

Abstract
Epileptic seizures constitute a significant comorbidity of Alzheimer’s Disease (AD), which are recapitulated in transgenic mouse models of amyloidogenesis. Here, we sought to evaluate the potential role of Tau pathology regarding seizure occurrence. To this end, we performed intra-hippocampal electroencephalogram (EEG) recordings and PTZ (pentylenetertrazol) seizure threshold tests in THY-Tau22 transgenic mice of AD-like Tau pathology. We demonstrate that despite a lack spontaneous epileptiform activity in Tau22 mice, the animals display increased PTZ-induced seizure susceptibility and mortality. The increased propensity for induced seizures in THY-Tau22 mutants correlates with astrogliosis and increased expression of adenosine kinase, consistent with increased network excitability. These data support an impact of Tau pathology towards AD-associated seizures and suggest that Tau pathology may contribute to seizure generation in AD independent of Aβ pathology.
Keywords: Alzheimer’s Disease, THY-Tau22, Tau, EEG, epilepsy, seizures
Introduction.
The microtubule-associated protein Tau is a scaffolding protein, largely expressed by neurons (Arendt et al., 2016). Under pathological conditions, Tau undergoes several modifications that are thought to impair brain function and to cause neurodegeneration. Neurofibrillary tangles, made of hyperphosphorylated and aggregated Tau proteins (“Tau pathology”) are hallmarks of Alzheimer’s disease (AD) and other dementia, particularly some subtypes of fronto-temporal lobar degenerations (FTLD; Lebouvier et al., 2018; Sergeant et al., 2008). In AD, neuronal Tau pathology initially appears at very circumscribed brain regions and subsequently progresses throughout the brain in a fairly predictive stereotypical pattern. Topographical spread of Tau pathology has been linked to progressive cognitive deterioration (Bejanin et al., 2017; Brier et al., 2016; Colin et al., 2020; Duyckaerts et al. 1997). All these observations support a pivotal contribution of neuronal Tau pathology to cognitive deficits. However, mechanisms underlying neuronal Tau pathology-induced cognitive deficits in AD remain poorly understood (Guo et al., 2017; Wang and Mandelkow, 2016).
Epileptic seizures and subclinical epileptiform activity constitute a frequent and severe comorbidity of AD, that might have an impact on cognitive status (Lam et al., 2017; reviewed in Vossel et al., 2017). The incidence of seizures has also been described in patients with different types of FTLD including those with MAPT mutations (reviewed in Sanchez et al., 2018). Obvious evidence speaks for a link between Tau and seizure occurrence. First, Tau hyperphophorylation and misfolding have been observed in patients with epilepsy as well as in different related experimental models (reviewed in Sanchez et al., 2018). Second, Tau reduction limits excessive network excitability in various experimental models of seizures (DeVos et al., 2013; Gheyara et al., 2014; Holth et al., 2013; Ittner et al., 2010; Roberson et al., 2007). Whether Tau pathology itself promotes the development of seizures independent of Aβ, and whether it correlates to memory deficits remains unclear. Few studies have been performed in models of tauopathy overexpressing human Tau protein bearing FTLD-MAPT related mutations (García-Cabrero et al., 2013; Maeda et al., 2016; Van Erum et al., 2020). Overall, these studies demonstrate a higher susceptibility to the proconvulsant GABAA receptor antagonist pentylenetetrazol (PTZ) as compared to wild-type littermate mice. However, presence of spontaneous epileptiform activity and occurrence of these latter phenomena earlier or concomitantly to memory impairments remain unclear. In the present study, we evaluated the impact of Tau pathology towards spontaneous electrographic seizure occurrence and the response of seizure thresholds to PTZ using the THY-Tau22 mouse model of Tauopathy, known to progressively develop hippocampal Tau pathology together with spatial memory impairments (Burnouf et al., 2013; Van der Jeugd et al., 2013).
Methods.
Male THY-Tau22 (Tau22) and littermate wild type (WT) mice, taken as controls [pure C57Bl6/J background], were kept in standard animal cages as described (Schindowski et al., 2006). Experiments were performed in an AAALAC-accredited facility in compliance with the principles outlined in the NIH Guide for the Care and Use of Laboratory animals and under approval by the Institutions Animal Care and Use Committee.
