

Abstract
Human induced pluripotent stem cells derived cardiomyocytes (hiPSC-CMs) have been extensively used for in vitro modeling of human cardiovascular disease, drug screening and pharmacotherapy, but little rigorous studies have been reported on their biophysical or Ca2+ signaling properties. There is also considerable concern as to the level of their maturity and whether they can serve as reliable models for adult human cardiac myocytes. Ultrastructural difference such as lack of t-tubular network, their polygonal shapes, disorganized sarcomeric myofilament, and their rhythmic automaticity, among others, have been cited as evidence for immaturity of hiPSC-CMs. In this review, we will deal with Ca2+ signaling, its regulation, and its stage of maturity as compared to the mammalian adult cardiomyocytes. We shall summarize the data on functional aspects of Ca2+ signaling and its parameters that include: L-type calcium channel (Cav1.2), ICa-induced Ca2+ release, CICR, and its parameters, cardiac Na/Ca exchanger (NCX1), the ryanodine receptors (RyR2), sarco-reticular Ca2+ pump, SERCA2a/PLB, and the contribution of mitochondrial Ca2+ to hiPSC-CMs excitation-contraction (EC)-coupling as compared with adult mammalian cardiomyocytes. The comparative studies suggest that qualitatively hiPSC-CMs have similar Ca2+ signaling properties as those of adult cardiomyocytes, but quantitative differences do exist. This review, we hope, will allow the readers to judge for themselves to what extent Ca2+ signaling of hiPSC-CMs represents the adult form of this signaling pathway, and whether these cells can be used as good models of human cardiomyocytes.
Keywords: hiPSC-CMs, mammalian cardiomyocytes, Ca2+ signaling, mitochondrial Ca2+
Introduction
The development of inducible pluripotent stem cells (iPSC) from human somatic cells [1] and the procedures to differentiate them into cardiac myocytes [2] could potentially become a milestone in advancing basic and translational cardiovascular research. Creating patient specific human cardiac myocytes from patient’s own tissue biopsy may also serve as a cell-source for in vitro modeling of human inherited cardiac pathologies, its pharmacotherapy, and drug screening [3–8]. There is nevertheless considerable concern as to the level of maturity of these cells and whether hiPSC-CMs can serve as a good model for primary human cardiomyocytes. Often cited examples of cellular immaturity are that hiPSC-CMs express atrial-, ventricular-, and SA-nodal-type action potentials, reflecting possibly their mixed ancestral origins [6, 9–11], and that these myocytes lack t-tubular network, have disorganized sarcomeric structures, inconsistent adrenergic responses, and beat rhythmically in cultures unlike adult cardiomyocytes [12, 13]. Since Ca2+ signaling plays a central role in initiation of action potential, regulation of pacemaking and cardiac contractility, evaluating the level of maturity of Ca2+ signaling pathway of hiPSC-CMs, as compared to primary mammalian cardiomyocytes, is crucial in determining whether these cells can serve as appropriate physiological models of adult human cardiomyocytes.
The studies reported thus far on Ca2+ signaling of hiPSC-CMs are mostly confined to measurements of intracellular calcium transients, without detailed analysis of the parameters of the calcium signaling pathway. Here, we shall examine the Ca2+ signaling parameters of hiPSC-CMs that include: biophysical properties of calcium channel (Cav1.2), the expression levels of ICa-induced Ca2+ release (CICR and its parameters: fractional release, gain, Ca2+ spark characteristics, the magnitude and source of calcium stores), the expression levels and regulation of cardiac Na/Ca exchanger (NCX1), the ryanodine receptors (RyR2), sarcoplasmic reticular Ca2+ pump, SERCA2a, and the contribution of mitochondrial Ca2+ in hiPSC-CMs excitation-contraction (EC)-coupling. To assess the level of maturity of Ca2+ signaling pathway in hiPSC-CMs, we shall compare their Ca2+ signaling properties to those of adult ventricular, atrial and neonatal rat cardiomyocytes. To this end and to facilitate comparisons of EC-coupling events between hiPSC-CMs and adult cardiomyocytes, we have chosen first to present an overview of Ca2+ signaling data of the adult mammalian cardiomyocytes in Section I, before discussing the published data on hiPSC-CMs in Section II. We hope this approach will allow the readers of this review to judge for themselves to what extent Ca2+ signaling of hiPSC-CMs represents the adult form of this signaling pathway.
I. Ca2+ signaling in primary mammalian cardiomyocytes
1.1. A historical retrospective of Cardiac EC-coupling
1.1a. EC-coupling and Ca2+-induced Ca2+ release.
Cardiac EC-coupling encompasses the molecular events that couple the depolarization of surface membrane to mechanical events of myofilament contraction. First attempts to determine the role of action potential in regulation of contraction in the mammalian heart, showed that the first 20–50ms of the action potential triggered over 70% of the contractile force, while the remaining 300ms of the action potential regulated the remaining fraction of the contraction [14]. These first voltage clamp studies in cat ventricular strips when compared to similar studies in the frog heart [15], suggested two types of regulatory mechanism for cardiac Ca2+ homeostasis. In the mammalian heart the action potential initially triggers and then in graded manner regulates the development of tension while in the frog heart the action potential continuously and linearly mediates the entry of Ca2+ into the myocytes that directly determine the magnitude and duration of contraction [15, 16]. This unique mammalian cardiac specific mechanism coincides with the activation of Ca2+ channels in the initial phase of the action potential [17], and suggests the possibility that the influx of calcium in early phase of the action potential triggers the release of intracellular Ca2+ causing the rapid component of phasic tension in the initial 50ms of the action potential [14, 18]. Despite such findings, significant controversy continued to exist about the mechanisms that coupled membrane depolarization to initiation of contraction. One popular idea was that depolarization of cardiomyocyte directly triggers the release of Ca2+ in a manner similar to skeletal muscle where the depolarization-induced capacitive charge movement in the surface membrane transfers the activating signal to the SR membrane, triggering the release of calcium [19]. Another view, consistent with 1830s frog heat reports of Sydney Ringer, held that cardiac contraction required extracellular calcium [15, 16], where influx of calcium either directly activated the contraction as in the frog heart, or triggered the release of larger Ca2+ from intracellular pools in the mammalian hearts. Two seminal reports in 1980s finally settled this issue: 1) in skinned mammalian cardiac muscle strips rapid applications of small Ca2+ concentrations caused larger releases of cytosolic Ca2+, a process called calcium induced calcium release, CICR [20–22] and 2) in intact voltage-clamped rat cardiomyocytes, 1–2 seconds of rapid withdrawal of extracellular Ca2+ prior to activation of calcium channels that fully suppressed ICa, but allowed large influx of Na+ to permeate through the Ca2+ channels, failed to trigger the release of Ca2+ from fully loaded SR stores, suggesting that influx of Ca2+ through the Ca2+ channels was mandatory for the release of Ca2+ [23]. This ICa-gated Ca2+ release mechanism showing that direct entry of Ca2+ and not membrane depolarization was responsible for the release of Ca2+ from the SR, was further strengthened by findings that the voltage dependence of intracellular Ca2+ release mirrored the bell-shaped voltage-dependence of ICa [24–26]. In sharp contrast, in skeletal muscle the bell-shaped voltage dependence of ICa was accompanied by a sigmoid voltage-dependence of Ca2+ release and developed tension, reflecting the voltage-dependence of charge movements associated with activation of Ca2+ channel, and consistent with a voltage-gated Ca2+ release mechanism [19]. Further, cardiac Ca2+ release could be triggered equally well by depolarizing pulses, or by repolarizing pulses (from positive potentials to resting potentials) that re-activated ICa “tail currents” [24, 26]. It was therefore concluded that mammalian cardiac EC-coupling is mediated by influx of Ca2+ through the L-type Ca2+ channels that triggers the release of 10–20 times larger Ca2+ concentrations from sarcoplasmic reticulum (SR) through the type-2 ryanodine receptor (RyR2) sufficient to activate the myofilaments.
