L. H., Yan Z.-X., et al. (2009). Induction of cerebral β-amyloidosis: intracerebral versus systemic Aβ inoculation. Proc. Natl. Acad. Sci. U S A 106, 12926–12931. 10.1073/pnas.0903200106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele Y. S., Obermüller U., Heilbronner G., Baumann F., Kaeser S. A., Wolburg H., et al. (2010). Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science 330, 980–982. 10.1126/science.1194516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esparza T. J., Zhao H., Cirrito J. R., Cairns N. J., Bateman R. J., Holtzman D. M., et al. (2013). Amyloid-β oligomerization in Alzheimer dementia versus high-pathology controls. Ann. Neurol. 73, 104–119. 10.1002/ana.23748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espuny-Camacho I., Arranz A. M., Fiers M., Snellinx A., Ando K., Munck S., et al. (2017). Hallmarks of Alzheimer’s disease in stem-cell-derived human neurons transplanted into mouse brain. Neuron 93, 1066.e8–1081.e8. 10.1016/j.neuron.2017.02.001 [DOI] [PubMed] [Google Scholar]
- Evans L. D., Wassmer T., Fraser G., Smith J., Perkinton M., Billinton A., et al. (2018). Extracellular monomeric and aggregated tau efficiently enter human neurons through overlapping but distinct pathways. Cell Rep. 22, 3612–3624. 10.1016/j.celrep.2018.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon B., Zhang W., Schweighauser M., Murzin A. G., Vidal R., Garringer H. J., et al. (2018). Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 136, 699–708. 10.1007/s00401-018-1914-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiandaca M. S., Kapogiannis D., Mapstone M., Boxer A., Eitan E., Schwartz J. B., et al. (2015). Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case-control study. Alzheimers Dement. 11, 600.e1–607.e1. 10.1016/j.jalz.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick A. W. P., Falcon B., He S., Murzin A. G., Murshudov G., Garringer H. J., et al. (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. 10.1038/nature23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavin W. P., Bousset L., Green Z. C., Chu Y., Skarpathiotis S., Chaney M. J., et al. (2017). Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 134, 629–653. 10.1007/s00401-017-1722-x [DOI] [PubMed] [Google Scholar]
- Fontaine S. N., Zheng D., Sabbagh J. J., Martin M. D., Chaput D., Darling A., et al. (2016). DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 35, 1537–1549. 10.15252/embj.201593489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forloni G., Artuso V., La Vitola P., Balducci C. (2016). Oligomeropathies and pathogenesis of Alzheimer and Parkinson’s diseases. Mov. Disord. 31, 771–781. 10.1002/mds.26624 [DOI] [PubMed] [Google Scholar]
- Fornari S., Schäfer A., Jucker M., Goriely A., Kuhl E. (2019). Prion-like spreading of Alzheimer’s disease within the brain’s connectome. J. R. Soc. Interface 16:20190356. 10.1098/rsif.2019.0356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen M., Meyer-Luehmann M. (2019). Aβ seeding as a tool to study cerebral amyloidosis and associated pathology. Front. Mol. Neurosci. 12:233. 10.3389/fnmol.2019.00233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B., Jacks R. L., Diamond M. I. (2009). Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852. 10.1074/jbc.m808759200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastard M. C., Troncoso J. C., Koliatsos V. E. (2003). Caspase activation in the limbic cortex of subjects with early Alzheimer’s disease. Ann. Neurol. 54, 393–398. 10.1002/ana.10680 [DOI] [PubMed] [Google Scholar]
- Gibbons G. S., Banks R. A., Kim B., Xu H., Changolkar L., Leight S. N., et al. (2017). GFP-mutant human tau transgenic mice develop tauopathy following cns injections of Alzheimer’s brain-derived pathological tau or synthetic mutant human tau fibrils. J. Neurosci. 37, 11485–11494. 10.1523/JNEUROSCI.2393-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G. (2011). Pathogenesis of the tauopathies. J. Mol. Neurosci. 45, 425–431. 10.1007/s12031-011-9593-4 [DOI] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G., Jakes R., Rutherford D., Crowther R. A. (1989). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3, 519–526. 10.1016/0896-6273(89)90210-9 [DOI] [PubMed] [Google Scholar]
- Goedert M., Wischik C. M., Crowther R. A., Walker J. E., Klug A. (1988). Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. U S A 85, 4051–4055. 10.1073/pnas.85.11.4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Billault C., Engelke M., Jiménez-Mateos E. M., Wandosell F., Cáceres A., Avila J. (2002). Participation of structural microtubule-associated proteins (MAPs) in the development of neuronal polarity. J. Neurosci. Res. 67, 713–719. 10.1002/jnr.10161 [DOI] [PubMed] [Google Scholar]