EEG electrodes were implanted in THY-Tau22 mice and age matched WT littermates at 6 (n=7/genotype) and 12 months of age (n=4 WT and n=9 Tau22). Briefly, each mouse was anesthetized (1.5–2% isofluorane) and equipped with a (i) a bipolar stainless steel electrode (insulated except for 80–100μm vertically exposed at the tip; tip diameter 5-μm; vertical tip separation 200–250μm; Plastics One Inc.) unilaterally implanted into the hippocampal CA1 subregion using stereotactic coordinates (AP = −2.1 mm; ML = −1.8 mm; DV = −1.7 mm with bregma as reference); (ii) a cortical monopolar screw electrode placed over the frontal cortex; (iii) and a monopolar screw ground electrode over the cerebellum. All electrodes were secured to the skull with dental cement. After recovery from surgery all animals were continuously recorded by EEG for at least 72 hours. Electrical brain activity was monitored using a Nervus EEG recording system connected with a Nervus Magnus 32/8 Amplifier. The digital EEG signal was band-pass filtered (high-pass filter 1.0 Hz cut off, low-pass 50 Hz) and analyzed using the LabChart version 7 software (AD Instruments). EEGs were assessed for seizure activity as defined as high-amplitude rhythmic discharges that represented a new pattern of tracing (repetitive spikes, spike-and-wave discharges, or slow waves) lasting at least 5 seconds.
The seizure thresholds of 6- and 12-month-old WT and Tau22 mice were assessed with escalating doses of PTZ (n=10/genotype/age). PTZ (Sigma P6500) was dissolved at 3.5 mg/kg in sterile 0.9% NaCl and delivered i.p. with a starting dose of 20 mg/kg, followed by additional doses of 5mg/kg every 5 minutes. During each 5min period, mice were evaluated for the presence of Straub’s tail, clonic convulsions, hindlimb extension, and death. Data are expressed as survival curves with the percentage of non-responding mice for each evaluation plotted relative to dose.
Western blots and histopathological evaluations and quantifications were performed in a separate group of animals as described (Gouder et al., 2004; Laurent et al., 2017; Leboucher et al., 2013). For Western blots, we used anti-GFAP (1:1000; Santa Cruz), anti-ADK (1:4000) and anti-GAPDH (1:10000; Santa Cruz) antibodies (n=3 genotype/age). For the histopathological evaluation mice were transcardially perfused with 0.9% saline and 4% paraformaldehyde, brains cut into 40 μm coronal sections. Immunohistochemical detection of either GFAP (1:15000; Chemicon International), ADK (1:5000) or pathological Tau (AT100; 1/400) was performed as previously published (Studer et al., 2006; Burnouf et al., 2013).
Quantitative real time PCR was performed as described (Burnouf et al., 2013). Primer sequences used are as following: 5’-tgtagctgacatctgcaaaaa-3’ (GAD65 forward), 5’-gggacatcagtaaccctcca-3’ (GAD65 reverse), 5’-agcatacaggtcctggcatc-3’ (cyclophilin A forward), 5’-ttcaccttcccaaagaccac-3’ (cyclophilin A reverse). Cyclophilin A was used as internal control. Amplifications were carried out in triplicate and the relative expression of target genes was determined by the ΔΔCT method.
For microdialysate analysis of hippocampal GABA, twelve-month old Tau22 mice (n=6) or WT (n=8) were anaesthetized with urethane (1.62 g/kg/i.p.) and placed in a stereotaxic frame (David Kopf, USA) with the body temperature maintained close to 37.5°C using a heated under-blanket (Harvard Instruments, USA). Homemade microdialysis probes with a 1-mm regenerated cellulose membrane (NeuroDialyTics facility, Lyon Neuroscience Research Center, France) was randomly implanted in the right or left dorsal hippocampus (AP −1.8 mm, ML ±1.5 mm, DV −2.4 mm). Probes were perfused at a rate of 1 μl/min with artificial cerebrospinal fluid (aCSF) (149 mM NaCl, 2.80 mM KCl, 1.2 mM MgCl2, 1.2 mM CaCl2, 2.78 mM phosphate buffer, pH 7.4). At least 2 hours elapsed before collection of basal samples (fractions 1, 2, and, 3), collected every five minutes. In situ depolarization was induced for 15 min (fractions 4, 5, and, 6) in both groups by switching the inlet of the probe with a aCSF containing a high concentration of potassium (51.8 mM NaCl, 100 mM KCl, 1.2 mM MgCl2, 1.2 mM CaCl2, 2.78 mM phosphate buffer, pH 7.4). Fractions 7 to 10 corresponded to perfusion with normal aCSF for washing and return to baseline. At the end of the experiment, the placement of the cannula was verified on the frozen hemisphere. Collected dialysates were stored and kept at −80°C before analyses, performed using capillary electrophoresis with laser-induced fluorescence detection (Sauvinet et al., 2003).