In a purely CICR controlled mechanism, however, only the charge carried by calcium channel would determine the magnitude of released calcium independent of the activating voltage of the Ca2+ channel. That is, there should be no voltage-dependence to the efficacy or gain of CICR. Several labs including ours [27], however, have found significant voltage-dependence to the gain of CICR at voltages between −30 to +10mVs [27, 28], suggesting that other mechanisms may also contribute to the release process. In our experiment CICR gain was ~40 at +10 to 30mVs (the plateau range of voltages of cardiac action potential) but increased to values exceeding 100 at −30mVs [27], inset fig.1. It has been suggested that the voltage dependence of CICR gain may arise from the greater efficacy of calcium release unites that are activated by the larger unitary ICa at −30 mV compared to +10 mV, despite the lower open probability of the channel at −30 mV. This possibility was tested by Bers and colleague who evaluated the role of open probability of the channel and its magnitude in the voltage dependence of CICR gain, and found that the decrease in the open probability of the channel rather than the magnitude of the current at −40mVs was responsible for the higher gain of CICR. They attributed this to the redundant openings of Ca-channels at +10 mV that would lower gain of CICR at the plateau range of potentials [29]. Another possible mechanism that could render voltage-dependence to the gain of CICR at negative potentials is a direct interaction of carboxyl tail of CaV1.2 with the RyR2 that would sensitize RyR2 to increase its spontaneous Ca2+ sparks frequency, resulting in enhanced ICa-gated Ca-transients at −30 to −40mVs [30–32]. This possibility may also underlie the often observed small differences in voltage dependence of ICa and ICa-triggered Ca-transients at voltages between −40 to −10mVs.
Fig.1.
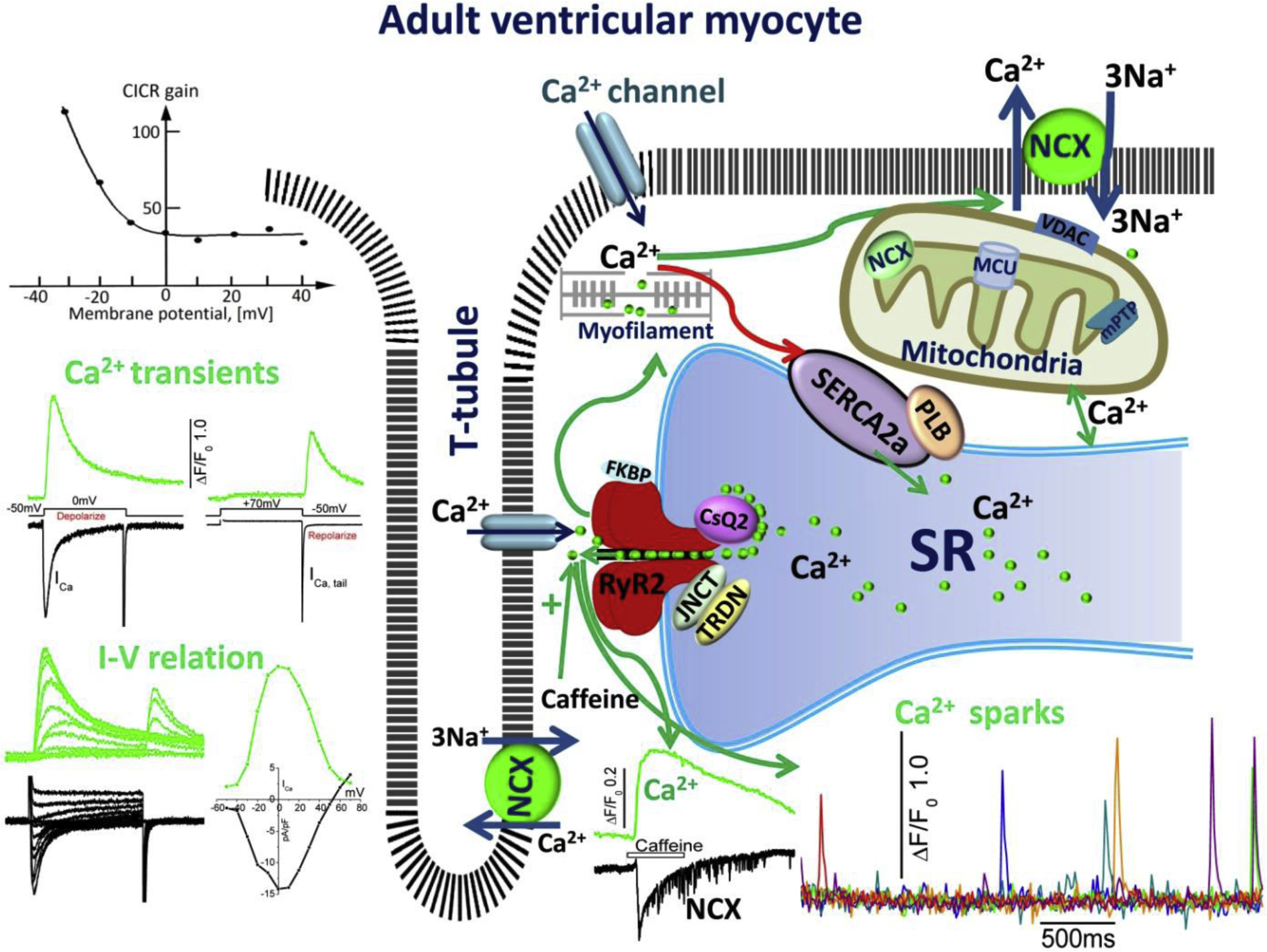
Excitation-contraction and Ca2+ cycling in adult ventricular myocytes. The schematic shows cell surface membrane and t-tubular expression of L-type Ca2+ channel, Na/Ca exchanger (NCX), and sarcoplasmic reticulum (SR) expression of Ryanodine receptors (RyR2) in dyadic couplon, as well as SERCA2a/PLB Ca2+ uptake, and calsequestrin the storage proteins of the SR. The green arrows depict the influx of Ca2+ via the Ca2+ channels, activating RyR2, the release of SR Ca2+ into the cytoplasm and its binding to myofilaments. Relaxation is initiated by reuptake of Ca2+ into the SR via the Ca2+ pump (red arrow) and extrusion by NCX across the membrane and possible sequestration by mitochondria. The figure also shows representative traces of ICa activated by depolarization to zero and by repolarization from +70mVs (ICa tail current), and the accompanying Ca2+ transients, the bell shaped I-V relation of ICa and Ca2+ transients, and voltage-dependence of CICR gain. Also shown are the time courses of Ca2+ sparks, activation of Ca2+ transients by caffeine and the accompanying INCX. Also depicted are: TRDN, triadine, JNCT, Junctin, associated with RyR2, and mitochondrial NCX, MCU, Ca2+ uniporter, and anion channel VDAC.