- Götz J., Chen F., van Dorpe J., Nitsch R. M. (2001). Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Aβ 42 fibrils. Science 293, 1491–1495. 10.1126/science.1062097 [DOI] [PubMed] [Google Scholar]
- Gouras G. K., Tsai J., Naslund J., Vincent B., Edgar M., Checler F., et al. (2000). Intraneuronal Aβ42 accumulation in human brain. Am. J. Pathol. 156, 15–20. 10.1016/s0002-9440(10)64700-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe M. J., Barthel H., Sepulcre J., Dyrba M., Sabri O., Teipel S. J., et al. (2017). in vivo staging of regional amyloid deposition. Neurology 89, 2031–2038. 10.1212/wnl.0000000000004643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. L., Lee V. M.-Y. (2011). Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 286, 15317–15331. 10.1074/jbc.M110.209296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. L., Lee V. M.-Y. (2013). Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett. 587, 717–723. 10.1016/j.febslet.2013.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. L., Narasimhan S., Changolkar L., He Z., Stieber A., Zhang B., et al. (2016). Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J. Exp. Med. 213, 2635–2654. 10.1084/jem.20160833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C., Kaether C., Thinakaran G., Sisodia S. (2012). Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2:a006270. 10.1101/cshperspect.a006270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton R. L. (2000). Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using α-synuclein immunohistochemistry. Brain Pathol. Zurich Switz. 10, 378–384. 10.1111/j.1750-3639.2000.tb00269.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. A., Higgins G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. 10.1126/science.1566067 [DOI] [PubMed] [Google Scholar]
- Hardy J., Selkoe D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- Harper J. D., Lansbury P. T., Jr. (1997). Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66, 385–407. 10.1146/annurev.biochem.66.1.385 [DOI] [PubMed] [Google Scholar]
- He Z., Guo J. L., McBride J. D., Narasimhan S., Kim H., Changolkar L., et al. (2018). Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 24, 29–38. 10.1038/nm.4443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z., McBride J. D., Xu H., Changolkar L., Kim S., Zhang B., et al. (2020). Transmission of tauopathy strains is independent of their isoform composition. Nat. Commun. 11:7. 10.1038/s41467-019-13787-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B. B., DeVos S. L., Kfoury N., Li M., Jacks R., Yanamandra K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U S A 110, E3138–3147. 10.1073/pnas.1301440110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopp S. C., Lin Y., Oakley D., Roe A. D., DeVos S. L., Hanlon D., et al. (2018). The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflammation 15:269. 10.1186/s12974-018-1309-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Crick S. L., Bu G., Frieden C., Pappu R. V., Lee J.-M. (2009). Amyloid seeds formed by cellular uptake, concentration and aggregation of the amyloid-β peptide. Proc. Natl. Acad. Sci. U S A 106, 20324–20329. 10.1073/pnas.0911281106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado D. E., Molina-Porcel L., Iba M., Aboagye A. K., Paul S. M., Trojanowski J. Q., et al. (2010). Aβ accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am. J. Pathol. 177, 1977–1988. 10.2353/ajpath.2010.100346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M., Guo J. L., McBride J. D., Zhang B., Trojanowski J. Q., Lee V. M.-Y. (2013). Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 33, 1024–1037. 10.1523/JNEUROSCI.2642-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising C., Venegas C., Zhang S., Scheiblich H., Schmidt S. V., Vieira-Saecker A., et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. 10.1038/s41586-019-1769-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner L. M., Götz J. (2011). Amyloid-β and tau—a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 12, 65–72. 10.1038/nrn2967 [DOI] [PubMed] [Google Scholar]