Data were analyzed using GraphPad Prism software (GraphPad Software, La Jolla, CA) using Student’s t, two-way ANOVA followed by Bonferroni post-hoc analysis and Mantel-Cox (Log-rank) test for the survival curves. The null hypothesis was rejected at the P = 0.05 level for all analyses.
Results.
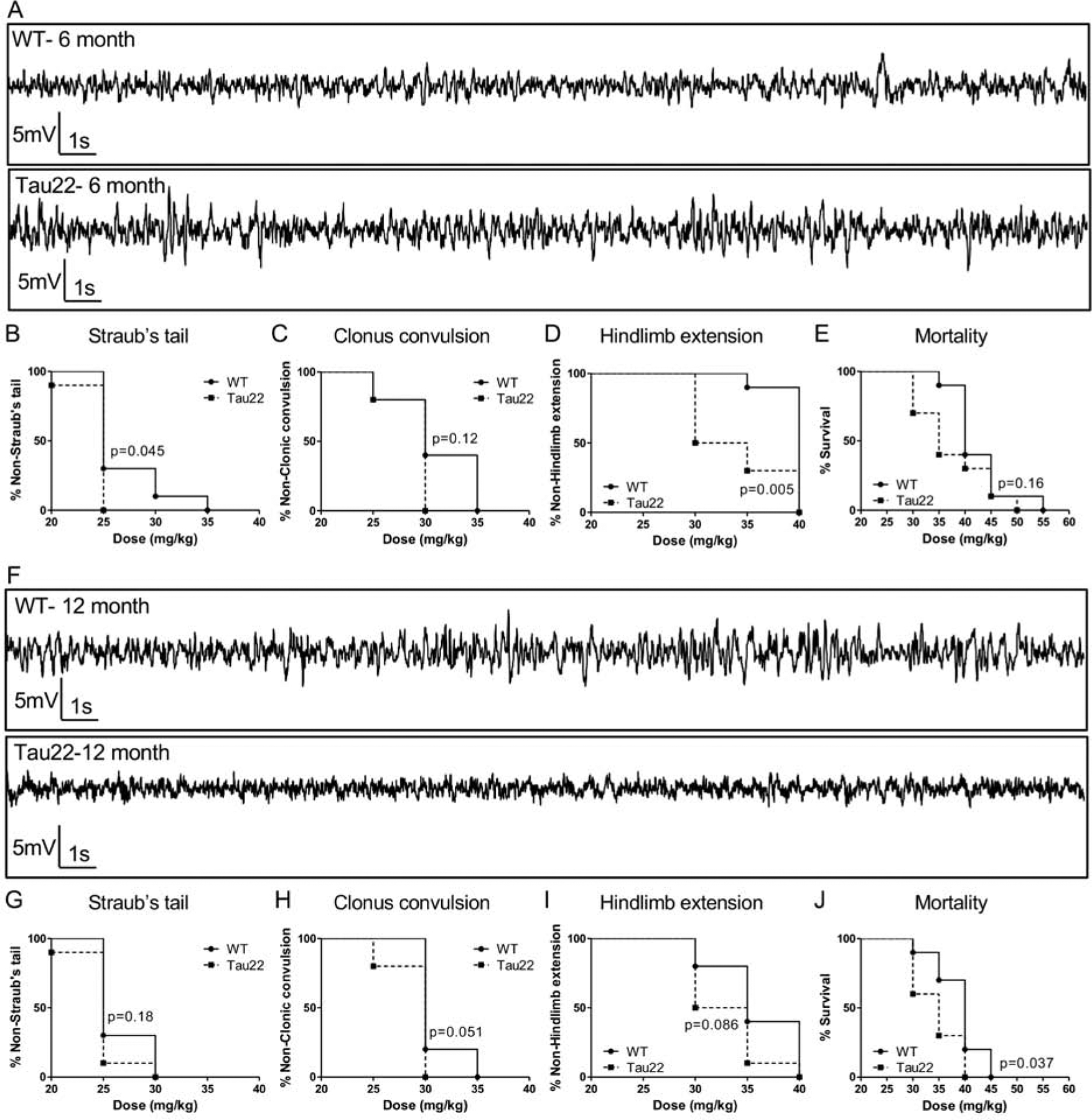
We first determined if Tau pathology is sufficient to elicit spontaneous seizures. In the THY-Tau22 strain, hippocampal pathology substantially progresses from 6 to 12 months of age (Schindowski et al., 2006). Importantly, at the age of 6 months, THY-Tau22 mice do not exhibit spatial memory impairments while at 12 months, these mice show significant alterations (Burnouf et al., 2013; Carvalho et al., 2019; Van der Jeugd et al., 2013). EEG recordings obtained from hippocampal depth electrodes implanted into the CA1 of 6 and 12-month-old mice were found to be similar to the brain activity of WT littermates (Fig. 1). Cumulative recording times of at least 500 hours did not reveal a single electrographic seizure. EEGs were without changes in the spike waveform that are indicative of spontaneous electrographic seizures or epileptiform activity (Fig. 1A and F). Despite a lack of spontaneous electrographic seizures in the mutant mice, Tau pathology led to a moderate increase in seizure susceptibility (Fig. 1B–D). Using the PTZ seizure threshold test, overall, THY-Tau22 mice progressed from Straub’s tail (Fig. 1B and G), to clonus extension (Fig. 1C and H) and finally hindlimb extension (Fig. 1D and I) at a lower dose of PTZ than WT littermates. There was also a trend towards increased mortality in the Tau22 mice (Fig. 1E and I). Therefore, Tau pathology increases seizure susceptibility, although insufficient to cause epileptic seizures by itself. Several mechanisms may be involved in the increased seizure susceptibility to PTZ. Here, we sought to investigate if Tau pathology modulates the expression of markers typically associated with network hyperexcitability and seizures such as astrocytic and GABA-related markers. First, we identified a