The released Ca2+ initiates contraction as Ca2+ binds to the myofilaments, allowing the cross-bridges to initiate their “rowing” action, allowing the sliding of actin over the myosin filaments and shortening the sarcomere. Cardiac relaxation begins as the released Ca2+ is sequestered by ATP-dependent Ca2+ uptake(SERCA2a) into the SR [33, 34], the activity of which is regulated, in turn, by phospholamban (PLB) phosphorylation [35–37] by protein kinase A/beta adrenergic agonists [38] or Ca2+/calmodulin-dependent protein kinase II (CaMKII) [39], thus increasing the affinity of SERCA2a to Ca2+ and enhancing the rate of Ca2+ uptake and relaxation of contraction, see figure 1.
1.1b. CICR and its inactivation
Fabiato et al. were first to suggest that influx of Ca2+ through CaV1.2 first activates the SR release mechanism, triggering the larger release of Ca2+ from the SR that in turn inactivates the release mechanism [20], somewhat similar to the L-type Ca2+ channel’s calcium-dependent inactivation mechanism, CDI [20]. This idea, when tested in intact myocytes by photo-releasing calcium from caged calcium compounds, at different times following the activation of release process by ICa, failed to show any evidence of inactivation or termination of Ca2+ release. Photo-released Ca2+ only enhanced the rise of cytosolic Ca2+ irrespective of its release time following ICa-gated Ca2+ release, i.e. photo-released Ca2+ failed to suppress or terminate the ICa-triggered Ca2+ release [40]. Studies in isolated cardiac SR vesicles expressing RyR2 further confirmed that Ca2+ release from the SR vesicles could only be suppressed by large un-physiological concentration of Ca2+. In skeletal muscle, while RyR1 is inhibited by ~1mM Ca2+, about 10-fold higher concentrations of Ca2+ are required to inhibit RyR2 channel [41, 42]. In canine vesicles of cardiac SR also, 1mM or higher Ca2+ concentrations were required to inhibit Ca2+ release in the presence of Mg2+ and adenine nucleotides [43]. Despite large differences in Ca2+ concentration required for inactivation of RyRs in skeletal and cardiac muscle (IC50 of RyR1, ~1mM, and RyR2 >10mM), the Ca2+ activation of Ca2+ release channels occurred with EC50 values of about 1μM.
Possible roles of other accessary proteins associated with RyR2, such as calmodulin, calsequestrin, triadin and junctin, in the inactivation of the release mechanism have also been considered [44,45]. Gyorke et al, for instance, suggested that the binding of Ca-free calsequestrin to the RyR/Junctin/Triadin complex may decrease the calcium sensitivity of RyR2 when the SR Ca2+ content decreases after SR release [46]. Considered also in the inactivation of RyR2 mechanism, was the possibility that the dyadic release couplons become fully emptied of their Ca2+ by the triggering ICa, thereby obviating the inactivation of Ca2+ release, and thereby suppressing also the inherent positive feedback of CICR [47]. This idea is consistent with the findings that the frequency of occurrence of activated Ca2+ sparks, but not their size, depended on the magnitude of activated ICa [48]. Considering the various likely mechanisms proposed and tested for the inactivation of the release mechanism, it is likely that multiple molecular processes may be involved in the Ca2+ dependent inactivation of the dyadic Ca2+ release.
1.1c. Dyadic Calcium release and RyR2 microdomains.
As it became generally accepted that ICa gates the release of calcium, ultrastructural and live cellular imaging were used to study the focal characteristics of dyadic Ca2+ release and its microdomains (20–50nm). Studies in voltage-clamped myocytes dialyzed with high concentrations of EGTA to buffer the global cytosolic Ca2+ concentrations showed that gating of dyadic Ca2+ release by ICa was resistant to 14–20mM intracellular EGTA concentrations, as if accessibility into the subsarcolemmal space surrounding the dyads was limited to the Ca2+ influx through the channel [49]. This and similar findings suggested that RyR2 proteins of dyadic junctions may have privileged access to dihydropyridine receptors of the surface membrane [49, 50]. These conclusions were later supported by confocal imaging data on intact cardiomyocytes, showing the development of spontaneous focal Ca2+ releases, sparks, associated with the dyadic structures that had durations in excess of 100ms and covered sarcomeric area of 1–2μm [51]. Since the morphology and time course of focal Ca2+ releases could be significantly altered by binding kinetics and affinity of the Ca2+ sensitive fluorescent dyes and cellular diffusion of the dye/Ca2+ complex, the measured sparks were unlikely to represent accurately the time course of dyadic Ca2+ release. In support of this possibility, rapid (240f/s) 2-D confocal imaging of 100s of sparks in patch-clamped rat ventricular myocytes, dialyzed with combination of EGTA and Fluo3 to limit the diffusion of dye/calcium complex, showed that spontaneous or triggered sparks had an average duration of ~7ms, carrying ~100,000 Ca2+ ions [48]. This finding and the fact that the sparks occurred at the same cellular location on repetitive activation of Ca2+ release ruled out that Ca2+ sparks represented the stochastic activity of a single RyR2, but rather the activation of single dyadic structure, with multiple RyR2s [48]. These studies also showed that the magnitude of Ca2+ sparks remained mostly constant, irrespective of the magnitude of ICa activated at different voltages, but the frequency of occurrence of sparks varied with the magnitude of ICa.
1.1d. Molecular determinants of Ca2+ signaling: Ca2+ and caffeine binding sites.
Ca2+ influx through the channel and its binding to the high affinity Ca2+-binding site on the ryanodine receptor is the initial step for the activation of RyR2 and Ca2+ release from the SR [43]. Two classes of Ca2+-binding sites have been identified on RyR protein: a high-affinity Ca2+ activation and a low-affinity Ca2+ inactivation site [41, 52]. Early studies of MacLennan group, using truncated forms of rabbit RyR1 with 45Ca2+ overlay [53, 54] and RyR1 site-specific antibodies purified by immobilized synthetic peptide [55], identified multiple Ca2+ regulatory domains in RyR1. Using site-directed mutagenesis in recombinant rabbit RyR3 expressed in HEK293, Chen et al showed that a glutamate-to-alanine mutation of E3885A in the M2 transmembrane sequence (now considered to be in cytosolic domain in high resolution structural model) resulted in decreases of both the caffeine response and the sensitivity to activation by Ca2+, implicating RyR3-E3885 as the potential calcium sensor residue [56]. The corresponding E3987A mutation in mouse RyR2 was later reported by the same group to markedly reduce the sensitivity of the channel to activation by Ca2+, indicating that the glutamic acid residue is the common Ca2+ activation site in all RyR isoforms [57]. Data from 4.3Å cryo-electron microscopic structure of RyR1, using 30 μM Ca2+ as activating stimulus, appears to implicate 5 amino acids E3893(3847), E3967(3921), T5001(4930), Q3970(3925) and H3895(3850) of RyR1/RyR2, located at the core domain just above the transmembrane domain to form the protein’s putative Ca2+-binding site [58, 59]. The site-directed mutagenesis of the recombinant RyRs and structural mapping of formerly identified E4032 of RyR1 (corresponding to E3987 of RyR2), not only showed that these residues were critical for Ca2+-dependent activation of RyRs, but also that E4032 may form an interface with carboxyl terminal that includes T5001, suggesting that mutation of E4032 could destabilize the Ca2+ binding conformation. In this model, the neighboring amino acid W4716 of RyR1 (corresponding to W4645 of RyR2), just below the Ca2+-binding site, was identified to be the putative caffeine-binding site. Our recent studies on human stem cell derived cardiomyocytes have confirmed some of these findings and further suggest possible interaction between the caffeine and Ca2+ binding sites, such that mutations of either Ca2+ or caffeine binding sites may alter each other’s sensitivity to caffeine binding and Ca2+-induced activation [60, 61]. Other studies using mutational analysis have shown that mutation or deletion of residues Δ4274–4535 of rabbit RyR1 leads to altered caffeine and Ca2+ sensitivity of RyR1 channel, suggesting that caffeine and Ca2+ activation sites may not be located within these residues [62]. In addition, several residues outside of these domains in RyR carboxyl terminal were found to be involved in Ca2+-dependent inactivation, which included two EF hand type Ca2+ binding sites (EF1: RyR1 aa 4081–4092; EF2: 4116–4127), with milli-molar affinity to Ca2+[42, 62], consistent with biochemical studies [63]. Gomez et al, in addition, showed that EF1 and the second transmembrane segment (S2) are critical for isoform-specific Ca2+-dependent inactivation of RyR [64]. Since EF hand domain and cytoplasmic loop between the second and the third transmembrane (S2-S3) loops are in close proximity of low affinity Ca2+ binding site [65], EF1 may transmit the signal for inactivation to the pore through S2-S3 loops. High resolution structural mappings of low affinity Ca2+ binding site may clarify this issue, but such studies remain to be carried out.