- Jack C. R., Wiste H. J., Botha H., Weigand S. D., Therneau T. M., Knopman D. S., et al. (2019). The bivariate distribution of amyloid-β and tau: relationship with established neurocognitive clinical syndromes. Brain J. Neurol. 142, 3230–3242. 10.1093/brain/awz268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs H. I. L., Hedden T., Schultz A. P., Sepulcre J., Perea R. D., Amariglio R. E., et al. (2018). Structural tract alterations predict downstream tau accumulation in amyloid-positive older individuals. Nat. Neurosci. 21, 424–431. 10.1038/s41593-018-0070-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P., Turola E., Ruiz A., Bergami A., Libera D. D., Benussi L., et al. (2014). Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 21, 582–593. 10.1038/cdd.2013.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M., Walker L. C. (2011). Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann. Neurol. 70, 532–540. 10.1002/ana.22615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M., Walker L. C. (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. 10.1038/nature12481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M. D., Lipinski W. J., Callahan M. J., Bian F., Durham R. A., Schwarz R. D., et al. (2000). Evidence for seeding of β -amyloid by intracerebral infusion of Alzheimer brain extracts in β -amyloid precursor protein-transgenic mice. J. Neurosci. 20, 3606–3611. 10.1523/JNEUROSCI.20-10-03606.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsinelos T., Zeitler M., Dimou E., Karakatsani A., Müller H.-M., Nachman E., et al. (2018). Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep. 23, 2039–2055. 10.1016/j.celrep.2018.04.056 [DOI] [PubMed] [Google Scholar]
- Katzman R. (1988). Alzheimer’s disease as an age-dependent disorder. Ciba Found. Symp. 134, 69–85. 10.1002/9780470513583.ch6 [DOI] [PubMed] [Google Scholar]
- Katzmarski N., Ziegler-Waldkirch S., Scheffler N., Witt C., Abou-Ajram C., Nuscher B., et al. (2020). Aβ oligomers trigger and accelerate Aβ seeding. Brain Pathol. Zurich Switz. 30, 36–45. 10.1111/bpa.12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman S. K., Sanders D. W., Thomas T. L., Ruchinskas A. J., Vaquer-Alicea J., Sharma A. M., et al. (2016). Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron 92, 796–812. 10.1016/j.neuron.2016.09.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie R. M., Meyer-Luehmann M., Hashimoto T., Adams K. W., Mielke M. L., Garcia-Alloza M., et al. (2009). Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U S A 106, 4012–4017. 10.1073/pnas.0811698106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachenal G., Pernet-Gallay K., Chivet M., Hemming F. J., Belly A., Bodon G., et al. (2011). Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell. Neurosci. 46, 409–418. 10.1016/j.mcn.2010.11.004 [DOI] [PubMed] [Google Scholar]
- LaFerla F. M., Green K. N., Oddo S. (2007). Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509. 10.1038/nrn2168 [DOI] [PubMed] [Google Scholar]
- Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., et al. (1998). Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U S A 95, 6448–6453. 10.1073/pnas.95.11.6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves C. A., Castillo-Carranza D. L., Sengupta U., Guerrero-Munoz M. J., Kiritoshi T., Neugebauer V., et al. (2012). Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2:700. 10.1038/srep00700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves C. A., Sengupta U., Castillo-Carranza D., Gerson J. E., Guerrero-Munoz M., Troncoso J. C., et al. (2014). The formation of tau pore-like structures is prevalent and cell specific: possible implications for the disease phenotypes. Acta Neuropathol. Commun. 2:56. 10.1186/2051-5960-2-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J., Dickson D. W., Lin W. L., Chisholm L., Corral A., Jones G., et al. (2001). Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491. 10.1126/science.1058189 [DOI] [PubMed] [Google Scholar]
- Leyssen M., Ayaz D., Hébert S. S., Reeve S., De Strooper B., Hassan B. A. (2005). Amyloid precursor protein promotes post-developmental neurite arborization in the Drosophila brain. EMBO J. 24, 2944–2955. 10.1038/sj.emboj.7600757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Drouet V., Wu J. W., Witter M. P., Small S. A., Clelland C., et al. (2012). Trans-synaptic spread of tau pathology in vivo. PLoS One 7:e31302. 10.1371/journal.pone.0031302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W., Liu W., Hu X., Hanna M., Caravaca A., Paul S. M. (2015). Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci. Rep. 5:11161. 10.1038/srep11161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maphis N., Xu G., Kokiko-Cochran O. N., Jiang S., Cardona A., Ransohoff R. M., et al. (2015). Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain J. Neurol. 138, 1738–1755. 10.1093/brain/awv081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari S. A., Wegmann S., Tepper K., Hyman B. T., Mandelkow E.-M., Mandelkow E., et al. (2018). Reversible cation-selective attachment and self-assembly of human tau on supported brain lipid membranes. Nano Lett. 18, 3271–3281. 10.1021/acs.nanolett.8b01085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini-Stoica H., Cole A. L., Swartzlander D. B., Chen F., Wan Y.-W., Bajaj L., et al. (2018). TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J. Exp. Med. 215, 2355–2377. 10.1084/jem.20172158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P. D., Pirttilä T., Mehta S. P., Sersen E. A., Aisen P. S., Wisniewski H. M. (2000). Plasma and cerebrospinal fluid levels of amyloid β proteins 1–40 and 1-42 in Alzheimer disease. Arch. Neurol. 57, 100–105. 10.1001/archneur.57.1.100 [DOI] [PubMed] [Google Scholar]
- Merezhko M., Brunello C. A., Yan X., Vihinen H., Jokitalo E., Uronen R.-L., et al. (2018). Secretion of tau via an unconventional non-vesicular mechanism. Cell Rep. 25, 2027.e4–2035.e4. 10.1016/j.celrep.2018.10.078 [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M., Coomaraswamy J., Bolmont T., Kaeser S., Schaefer C., Kilger E., et al. (2006). Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science 313, 1781–1784. 10.1126/science.1131864 [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M., Mielke M., Spires-Jones T. L., Stoothoff W., Jones P., Bacskai B. J., et al. (2009). A reporter of local dendritic translocation shows plaque- related loss of neural system function in APP-transgenic mice. J. Neurosci. 29, 12636–12640. 10.1523/JNEUROSCI.1948-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M., Spires-Jones T. L., Prada C., Garcia-Alloza M., de Calignon A., Rozkalne A., et al. (2008). Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724. 10.1038/nature06616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M., Stalder M., Herzig M. C., Kaeser S. A., Kohler E., Pfeifer M., et al. (2003). Extracellular amyloid formation and associated pathology in neural grafts. Nat. Neurosci. 6, 370–377. 10.1038/nn1022 [DOI] [PubMed] [Google Scholar]
- Mirbaha H., Chen D., Morazova O. A., Ruff K. M., Sharma A. M., Liu X., et al. (2018). Inert and seed-competent tau monomers suggest structural origins of aggregation. eLife 7:e36584. 10.7554/eLife.36584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova V., Cohen L. S., Makki A. E.-H., Shur A., Pilar G., El Idrissi A., et al. (2019). Normal and pathological tau uptake mediated by M1/M3 muscarinic receptors promotes opposite neuronal changes. Front. Cell. Neurosci. 13:403. 10.3389/fncel.2019.00403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudher A., Colin M., Dujardin S., Medina M., Dewachter I., Alavi Naini S. M., et al. (2017). What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol. Commun. 5:99. 10.1186/s40478-017-0488-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller M. C., Baranowski B. J., Hayward G. C. (2018). New insights on the role of residue 673 of APP in Alzheimer’s disease. J. Neurosci. 38, 515–517. 10.1523/JNEUROSCI.2710-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan S., Guo J. L., Changolkar L., Stieber A., McBride J. D., Silva L. V., et al. (2017). Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci. 37, 11406–11423. 10.1523/JNEUROSCI.1230-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N., Ramachandran S., Azimov R., Kagan B. L., Lal R. (2015). Ion channel formation by tau protein: implications for Alzheimer’s disease and tauopathies. Biochemistry 54, 7320–7325. 10.1021/acs.biochem.5b00988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea J. R., López E., Díez-Ballesteros J. C., Ávila J., Hernández F., Bolós M. (2019). Extracellular monomeric tau is internalized by astrocytes. Front. Neurosci. 13:442. 10.3389/fnins.2019.00442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett E. K., Herrmann A. G., McQueen J., Abt K., Dando O., Tulloch J., et al. (2019). Amyloid β and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of Alzheimer’s disease. Cell Rep. 29, 3592.e5–3604.e5. 10.1016/j.celrep.2019.11.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman A. M., Fung H.-C., de Silva R. (2006). Untangling the tau gene association with neurodegenerative disorders. Hum. Mol. Genet. 15, R188–R195. 10.1093/hmg/ddl190 [DOI] [PubMed] [Google Scholar]