significant hippocampal increase of the astrogliosis marker GFAP at both 6 and 12 months of age in Tau mice (Fig. 2A–C). This coincided with a delayed increase in adenosine kinase (ADK) expression in transgenic animals (Fig. 2D–F). Noteworthy, Tau pathology, as marked by the conformational AT100 antibody, is significantly enhanced between these two time-points (Fig. 2G–I). In addition, while we found a moderate increase in GAD65 expression in THY-Tau22 as compared to WT (Fig. 2J), basal GABA levels revealed no statistical difference between WT and Tau22 mice (Fig. 2K). Further, responses to potassium-induced depolarization were also examined; and in spite of a downward trend, the release of GABA induced by 100 mmol/L K+ for Tau 22 mice was not statistically different from that in WT mice (Fig. 2L).
Figure 1.

Tau pathology decreases seizure threshold but is not sufficient to cause spontaneous electrographic seizures. (A) Representative hippocampal electroencephalographic (EEG) traces from a 6 month-old mutant Tau22 mouse and wildtype (WT) littermate are both indicative of normal brain activity. (B-D) The Straub’s tail (B); clonus extension (C); and hindlimb extension (D) responses of 6 month-old WT and Tau22 mice to escalating pentylenetertrazol (PTZ) doses. The percent of Tau22 mice that elicit Straub’s tail (B) and hindlimb extension (D) at a lower PTZ dose is significantly different from the response in WT mice. (E) Percent mortality in response to PTZ dose is equivalent between 6-month-old Tau22 and control mice. (F) Representative hippocampal EEG traces from a 12-month-old Tau22 mutant and WT control are both indicative of normal brain activity. (G-I) The Straub’s tail (G); clonus extension (H); and hindlimb extension (I) responses of 12-month-old WT and Tau22 mice to escalating PTZ doses. (J) The percent mortality in response to increasing doses of PTZ is significantly increased in 12-month-old Tau22 mice, compared to controls. PTZ threshold data are plotted as percent survival curves (n = 10 / genotype) and analyzed with a Mantel-Cox (Log-rank) test.
Figure 2.