1.2. Physiological modulation of EC-coupling
1.2a. NCX1 and the modulation of CICR.
To achieve Ca2+ homeostasis, the additional per beat entry of Ca2+ through the Ca2+ channels is extruded by the sarcolemmal Na+-Ca2+ exchanger (NCX1), which exchanges one calcium for three sodium ions, generating an inward current [66–68]. Since NCX1 protein can function both in Ca2+ influx or efflux modes, a number of reports in 1990s suggested that during early phases of the plateau of the action potential Ca2+ could enter the cell via the Ca2+ influx, reverse mode, of the NCX1 and thereby trigger Ca2+ release from the SR [69, 70]. Such findings were, somewhat, controversial as other studies that examined Ca2+ influx on NCX1 failed to show significant role for the entry of Na+ or the exchanger in release of Ca2+ under physiological conditions [71]. Considering the stoichiometry, kinetics and voltage-dependence of NCX1, it is also unlikely that NCX1 could operate in the Ca2+ influx mode within the first 10ms of depolarization of the action potential, because Ca2+ concentrations delivered by the Ca2+ channel into the nano-domains of dyadic RyR2, in the first few ms of action potential, would reach micro-molar levels, thereby shifting the electro-chemical potential for NCX1 (ENa-Ca) to potential positive to +30mVs, forcing the exchanger to operate in its Ca2+ efflux mode. In support of this idea, high-resolution immunofluorescence imaging of NCX1, CaV1.2 and RyR2, showed that while CaV1.2 and RyR2 were co-expressed in the same microdomains in the t-tubules, there was little co-expression of NCX1 and RyR2 in the same microdomains [72, 73]. Since the proximity of RyR2 proteins to CaV1.2 in dyadic junctions is paramount for effective activation of RyR2 to release Ca2+, the absence of NCX1 from the dyadic microdomains would further minimize the role of the exchanger in gating Ca2+ release from the SR, under physiological conditions. The sub-cellular microarchitecture, where dihydropyridine receptors and RyR2s are co-expressed in the same microdomains, and are surrounded by clusters of NCX1 proteins in the inter-dyadic spaces, would tend to minimize the diffusion of Ca2+ from neighboring dyads that could trigger Ca2+ release, thus suppressing the inherent positive feedback of CICR.
1.2b. The evolutionary role of NCX1 in cardiac EC-coupling
The role of NCX1 in Ca2+ homeostasis of mammalian myocardium, where there is both triggered release and entry of Ca2+, appears to have diverged significantly from that expressed in hearts of species that have not developed Ca2+ uptake and release components of the reticulum. In Rana pippins frog and Squalus acanthus shark hearts, where SR is poorly developed and dyadic junctions and caffeine-triggered pools are absent, NCX1 appears to mediate both the influx of Ca2+ for contraction and extrusion of Ca2+ mediating the relaxation [74–77]. Surprisingly, in frog heart, which lacks SERCA2a/PLB entities of SR [78, 79] that mediate the adrenergic enhancement of Ca2+ uptake and relaxation in the mammalian hearts, beta-adrenergic agonists still produce both their positive inotropic and relaxant effects. In these hearts, calcium enters the myocardium during the action potential continuously via the Ca2+ channels and Na/Ca exchanger and relaxation is mediated by efflux of Ca2+ on the exchanger [15, 80]. Fan et al, were first to report that the beta-adrenergic induced enhancement of relaxation was mediated by adrenergic modulation of NCX1 activity [76], where NCX1 has an additional P-loop with Walker A sequence [81], which endows the protein with PKA-mediated regulation. Consistent with this possibility, while the Na+-dependent Ca2+ uptake in oocytes expressing the frog exchanger was enhanced by cAMP, this effect was absent in the dog exchanger, or the recombinant frog exchanger lacking the p-loop [81]. Further, insertion of the p-loop in the dog exchanger conferred the cAMP-dependent regulation to HEK cells expressing the chimera NCX1 expressing the frog exon [82]. The molecular data, therefore, suggests that the 9-amino acid of P-loop containing the Walker A nucleotide binding motif maybe responsible for the observed cAMP-dependent functional differences between the frog and the mammalian heart NCX1 [81].
In shark heart too, reduction of [Na+]i suppressed while elevation of [Na+]i enhanced a tonic component of contraction as it activated an outward current reflecting the influx of Ca2+ via the electrogenic Na+-Ca2+ exchanger, thus regulating the contraction/relaxation cycle [77]. A bimodal adrenergic regulation of NCX1 appears to have evolved in the shark heart, where adrenergic agonists suppress greatly the Ca2+ influx on NCX1 as they enhance the efflux mode of the exchanger [83]. Cloning and sequencing of shark NCX revealed an insertion of A/P rich region-2 domain, not present in the mammalian NCX1 [84]. The deletion of this domain removed cAMP mediated bimodal regulation of the shark exchanger, suggesting that it mediates the beta-adrenergic modulation by PKA phosphorylation. The bi-modal beta-adrenergic/PKA mediated phosphorylation of NCX1 may have evolved to satisfy the evolutionary needs for accelerated contraction and relaxation during a fight or flight response in hearts of animals missing the SR Ca2+ release and uptake molecular machinery. The development Ca2+ release and uptake stores of SR and their regulation by PKA phosphorylation of PLB/SERCA2a protein complex, appears to have resulted in pruning the molecular motifs that were evolved for adrenergic regulation of NCX1. This evolutionary pruning provides the cell with only one regulated Ca2+ efflux or uptake mechanism. With the development of SR expressing SERCA2a/PLB, beta-adrenergic phosphorylation of PLB enhances the sequestration of Ca2+ into the SR leading to enhancement of the Ca2+ store, the contractile force, and relaxation, obviating the evolutionary needs of the heart to have PKA-regulation of NCX1.
1.3. Transgenic mice with deletions or overexpression of NCX1, ICa, PLB, CSQ, and their pathophysiological consequences
Creation of transgenic mice with overexpression or deletion of Ca2+ signaling pathway proteins have provided unexpected insights into pathophysiology of Ca2+ signaling.
1.3a. Na/Ca exchanger, NCX1.