- Plouffe V., Mohamed N.-V., Rivest-McGraw J., Bertrand J., Lauzon M., Leclerc N. (2012). Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One 7:e36873. 10.1371/journal.pone.0036873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanco J. C., Scicluna B. J., Hill A. F., Götz J. (2016). Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J. Biol. Chem. 291, 12445–12466. 10.1074/jbc.m115.709485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnambalam S., Baldwin S. A. (2003). Constitutive protein secretion from the trans-Golgi network to the plasma membrane. Mol. Membr. Biol. 20, 129–139. 10.1080/0968768031000084172 [DOI] [PubMed] [Google Scholar]
- Pooler A. M., Phillips E. C., Lau D. H. W., Noble W., Hanger D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. 10.1038/embor.2013.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler A. M., Polydoro M., Maury E. A., Nicholls S. B., Reddy S. M., Wegmann S., et al. (2015). Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease. Acta Neuropathol. Commun. 3:14. 10.1186/s40478-015-0199-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler A. M., Usardi A., Evans C. J., Philpott K. L., Noble W., Hanger D. P. (2012). Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol. Aging 33, 431.e27–431.e38. 10.1016/j.neurobiolaging.2011.01.005 [DOI] [PubMed] [Google Scholar]
- Priller C., Bauer T., Mitteregger G., Krebs B., Kretzschmar H. A., Herms J. (2006). Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci. 26, 7212–7221. 10.1523/JNEUROSCI.1450-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W., Yau W.-M., Lu J.-X., Collinge J., Tycko R. (2017). Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature 541, 217–221. 10.1038/nature20814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A., LoCastro E., Kuceyeski A., Tosun D., Relkin N., Weiner M., et al. (2015). Network diffusion model of progression predicts longitudinal patterns of atrophy and metabolism in Alzheimer’s disease. Cell Rep. 10, 359–369. 10.1016/j.celrep.2014.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L., Honsho M., Zahn T. R., Keller P., Geiger K. D., Verkade P., et al. (2006). Alzheimer’s disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U S A 103, 11172–11177. 10.1073/pnas.0603838103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch J. N., Chen J. J., Sorum A. W., Miller G. M., Sharf T., See S. K., et al. (2018). Tau internalization is regulated by 6-O sulfation on heparan sulfate proteoglycans (HSPGs). Sci. Rep. 8:6382. 10.1038/s41598-018-24904-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch J. N., Luna G., Guzman E., Audouard M., Challis C., Sibih Y. E., et al. (2020). LRP1 is a master regulator of tau uptake and spread. Nature 580, 381–385. 10.1038/s41586-020-2156-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribé E. M., Pérez M., Puig B., Gich I., Lim F., Cuadrado M., et al. (2005). Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol. Dis. 20, 814–822. 10.1016/j.nbd.2005.05.027 [DOI] [PubMed] [Google Scholar]
- Ricciarelli R., Fedele E. (2017). The amyloid cascade hypothesis in Alzheimer’s disease: it’s time to change our mind. Curr. Neuropharmacol. 15, 926–935. 10.2174/1570159X15666170116143743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson E. D., Scearce-Levie K., Palop J. J., Yan F., Cheng I. H., Wu T., et al. (2007). Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. 10.1126/science.1141736 [DOI] [PubMed] [Google Scholar]
- Rönnbäck A., Sagelius H., Bergstedt K. D., Näslund J., Westermark G. T., Winblad B., et al. (2012). Amyloid neuropathology in the single Arctic APP transgenic model affects interconnected brain regions. Neurobiol. Aging 33, 831.e11–831.e19. 10.1016/j.neurobiolaging.2011.07.012 [DOI] [PubMed] [Google Scholar]
- Saman S., Kim W., Raya M., Visnick Y., Miro S., Saman S., et al. (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. 10.1074/jbc.m111.277061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders D. W., Kaufman S. K., DeVos S. L., Sharma A. M., Mirbaha H., Li A., et al. (2014). Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288. 10.1016/j.neuron.2014.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayas C. L., Medina M., Cuadros R., Ollá I., García E., Pérez M., et al. (2019). Role of tau N-terminal motif in the secretion of human tau by End Binding proteins. PLoS One 14:e0210864. 10.1371/journal.pone.0210864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J., Hardy J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A., Frosch M. P., Masliah E., Hyman B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. 