Tau pathology is associated with increased astrogliosis and expression of ADK and GAD65. (A-F). The astrogliosis markers GFAP and ADK are increased in the hippocampus of Tau22 mutants compared to age-matched controls. (A) Upper panel: representative GFAP and GAPDH Western blots. Lower panel: densitometry analysis of the GFAP / GAPDH ratio identifies a significant increase in hippocampal GFAP protein levels in the 6 and 12-month-old Tau22 mutants (black bars), compared to age-matched WT littermates (white bars). GFAP measurements are normalized to GAPDH and expressed as percent of control (mean ± SEM, n = 3 per age and genotype). (B,C) Representative images of GFAP immunohistofluorescence in the hippocampal formation of 12 month old WT (B) and Tau22 (C) mice. Insets are higher magnification images of astrocytes obtained from the respective fields in panels B and C. (D) Upper panel: representative ADK and GAPDH Western blots. Lower panel: densitometry analysis of the ADK / GAPDH Western blot indicates a significant increase in hippocampal ADK protein levels at 12 months of age in the Tau22 mutants (black bars), compared to controls (white bars). ADK measurements are normalized to GAPDH and expressed as percent of control (mean ± SEM, n = 3 per age and genotype). (E,F) Representative images of ADK immunohistochemistry in the hippocampal formation of 12 month old WT (E) and Tau22 (F) mice. (G,H, I) Representative images of AT100 tau immunohistochemistry in the hippocampal formation of 6 (G) and 12 month-old (H) Tau22 mice and related quantification (I). (J) Quantitative PCR for GAD65 mRNA indicates a significant genotype effect (p=0.0045) with a significant increase at 6 months of age and a trend towards increased expression at 12 months of age within the Tau22 hippocampus, compared to the age matched controls. Data are normalized to cyclophilin A and expressed as percent of control (mean ± SEM, n = 5 Tau22 and n = 6 WT per age). (K,L) GABA concentrations in microdialysates obtained in the dorsal hippocampus of anesthetized 12 month old Tau22 (n=6, in black) and WT mice (n=8, in white) are equivalent. (K) The basal levels represented as histograms were calculated as the mean ± SEM of the three consecutive 5-min microdialysates following the 2-h equilibration period and preceding K+ depolarization. (L) The effect of a 15-min potassium-induced depolarization (black line) on GABA extracellular concentrations (circles) monitored at a 5-min sampling rate. Data are expressed as percent (mean ± SEM) of the three baseline values preceding K+ depolarization. No statistical difference was revealed. All data were analyzed by two-way ANOVA followed by Bonferroni post-hoc analysis when appropriate (* < 0.05; ** < 0.01, *** < 0.0001).
Discussion.
Accumulation of Aβ into plaques and Tau in neurofibrillary degeneration are the two hallmarks of AD. While the role of Aβ into the development of AD-related seizures has been well investigated (reviewed in Vossel et al., 2017), the involvement of Tau pathology and relationship to memory impairments remained ill-defined. Here, we used the progressive THY-Tau22 strain, to investigate the link between Tau pathology development, hippocampal memory deficits and seizure occurrence, as well as susceptibility to PTZ. Evaluations were performed at 6 months of age, when pathology is present, but memory unaffected, and 12 months of age when hippocampal pathology and memory impairments culminate (Van der Jeugd et al., 2013). Moreover, during this period, neuro-inflammation, a known contributor to the pathophysiology of epilepsy (Patel et al., 2019), is ongoing and participate to memory deficits (Ising et al., 2019; Laurent et al., 2016; Laurent et al., 2017).
Our results suggest that established Tau pathology unlikely leads to spontaneous seizure occurrence. However, the PTZ data indicates that Tau pathology reduced seizure threshold as early as 6 months, when hippocampal-dependent memory is not impaired. It seems unlikely that susceptibility to PTZ relates to Tau pathology itself. Indeed, while the latter is substantially enhanced between 6 and 12 months of age in the THY-Tau22 strain, this does not lead to a particular enhancement of susceptibility to PTZ. Excitability changes might therefore relate to either soluble toxic species and/or Tau overexpression as previously suggested in another transgenic Tau models (Maeda et al., 2016). Several mechanisms could be involved in the observed increased seizures susceptibility (reviewed in Vossel et al., 2017), such as glutamate spillover that may occur through impaired glial transporters and/or impaired presynaptic release, alterations of GABA or GABAergic interneurons, or metabolism of the endogenous anticonvulsant adenosine through astrocytic ADK. In a former study, we demonstrated that THY-Tau22 mice exhibit increased hippocampal levels of glutamate (Laurent et al., 2016) and an increase in the glutamatergic drive in CA1 (Zappettini et al., 2019), which might be implicated in the increased susceptibility to PTZ found in Tau mice. Conversely, while Tau mice exhibit a slight change in GAD mRNA expression, steady-state and induced GABA levels were not found altered in Tau mice, presumably excluding an involvement in the PTZ phenotype.