Over expression of cardiac NCX1, for instance, while producing 3–5 times larger exchanger current on releasing the SR Ca2+ pools by caffeine, did not produce significant hypertrophy or cardiac pathology in transgenic mice [85, 86]. In myocytes isolated from mice hearts overexpressing the exchanger, the intracellular Ca2+ pools of SR remained either unchanged or were slightly enhanced. The myocytes also showed no changes in their ICa density, Ca-transients magnitude, level of expression of SERCA2a, phospholamban or calsequestrin [28, 85–87]. Wang et al confirmed significantly larger SR Ca2+ content and no cardiac hypertrophy in NCX overexpressing myocytes [88]. The enhanced SR calcium content is unexpected in the light of larger Ca2+ efflux on the exchanger, suggesting possible compensatory mechanisms involving enhanced SERCA2a calcium reuptake into the SR. Homozygous overexpression of NCX1, on the other hand, caused 40% cardiac hypertrophy with contractile dysfunction and clinical evidence of heart failure [89]. Homozygous knock out of NCX1 was embryonically lethal. Heart tubes at 9.5 day post coitum had normal Ca2+ transients but were unable to maintain Ca2+ homeostasis when subjected to Ca2+ challenges [90]. Conditionally-deleted NCX1 mice appeared to live into adulthood, had normal cardiac functions at seven-weeks and somewhat higher CICR gain [91]. In sum, although deletion of NCX1 is embryonically lethal, either its conditional deletion or overexpression in adult mice appears not to produce significant cardiac pathology.
1.3b. L-type Ca2+ channel.
Over expression of α1C subunit of calcium channel is reported to generate a slowly developing cardiac hypertrophy within 8 months and 20% increase in cell capacitance at 4 month [92, 93]. While ICa density and its inactivation kinetics increased in 2–8 months, Ca2+ transients increased significantly only in 4-month-old mice. Although the larger Ica of overexpressed hearts triggered larger Ca2+ release from the SR, the caffeine sensitive Ca2+ store, diastolic Ca2+ levels and Ca2+ spark properties remained mostly unchanged. The enhanced ICa entry and SR Ca2+ release appeared to have been compensated for by upregulation of the Na-Ca exchanger activity. Somewhat unexplained was the finding that the inotropic and lusitropic responses of isoproterenol and forskolin were significantly reduced in ICa overexpressing myocytes [92–94].
1.3c. Calsequestrin, CSQ2 the SR Ca2+-binding and storage protein.
Overexpression of calsequestrin in mice hearts leads to marked cardiac hypertrophy, arrhythmias and heart failure. Electrophysiologically, myocyte action potentials were prolonged and ICa, Ito, and IK1 were suppressed [95]. CSQ2 overexpressing mice hearts with 10-fold higher levels of calsequestrin survived into adulthood, but had severe cardiac hypertrophy. The impaired Ca2+ signaling in isolated myocytes appeared to result from structural uncoupling of dyadic couplons from the sarcolemma, resulting in suppressed ICa-gated Ca2+ release and marked reduction in the rate of inactivation of ICa, even though caffeine-triggered Ca2+ release was markedly enhanced [96]. Since the Ca2+ storage capacity of SR was increased by overexpression of CSQ2, caffeine releasable Ca2+ pools and the resultant exchanger currents were also highly enhanced. Ca2+ channel gated Ca2+ release, however, and the frequency of spontaneously occurring sparks were suppressed despite the high Ca2+ content of SR, implicating structural deformation of dyadic/sarcolemmal couplings [95, 96].
CSQ2 null mice myocytes, showing significantly decreased triadin and junction levels, unexpectedly displayed normal caffeine-triggered Ca2+ release and unchanged contractile function under basal conditions [97]. In another study, CSQ2 knockout mice showed accelerated intra-SR Ca2+ refilling kinetics, reduced refractoriness of Ca2+ release and reduced rate-dependent Ca2+ alternans in intact mouse hearts [98]. The loss of SR Ca2+ release refractoriness appeared to have resulted from impaired Ca2+-CaM-dependent inactivation of L-type Ca2+current [99]. Catecholamines appears to increase the diastolic SR Ca2+ leak in CSQ2 knockout myocytes, triggering premature spontaneous Ca2+ releases and aberrant contractions [97]. Heterozygous CSQ2 deleted mice, with 25% decrease in protein levels and no significant decrease in junctin and triadin-1 levels, had unchanged field-stimulated Ca2+ transients, L-type Ca2+ currents, and SR volume [100]. Adult rat myocytes transfected with adenoviral gene expressing antisense for canine calsequestrin cDNA, reduced CSQ2 levels by >50% and caffeine-releasable pools by 30%. These myocytes had markedly reduced ICa-gated Ca2+ release [101], supporting the idea that the most critical Ca2+ signaling defect of either suppression or overexpression of CSQ2 is the defective signaling between CaV1.2 and RyR2, most likely caused by structural disruptions between t-tubules and SR membranes. It is likely that the observed increase in SR Ca2+ leak is responsible for the reported triggered ventricular arrhythmias in patients expressing CSQ2 mutations [100]. It should be noted that although it has long been known that CSQ2 was the major Ca2+ storage protein of the SR, the creation of transgenic mice made it clear that CSQ2 in addition critically regulates CICR and the normal function of the heart. It still remains somewhat puzzling why the deletion of CSQ2 in mice does not reduce the magnitude of Ca2+ release from the SR as it does in CSQ antisense experiments, and whether the increased volume of SR in CSQ2 null mice compensate for the lack of this protein in the SR.
1.3d. Phospholamban (PLB).
Phospholamban is the key inhibitory protein associated with the cardiac Ca2+ pump (SERCA2a) that regulates the rate of Ca2+ uptake into the SR under baseline conditions. On beta-adrenergic stimulation, PLB is phosphorylated decreasing its affinity to bind and inhibit SERCA2a, thus enhancing the rate SERCA2a sequestration of Ca2+, thereby enhancing the rate of decay of Ca2+ transients and relaxation.
Mice hearts overexpressing PLB show significant suppression of electrically triggered Ca2+ transients, cardiac contractility and rate of relaxation with no hypertrophy or heart rate changes, resulting from suppressed SERCA2a activity [102–104]. In contrast, genetic deletion of PLB causes enhanced myocardial contractility, increased Ca2+ ATPase activity and Ca uptake resulting in larger Ca2+ content of SR [105–108]. As expected PLB-deficient myocytes lose or respond poorly to β-agonist stimulation [107]. Phospholamban deletion, using CRISPR/Cas9 technique, appeared to improve the mortality of mice suffering from heart failure caused by CSQ-overexpression [109]. Recent finding in saponin-permeablized cardiomyocytes isolated from wild-type or PLB-knockout mice suggest that PLB is also highly expressed in the nuclear envelope where it regulates the nuclear Ca2+ handling and IP3R-mediates nuclear Ca2+ release [110]. PLB engineered mice appear to reflect the functional consequences of altered SERCA2a activity on Ca2+ uptake and Ca2+ content of SR.