10.1101/cshperspect.a006189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P., Lal N., Leung E., Traul D. E., Gonzalo-Ruiz A., Geula C. (2010). Neuronal and axonal loss are selectively linked to fibrillar amyloid-β within plaques of the aged primate cerebral cortex. Am. J. Pathol. 177, 325–333. 10.2353/ajpath.2010.090937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar G. M., Li S., Mehta T. H., Garcia-Munoz A., Shepardson N. E., Smith I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. 10.1038/nm1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples R. A., Vella L. J., Nisbet R. M., Naylor R., Perez K., Barnham K. J., et al. (2008). Inhibition of γ-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 22, 1469–1478. 10.1096/fj.07-9357com [DOI] [PubMed] [Google Scholar]
- Siman R., Card J. P., Nelson R. B., Davis L. G. (1989). Expression of β-amyloid precursor protein in reactive astrocytes following neuronal damage. Neuron 3, 275–285. 10.1016/0896-6273(89)90252-3 [DOI] [PubMed] [Google Scholar]
- Simón D., García-García E., Gómez-Ramos A., Falcón-Pérez J. M., Díaz-Hernández M., Hernández F., et al. (2012). Tau overexpression results in its secretion via membrane vesicles. Neurodegener. Dis. 10, 73–75. 10.1159/000334915 [DOI] [PubMed] [Google Scholar]
- Sinha A., Principe S., Alfaro J., Ignatchenko A., Ignatchenko V., Kislinger T. (2018). Proteomic profiling of secreted proteins, exosomes, and microvesicles in cell culture conditioned media. Methods Mol. Biol. 1722, 91–102. 10.1007/978-1-4939-7553-2_6 [DOI] [PubMed] [Google Scholar]
- Smolek T., Cubinkova V., Brezovakova V., Valachova B., Szalay P., Zilka N., et al. (2019a). Genetic background influences the propagation of tau pathology in transgenic rodent models of tauopathy. Front. Aging Neurosci. 11:343. 10.3389/fnagi.2019.00343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolek T., Jadhav S., Brezovakova V., Cubinkova V., Valachova B., Novak P., et al. (2019b). First-in-rat study of human Alzheimer’s disease tau propagation. Mol. Neurobiol. 56, 621–631. 10.1007/s12035-018-1102-0 [DOI] [PubMed] [Google Scholar]
- Sokolow S., Henkins K. M., Bilousova T., Gonzalez B., Vinters H. V., Miller C. A., et al. (2015). Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J. Neurochem. 133, 368–379. 10.1111/jnc.12991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg E., Severson P. L., Hohsfield L. A., Crapser J., Zhang J., Burton E. A., et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 10:3758. 10.1038/s41467-019-11674-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M., Miyata H., Kametani F., Nonaka T., Akiyama H., Hisanaga S., et al. (2015). Extracellular association of APP and tau fibrils induces intracellular aggregate formation of tau. Acta Neuropathol. 129, 895–907. 10.1007/s00401-015-1415-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei K., Mundigl O., Daniell L., De Camilli P. (1996). The synaptic vesicle cycle: a single vesicle budding step involving clathrin and dynamin. J. Cell Biol. 133, 1237–1250. 10.1083/jcb.133.6.1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardivel M., Bégard S., Bousset L., Dujardin S., Coens A., Melki R., et al. (2016). Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 4:117. 10.1186/s40478-016-0386-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal D. R., Rüb U., Orantes M., Braak H. (2002). Phases of A β-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. 10.1212/wnl.58.12.1791 [DOI] [PubMed] [Google Scholar]
- Turner R. S., Suzuki N., Chyung A. S., Younkin S. G., Lee V. M. (1996). Amyloids β40 and β42 are generated intracellularly in cultured human neurons and their secretion increases with maturation. J. Biol. Chem. 271, 8966–8970. 10.1074/jbc.271.15.8966 [DOI] [PubMed] [Google Scholar]
- Van den Heuvel C., Blumbergs P. C., Finnie J. W., Manavis J., Jones N. R., Reilly P. L., et al. (1999). Upregulation of amyloid precursor protein messenger RNA in response to traumatic brain injury: an ovine head impact model. Exp. Neurol. 159, 441–450. 10.1006/exnr.1999.7150 [DOI] [PubMed] [Google Scholar]
- van der Kant R., Goldstein L. S. B., Ossenkoppele R. (2020). Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 21, 21–35. 10.1038/s41583-019-0240-3 [DOI] [PubMed] [Google Scholar]