Interestingly, we found that PTZ sensitivity was associated with a significant increase in astroglial ADK levels at 12 months of age. Increased levels of ADK are a pathological hallmark of both epilepsy and AD (Aronica, et al., 2015). It has also been documented that increased levels of ADK can be a direct cause for the generation of spontaneous electrographic seizures (Li et al, 2008). The lack of spontaneous electrographic seizures in the THY-Tau22 mice was therefore an unexpected finding. However, we found a reduction of PTZ seizure thresholds, which is in line with increased ADK expression and a concomitant reduction of the inhibitory adenosinergic tone.
Overall, our results show that THY-Tau22 pathology is not sufficient to generate spontaneous subclinical seizures, but our results clearly show that the THY-Tau22 pathology can reduce seizure thresholds independently of Aβ. In contrast to Tau models, previous data from transgenic amyloidogenesis models clearly demonstrate the occurrence of spontaneous seizures (Palop et al., 2007; Minkeviciene et al., 2009). Reasons for such discrepancy remain unclear, but it may potentially depend on the enhanced ability of amyloid peptides and plaques to promote microglia and astrocytic neuroinflammation (Sierksma et al., 2020) known to strongly contribute to seizure development (Vezzani et al., 2010), as well as to memory impairments (Laurent et al., 2017; Ising et al., 2019). Further, amyloid-β induced seizures depend on the presence of tau (Roberson et al., 2007), suggesting that the development of Tau pathology, may may further aggravate Aβ-driven seizures in AD.
Highlights.
Tau pathology development is not associated with spontaneous seizures
Tau pathology increases propensity for PTZ-induced seizures
Increased network excitability is associated with enhanced adenosine kinase expression
Verifications.
We do not have conflicts of interest regarding the present work.
This work was supported by grants from Hauts-de-France (PARTEN-AIRR, COGNADORA), ANR (ADORASTrAU to DBl), CoEN and Programs d’Investissements d’Avenir LabEx (excellence laboratory) DISTALZ (Development of Innovative Strategies for a Transdisciplinary approach to ALZheimer’s disease). Our laboratories are also supported by Fondation pour la Recherche Médicale, France Alzheimer/Fondation de France, FHU VasCog research network (Lille, France), Fondation Vaincre Alzheimer, Fondation Plan Alzheimer as well as Inserm, CNRS, Université Lille, Lille Métropole Communauté Urbaine, DN2M. VG-M was supported by Fondation pour la Recherche Médicale (SPF20160936000). DBo is funded through grants from the US National Institutes of Health (NIH, NS103740, NS065957), the CURE foundation (CURE Catalyst Award) and the New Jersey Commission for Brain Injury Research.
Data contained in the manuscript being submitted have not been previously published, have not been submitted elsewhere and will not be submitted elsewhere while under consideration at Neurobiology of Aging.
Concerning animals, all protocols were approved by an ethical committee.
All the authors have read and approved submission.
Acknowledgements.
We thank the Animal Facility (F-59000 Lille, France) and Melanie Besegher, Cyrille Degraeve, Caroline Declerck, Kim Letten, Yann Lepage, Benjamin Guerrin, Didier Montignies, Christian Meunier, Quentin Dekeyser and Romain Dehaynin for animal care.
Funding. This work was supported by grants from Hauts-de-France (PARTEN-AIRR, COGNADORA), ANR (ADORASTrAU to DBl), CoEN and Programs d’Investissements d’Avenir LabEx (excellence laboratory) DISTALZ (Development of Innovative Strategies for a Transdisciplinary approach to ALZheimer’s disease). Our laboratories are also supported by Fondation pour la Recherche Médicale, France Alzheimer/Fondation de France, FHU VasCog research network (Lille, France), Fondation Vaincre Alzheimer, Fondation Plan Alzheimer as well as Inserm, CNRS, Université Lille, Lille Métropole Communauté Urbaine, DN2M. VG-M was supported by Fondation pour la Recherche Médicale (SPF20160936000). DBo is funded through grants from the US National Institutes of Health (NIH, NS103740, NS065957), the CURE foundation (CURE Catalyst Award) and the New Jersey Commission for Brain Injury Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests.
The authors declare no conflict of interest.
References.