1.4. Ca2+ signaling in Atrial myocytes
Atrial myocytes are significantly smaller than ventricular myocytes averaging in diameter (5–10 vs 20–40 um), are often devoid of t-tubules, especially in smaller mammals [111–113]. Electrophysiologically, also there are differences in expression levels of K+ and chloride channels, resulting in shorter atrial action potentials. Generally, atrial Ca2+ signaling is initiated by activation of ICa that triggers a rise in cytosolic Ca2+ in cell periphery that diffuses to cell center activating release of Ca2+ from the corbular SR and giving rise to the global Ca-transients [111, 114, 115]. Such a Ca2+ diffusion-dependent signaling pathway and sparse expression of t-tubules requires time delays for development of Ca2+ transients and generation of contraction, consistent with the finding that action potential induced Ca2+ releases in cat atrial myocytes are spatially inhomogeneous [116]. Rapid 2-D (240 f/s) confocal imaging also shows two groups of Ca2+ sparks in rat atrial myocytes, those near the surface membrane co-localized with dihydropyridine receptors (DHPRs), having significant voltage-dependence to the frequency of their occurrence, and those centrally located (not associated with DHPRs), showing no voltage-dependence [117]. Peripheral sparks were brighter, flatter, had ~5-fold higher frequency of occurrence, ~2 times faster diffusion coefficient, and extinguished abruptly. Central sparks, in contrast, occurred less frequently, were elongated along the cellular longitudinal axis, and dissipated slowly [117]. Interestingly, the magnitude of Ca2+ sparks was about 3 times larger in rat atrial cells, ranging ~300,000 Ca2+ ions, as compared to rat ventricular cells. Surprisingly, central sparks and peripheral sparks appeared often to activate within 4ms of each other (2-D confocal imaging at 240 frames/s), even in myocytes dialyzed with 5mM EGTA, suggesting that either expression of vestigial t-tubular system mediates the rapid activation of central sparks, or an as yet unknown signaling pathway couples the activation of central sparks to calcium channel current. A sparse t-tubular network was also reported in dog hearts, in both right (<25%) and left (~12.5%) atria [118], but despite the sparsity of t-tubules the rate of developed tension in dog atrial strips was faster than ventricular strips [119], suggesting that either an alternate, Ca2+ signaling pathway is expressed in the dog atria, or that atrial myofilament sensitivity to Ca2+ is significantly higher than in the ventricles.
There seems to be little qualitative difference in the fundamental steps that regulate Ca2+ signaling pathway when comparing the atrial and ventricular EC-coupling. Variability in size of Ca2+ sparks and their morphology in atria seems to be related to the location of SR Ca2+ release structures (couplons), vis-à-vis the sarcolemma or vestigial t-tubules, and the consequence of calcium dependent inactivation of sarcolemmal CaV1.2. Development of t-tubules seems to be strictly cell size dependent, as larger mammals (sheep, pigs, etc.) with larger hearts appear to have a higher fraction of their atrial cells expressing t-tubular network [120–122]. Despite these differences dictated by ultrastructural differences of atrial myocytes, the fundamental aspects of CICR, the gain, the fractional release, the magnitude of SR calcium stores and their sensitivity to caffeine and adrenergic agonists are nearly identical to those of ventricular myocytes. Physiologically, however, atrial roles as secretory organ, mechanical stretch or sheer stress sensor, its differential cholinergic and purinergic innervation, and its role in regulation of blood pressure makes it a unique myocardial tissue with multiple functions. Consistent with multiplicity of atrial function, these myocytes express significantly higher levels of IP3Rs (both type 1 and type 2) as compared to ventricular cells [123, 124]. The parallel IP3 Ca2+ signaling pathway may have evolved to regulate the signaling pathways associated to other functions of atria, as well as expression of transcription factors related to development and growth of the myocytes [125].
II. Ca2+ signaling in hiPSC-CMs
As discussed in the first section of this review, cellular evaluation of Ca2+ signaling requires critical measurements of amount of charge carried by ICa, its inactivation kinetics, the magnitude of Ca2+ released from the SR by caffeine, the gain of CICR, and the fractional release of SR Ca2+ by ICa.[126] The gain of CICR. measured as the degree to which ICa triggers Ca2+ release normalized to the Ca2+ content of the SR store and the charge carried by ICa [4, 127], shows unexpected voltage-dependence in adult cardiomyocytes, as discussed above.
To determine whether hiPSC-CMs represent a good model of adult human cardiomyocytes or merely replicate the physiology of immature cardiomyocytes, it is critical to compare and quantitate the structural and functional differences of the two tissues, as they pertain to the different cellular physiological signaling pathways. On Structural level, hiPSC-CMs have flat rounded or polygonal/spindle shapes with large surface to volume ratios [13, 129], low levels of myofibrils, and disorganized z-lines and sarcomeres [12, 130, 131]. They also lack t-tubular network [132–134] and have non-uniform distribution of calcium release units [12, 130]. Functionally, hiPSC-CMs beat spontaneously in culture, unlike adult cardiomyocytes [13, 128], express variable levels of If, lower levels of IK1, and T-type Ca2+channels [13, 128, 135]. In addition, hiPSC-CMs show mixture of atrial, ventricular, and nodal action potentials, possibly related to their varied ancestral origins. These signaling parameter may result in the observed lower rate of rise and decay of cellular Ca2+ transients [10, 129], and lower fractional Ca2+ release [4] as compared to adult cardiomyocytes. Nevertheless, the fundamental Ca2+ signaling parameters of hiPSC-CMs are almost identical to neonatal rat cardiomyocytes and similar to adult rat cardiomyocytes [13]. For instance, their Ca2+ release is gated by L-type Ca2+ channel, the voltage dependence of ICa and Ca-transients are similar and bell-shaped and indistinguishable from those of neonatal or adult rat cardiomyocytes, Fig.2. [4]. HiPSC-CMs also express large SR Ca2+ pools that are effectively released by 5–10mM caffeine generating significant inward NCX1 currents in voltage-clamped myocytes [13, 129]. The magnitude of INCX was significantly larger in hiPSC-CMs as compared to rat neonatal and adult cardiomyocytes (2–3 vs 1–1.5 pA/pF)[13], suggestive of either larger levels of expression of NCX1 protein in hiPSC-CMs, or more effective rise of intracellular Ca2+ in hiPSC-CMs that have small cell volumes but large surface areas [13, 129]. As to the proteins of Ca2+ signaling pathway, hiPSC-CMs express significant levels of RyR2, calcium-ATPase (SERCA2a), phospholamban, PLB, Junctin (Jun), Triadin (TRDN), calsequestrin (CSQ2) and inositol tris phosphate receptor (IP3R) [130, 136–139]. Even though the expression levels of these proteins vary greatly in reports from different labs, most likely related to their developmental stage or their in vitro culture conditions, it is clear that hiPSC-CMs express all the molecular components of cardiac Ca2+ signaling pathway.
Fig.2.