- Vasconcelos B., Stancu I.-C., Buist A., Bird M., Wang P., Vanoosthuyse A., et al. (2016). Heterotypic seeding of Tau fibrillization by pre-aggregated Aβ provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo. Acta Neuropathol. 131, 549–569. 10.1007/s00401-015-1525-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venegas C., Kumar S., Franklin B. S., Dierkes T., Brinkschulte R., Tejera D., et al. (2017). Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355–361. 10.1038/nature25158 [DOI] [PubMed] [Google Scholar]
- Vergara C., Houben S., Suain V., Yilmaz Z., De Decker R., Vanden Dries V., et al. (2019). Amyloid-β pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 137, 397–412. 10.1007/s00401-018-1953-5 [DOI] [PubMed] [Google Scholar]
- Villemagne V. L., Rowe C. C. (2011). Amyloid imaging. Int. Psychogeriatr. 23, S41–S49. 10.1017/S1041610211000895 [DOI] [PubMed] [Google Scholar]
- Vogel J. W., Iturria-Medina Y., Strandberg O. T., Smith R., Levitis E., Evans A. C., et al. (2020). Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 11:2612. 10.1038/s41467-020-15701-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L. C., Callahan M. J., Bian F., Durham R. A., Roher A. E., Lipinski W. J. (2002). Exogenous induction of cerebral β-amyloidosis in βAPP-transgenic mice. Peptides 23, 1241–1247. 10.1016/s0196-9781(02)00059-1 [DOI] [PubMed] [Google Scholar]
- Wang Y., Balaji V., Kaniyappan S., Krüger L., Irsen S., Tepper K., et al. (2017). The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 12:5. 10.1186/s13024-016-0143-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann S., Bennett R. E., Delorme L., Robbins A. B., Hu M., McKenzie D., et al. (2019). Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 5:eaaw6404. 10.1126/sciadv.aaw6404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisová P., Cehlár O., Škrabana R., Žilková M., Filipčík P., Kováčech B., et al. (2019). Therapeutic antibody targeting microtubule-binding domain prevents neuronal internalization of extracellular tau via masking neuron surface proteoglycans. Acta Neuropathol. Commun. 7:129. 10.1186/s40478-019-0770-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertkin A. M., Turner R. S., Pleasure S. J., Golde T. E., Younkin S. G., Trojanowski J. Q., et al. (1993). Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular β-amyloid or A4 peptides. Proc. Natl. Acad. Sci. U S A 90, 9513–9517. 10.1073/pnas.90.20.9513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston C. N., Goetzl E. J., Akers J. C., Carter B. S., Rockenstein E. M., Galasko D., et al. (2016). Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement. 3, 63–72. 10.1016/j.dadm.2016.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski H. M., Vorbrodt A. W., Wegiel J., Morys J., Lossinsky A. S. (1990). Ultrastructure of the cells forming amyloid fibers in Alzheimer disease and scrapie. Am. J. Med. Genet. Suppl. 7, 287–297. 10.1002/ajmg.1320370757 [DOI] [PubMed] [Google Scholar]
- Wood J. G., Zinsmeister P. (1989). Immunohistochemical evidence for reorganization of tau in the plaques and tangles in Alzheimer’s disease. Histochem. J. 21, 659–662. 10.1007/bf01002486 [DOI] [PubMed] [Google Scholar]
- Wu J. W., Herman M., Liu L., Simoes S., Acker C. M., Figueroa H., et al. (2013). Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. 10.1074/jbc.m112.394528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K., Holth J. K., Liao F., Stewart F. R., Mahan T. E., Jiang H., et al. (2014). Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211, 387–393. 10.1084/jem.20131685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L., Hamaguchi T., Fritschi S. K., Eisele Y. S., Obermüller U., Jucker M., et al. (2015). Progression of seed-induced Aβ deposition within the limbic connectome. Brain Pathol. 25, 743–752. 10.1111/bpa.12252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young-Pearse T. L., Chen A. C., Chang R., Marquez C., Selkoe D. J. (2008). Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin β1. Neural Dev. 3:15. 10.1186/1749-8104-3-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Higuchi M., Yoshiyama Y., Ishihara T., Forman M. S., Martinez D., et al. (2004). Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J. Neurosci. 24, 4657–4667. 10.1523/JNEUROSCI.0797-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler-Waldkirch S., d’Errico P., Sauer J.-F., Erny D., Savanthrapadian S., Loreth D., et al. (2018). Seed-induced Aβ deposition is modulated by microglia under environmental enrichment in a mouse model of Alzheimer’s disease. EMBO J. 37, 167–182. 10.15252/embj.201797021 [DOI] [PMC free article] [PubMed] [Google Scholar]