- Arendt T, Stieler JT, Holzer M Tau and tauopathies. Brain Res Bull. 2016. Sep;126(Pt 3):238–292. doi: 10.1016/j.brainresbull.2016.08.018. [DOI] [PubMed] [Google Scholar]
- Boison D, Aronica E Comorbidities in Neurology: Is adenosine the common link? Neuropharmacology. 2015. Oct;97:18–34. doi: 10.1016/j.neuropharm.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejanin A, Schonhaut DR, La Joie R, Kramer JH, Baker SL, Sosa N, Ayakta N, Cantwell A, Janabi M, Lauriola M, O’Neil JP, Gorno-Tempini ML, Miller ZA, Rosen HJ, Miller BL, Jagust WJ, Rabinovici GD Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain. 2017. Dec1;140(12):3286–3300. doi: 10.1093/brain/awx243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, Owen C, Aldea P, Su Y, Hassenstab J, Cairns NJ, Holtzman DM, Fagan AM, Morris JC, Benzinger TL, Ances BM Tau and Aβ imaging., CSF measures., and cognition in Alzheimer’s disease. Sci Transl Med. 2016. May 11;8(338):338ra66. doi: 10.1126/scitranslmed.aaf2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnouf S, Martire A, Derisbourg M, Laurent C, Belarbi K, Leboucher A, Fernandez-Gomez FJ., Troquier L, Eddarkaoui S, Grosjean ME, Demeyer D, Muhr-Tailleux A, Buisson A, Sergeant N, Hamdane M, Humez S, Popoli P, Buée L, Blum D NMDA receptor dysfunction contributes to impaired brain-derived neurotrophic factor-induced facilitation of hippocampal synaptic transmission in a Tau transgenic model. Aging Cell. 2013. Feb;12(1):11–23. doi: 10.1111/acel.12018. [DOI] [PubMed] [Google Scholar]
- Colin M, Dujardin S, Schraen-Maschke S, Meno-Tetang G, Duyckaerts C, Courade JP., Buée, L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020. Jan;139(1):3–25. doi: 10.1007/s00401-019-02087-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, Schuler DR, Maloney SE, Wozniak DF, Rigo F, Bennett CF, Cirrito JR, Holtzman DM, Miller TM Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013. Jul 31;33(31):12887–97. doi: 10.1523/JNEUROSCI.2107-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Bennecib M, Grignon Y, Uchihara T, He Y, Piette F, Hauw JJ Modeling the relation between neurofibrillary tangles and intellectual status. Neurobiol Aging. 1997. May-Jun;18(3):267–73. [DOI] [PubMed] [Google Scholar]
- García-Cabrero AM, Guerrero-López R, Giráldez BG, Llorens-Martín M, Avila J, Serratosa JM, Sánchez MP Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol Dis. 2013. Oct;58:200–8. doi: 10.1016/j.nbd.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Gouder N., Scheurer L., Fritschy J-M., and Boison D (2004). Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci 24, 692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheyara AL, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W, Finucane MM, Sanchez PE, Mucke L Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann Neurol. 2014. Sep;76(3):443–56. doi: 10.1002/ana.24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Noble W, Hanger DP Roles of tau protein in health and disease. Acta Neuropathol. 2017. May;133(5):665–704. doi: 10.1007/s00401-017-1707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J, and Noebels JL (2013). Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci 33, 1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, Griep A, Santarelli F, Brosseron F, Opitz S, Stunden J, Merten M, Kayed R, Golenbock DT, Blum D, Latz E, Buée L, Heneka MT NLRP3 inflammasome activation drives tau pathology. Nature. 2019. Nov;575(7784):669–673. doi: 10.1038/s41586-019-1769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010. Aug 6;142(3):387–97. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Lam AD, Deck G, Goldman A, Eskandar EN, Noebels J, Cole AJ Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat Med. 2017. Jun;23(6):678–680. doi: 10.1038/nm.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent C, Dorothée G, Hunot S, Martin E, Monnet Y, Duchamp M, Dong Y, Légeron FP, Leboucher A, Burnouf S, Faivre E, Carvalho K, Caillierez R, Zommer N, Demeyer D, Jouy N, Sazdovitch V, Schraen-Maschke S, Delarasse C, Buée L, Blum D Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain. 2017. Jan;140(1):184–200. doi: 10.1093/brain/aww270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent C, Burnouf S, Ferry B, Batalha VL, Coelho JE, Baqi Y, Malik E, Mariciniak E, Parrot S, Van der Jeugd A, Faivre E, Flaten V, Ledent C, D’Hooge R, Sergeant N, Hamdane M, Humez S, Müller CE, Lopes LV, Buée L, Blum D A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol Psychiatry. 2016. Jan;21(1):97–107. doi: 10.1038/mp.2014.151. [DOI] [PubMed] [Google Scholar]