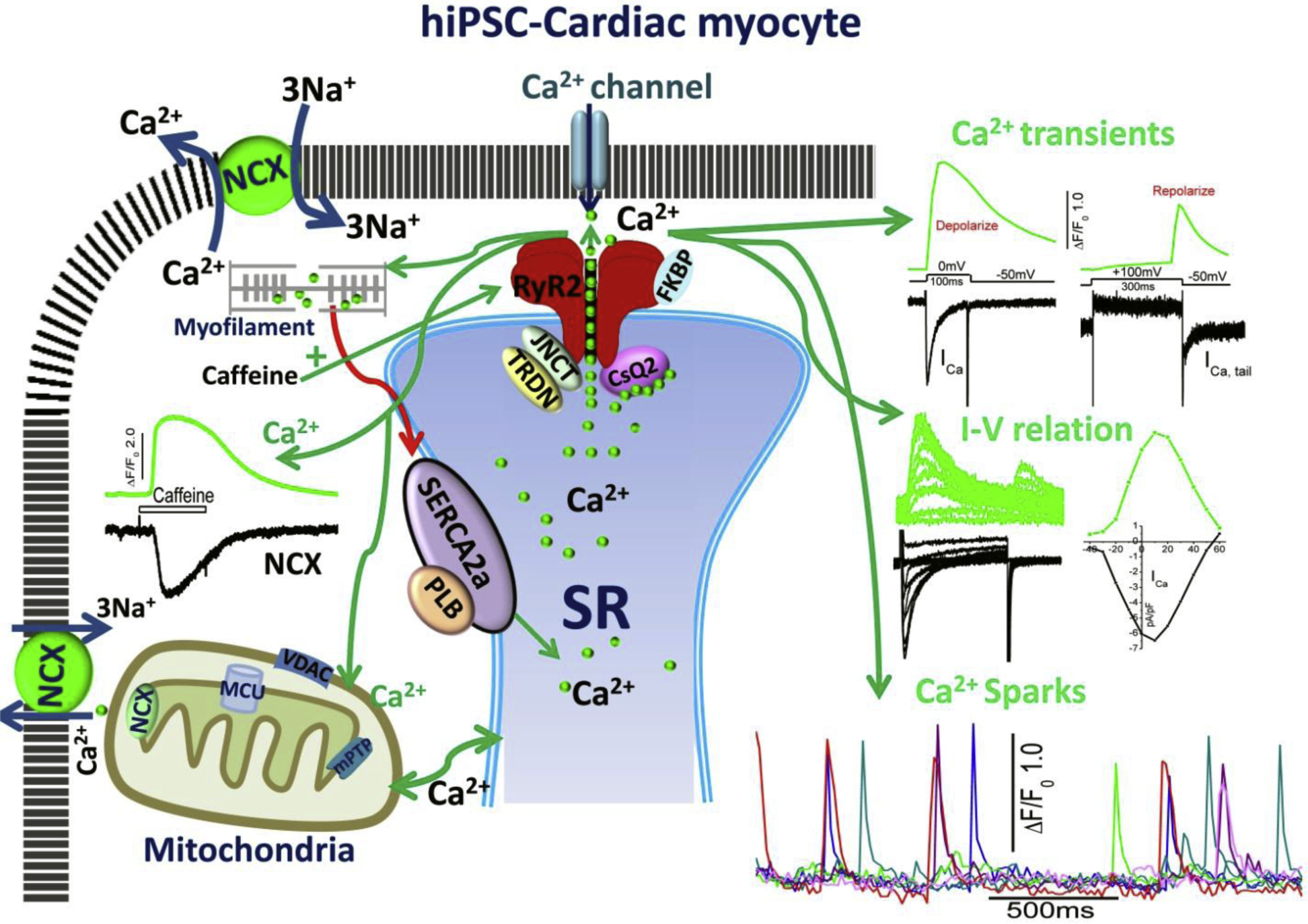
Excitation-contraction and Ca2+ cycling in hiPSC-CMs. The schematic shows cell surface membrane L-type Ca2+ channel, Na/Ca exchanger (NCX), and sarcoplasmic reticulum (SR) expression of Ryanodine receptors (RyR2) in dyadic couplon, as well as SERCA2a/PLB Ca2+ uptake, and calsequestrin the storage proteins of the SR. The green arrows depict the influx of Ca2+ via the Ca2+ channels, activating RyR2, the release of SR Ca2+ into the cytoplasm and its binding to myofilaments. Relaxation is initiated by reuptake of Ca2+ into the SR via the Ca2+ pump and extrusion (red arrow) by NCX across the membrane and possible uptake by mitochondria. The figure also shows representative traces of ICa activated by depolarization to zero and by repolarization from +100mVs (ICa tail current), and the accompanying Ca2+ transients, and the bell shaped I-V relation of ICa and Ca2+ transients. Also shown are the time courses of Ca2+ sparks, activation of Ca2+ transients by caffeine and the accompanying INCX. Also depicted are: TRDN, triadine, JNCT, Junctin, associated with RyR2, and mitochondrial NCX, MCU, Ca2+ uniporter, and anion channel VDAC. The major difference between hiPSC-CM and adult ventricular myocytes is the absence of t-tubules.
2.1. Functional components of Ca2+ signaling: Ca2+ current, NCX1 current, Ca2+ sparks, and SR Ca2+ store
2.1a. Calcium channels.
In adult ventricular myocytes L-type calcium channels are highly expressed both on the sarcolemma and t-tubular network. Despite the paucity of t-tubular network in hiPSC-CMs, the density of ICa in these cells appears to be comparable to those of adult human cardiomyocytes averaging around 8 pA/pF [4] and significantly higher, 10–12pA/pF, in monolayers of engineered heart tissue [140]. The pharmacology and voltage-dependence/kinetics of ICa in hiPSC-CMs suggest that the current is carried by the L-type Ca2+ channels (CaV1.2), where ICa activates at −30mVs, reaches its peak values around zero, reverses at potentials positive to 80mVs, and dihydropyridine blockers and divalent cations like Cd2+ and Ni2+ block, while BayK 8644 effectively enhances the current [13, 140]. Direct measurements of Ca2+ channel (CaV1.2) gene and proteins, or α−1C subunit of L-type Ca2+ channels, also confirm similar expression levels of calcium channel protein in hiPSC-CMs and adult human heart [130, 135, 141]. These findings suggest that irrespective of their originating lab and differentiation procedures used to create them, hiPSC derived cardiomyocytes express robust and similar levels of Ca2+ channels, comparable to those of adult human cardiomyocytes, compare Fig.1 of 2.
Calcium dependent inactivation (CDI), a biophysical property specific to the dihydropyridine sensitive CaV1.2 channel, allows the more Ca2+ permeating through the channel to regulate the time course of the current. It has been consistently shown, for instance, that intracellular Ca2+ buffers like EGTA or BAPTA or replacement of Ca2+ by Ba2+ as the permeating cation, markedly slows the kinetics of inactivation of only CaV1.2 channel (L-type Ca2+ current). Although the kinetics of inactivation of ICa seem to vary greatly in different hiPSC-CM cell populations, intracellular Ca2+ buffers or extracellular replacement of Ca2+ by Ba2+ have the same effects on the kinetics of channel inactivation as those reported for those expressed in the adult or neonatal cardiomyocytes. A more recent report suggests that hiPSC-CMs may express two types of nifedipine-sensitive Ca2+ channel cells, based on the kinetics of inactivation of ICa [142]. The rapidly inactivating ICa cells (tau<40ms) appeared to be dominant in 3–5 days post-dissociation hiPSC-CM cultures, while the slowly inactivating ICa cells (tau≥ 40ms) were more prevalent in 6–8 days post-dissociation cultures. The authors suggest that differences in kinetics of inactivation of ICa were in part determined by the proximity of the L-type channels to dyadic calcium release structures, such that in younger cells where dyadic Ca2+ stores are in close proximity of sarcolemmal calcium channels, the cells show rapidly inactivating ICa. In older and much larger cells, on the other hand, the dyadic Ca2+ pools distributed in much larger cellular volumes maybe at a distance from the sarcolemma, thus decreasing the effectiveness of released Ca2+ and CDI in regulating the inactivation kinetics of ICa [142]. These studies also showed that slowly inactivating ICa cells had longer ventricular-like action potentials, while the rapidly inactivating ICa cells had shorter atrial-like action potentials, a finding also confirmed in rat neonatal cardiomyocytes, see fig 3. If these findings are consistently confirmed in hiPSC-CMs created in different labs, it might resolve the issue of whether diversity in the shapes and durations of action potentials in hiPSC-CM cultures represents different ancestral origins of hiPSC-CMs, or whether they are a consequence of proximity-dependence of dyadic Ca2+ release pools to sarcolemmal Ca2+ channels caused by cell growth in culture. Some researchers, but not all, have reported the expression of T-type Ca2+ currents in hiPSC-CMs [135, 140, 143].