- Lebouvier T, Pasquier F, Buée L Update on tauopathies. Curr Opin Neurol. 2017. Dec;30(6):589–598. doi: 10.1097/WCO.0000000000000502. [DOI] [PubMed] [Google Scholar]
- Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest. 2008. Feb;118(2):571–82. doi: 10.1172/JCI33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Djukic B, Taneja P, Yu GQ, Lo I, Davis A, Craft R, Guo W, Wang X, Kim D, Ponnusamy R, Gill TM, Masliah E, Mucke L Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 2016. Apr;17(4):530–51. doi: 10.15252/embr.201541438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009. Mar 18;29(11):3453–62. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO., Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007. Sep 6;55(5):697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, and Mucke L (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. [DOI] [PubMed] [Google Scholar]
- Sánchez MP, García-Cabrero AM, Sánchez-Elexpuru G, Burgos DF, Serratosa JM Tau-Induced Pathology in Epilepsy and Dementia: Notions from Patients and Animal Models. Int J Mol Sci. 2018. Apr 5;19(4). pii: E1092. doi: 10.3390/ijms19041092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvinet V, Parrot S, Benturquia N, Bravo-Morato E, Renaud B, and Denoroy L (2003). In vivo simultaneous monitoring of gamma-aminobutyric acid., glutamate., and L-aspartate using brain microdialysis and capillary electrophoresis with laser-induced fluorescence detection: Analytical developments and in vitro/in vivo validations. Electrophoresis 24, 3187–3196. [DOI] [PubMed] [Google Scholar]
- Schindowski K, Bretteville A, Leroy K, Begard S, Brion JP, Hamdane M, and Buee L (2006). Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol 169, 599–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N, Bretteville A, Hamdane M, Caillet-Boudin ML, Grognet P, Bombois S, Blum D, Delacourte A, Pasquier F, Vanmechelen E, et al. (2008). Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Expert Rev Proteomics 5, 207–224. [DOI] [PubMed] [Google Scholar]
- Sierksma A, Lu A, Mancuso R, Fattorelli N, Thrupp N, Salta E, Zoco J, Blum D, Buée L, De Strooper B, Fiers M. Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. EMBO Mol Med. 2020. Mar 6;12(3):e10606. doi: 10.15252/emmm.201910606. Epub 2020 Jan 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer FE, Fedele DE, Marowsky A, Schwerdel C, Wernli K, Vogt K, Fritschy JM and Boison D (2006). Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience 142, 125–137. [DOI] [PubMed] [Google Scholar]
- Van der Jeugd A, Vermaercke B, Derisbourg M, Lo AC, Hamdane M, Blum D, Buée L, D’Hooge R Progressive age-related cognitive decline in tau mice. J Alzheimers Dis. 2013;37(4):777–88. doi: 10.3233/JAD-130110. [DOI] [PubMed] [Google Scholar]
- Van Erum J, Valkenburg F, Van Dam D, De Deyn PP Pentylenetetrazole-induced Seizure Susceptibility in the Tau58/4 Transgenic Mouse Model of Tauopathy. Neuroscience. 2020. Jan 15;425:112–122. doi: 10.1016/j.neuroscience.2019.11.007. [DOI] [PubMed] [Google Scholar]
- Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011. January;7(1):31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL Epileptic activity in Alzheimer’s disease: causes and clinical relevance. Lancet Neurol. 2017. Apr;16(4):311–322. doi: 10.1016/S1474-4422(17)30044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E Tau in physiology and pathology. Nat Rev Neurosci. 2016. Jan;17(1):5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- Zappettini S, Faivre E, Ghestem A, Carrier S, Buée L, Blum D, Esclapez M, Bernard C Caffeine Consumption During Pregnancy Accelerates the Development of Cognitive Deficits in Offspring in a Model of Tauopathy. Front Cell Neurosci. 2019. Oct 1;13:438. doi: 10.3389/fncel.2019.00438. [DOI] [PMC free article] [PubMed] [Google Scholar]