Fig.3.
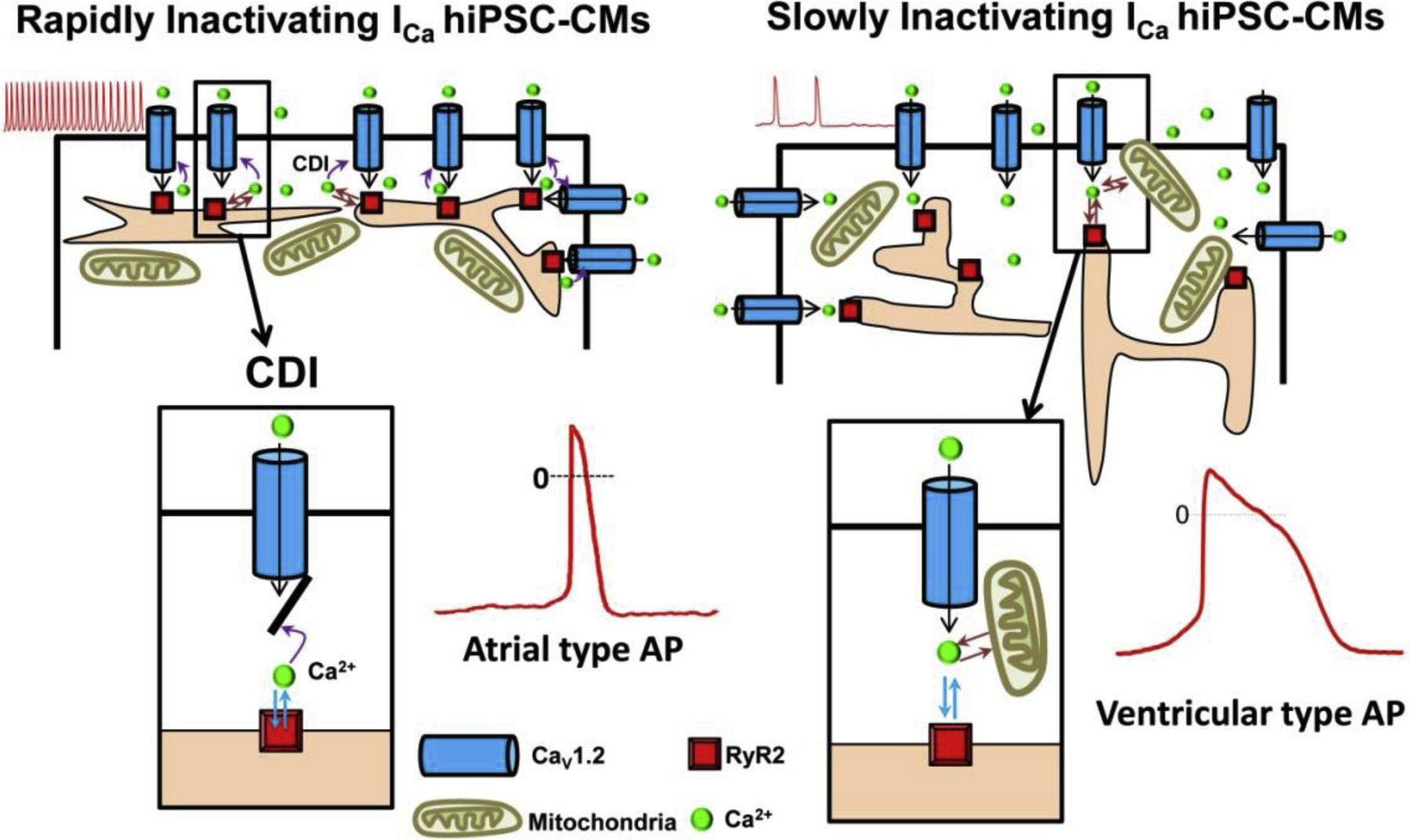
Two cell-type populations of hiPSC-CMs were identified based on the kinetics of ICa inactivation. The 3–4 day post-dissociation smaller hiPSC-CMs, expressed predominantly rapidly inactivating ICa (left panel), shorter action potentials (AP), “atrial-type” and rapid rates of spontaneous activity (red traces) whereas the 6–8 day post-dissociation hiPSC-CMs were larger, expressed mostly slowly inactivating ICa (right panel), longer action potential durations (“Ventricular-type”), and slower rates of spontaneous activity. Enlarged cartoon of calcium dependent inactivation, CDI, responsible for the differential rates of ICa inactivation and durations of APs, showing close proximity of Ca2+ channel with SR release machinery in atrial type APs (left box), and at larger distances in ventricular APs, (right box).
2.1b. β-Adrenergic regulation of ICa
Cardiac myocytes respond to β-adrenergic agonists by enhancing their ICa, global Ca-transients, the rate of development of contraction and relaxation. Similarly, hiPSC-CMs when exposed to β-adrenergic agonists also show both positive chronotropic and inotropic effects in a manner similar to adult or neonatal cardiomyocytes [128, 135, 137, 141, 144, 145]. The expression of β-adrenergic receptors (ARs) and their functional regulation of ICa in hiPSC-CMs, however, appears to be cell maturation dependent [146]. In younger cell cultures of hiPSC-CMs, (<~30 days), β2-ARs are the primary source of cAMP/PKA signaling, while in older cultures (30–60 days) β1-AR signaling appears to increase markedly, reaching cAMP generating values of 56.8 ± 6.6%. PKA signaling also shows a similar pattern of expression, increasing from day 30 to 90 [146]. Consistent with this idea, the positive inotropic and lusitropic effects of both isoproterenol and forskolin (%) appear to be more prevalent in older 90-day cultures of hiPSC-CMs [147]. HiPSC-CMs or micro-tissues derived from them show robust β-adrenergic positive chronotropic but not the positive inotropic response [148]. In such studies PLB expression by recombinant adenoviruses appears to restore the positive inotropic response of β-adrenergic stimulation [148], suggesting that the positive inotropic effect is mostly dependent on the SERCA2a/PLB activation.
2.1c. Caffeine-induced Ca2+ release, Na/Ca-exchanger, and Fractional Ca2+ release.
Sarcoplasmic reticulum is the major intracellular Ca2+ pool of the mammalian hearts that is generally gated by RyR2 channel, and is activated by rise of intracellular calcium exceeding 10−7 M or by caffeine which sensitizes RyR2 to Ca2+ such that resting cytoplasmic concentrations (~10−7M) of Ca2+ triggers the release of Ca2+ from the SR [41, 149]. Caffeine-releasable Ca2+ stores of hiPSC-CMs were reported to be comparable to those of adult rabbit and mouse cardiomyocytes [129]. Our studies show that hiPSC-CMs have rapid and robust releases of Ca2+ from the SR on rapid application of caffeine that in turn also activate the sarcolemmal Na/Ca exchanger. The caffeine induced INCX current density was significantly larger in hiPSC-CMs than in rat neonatal cardiomyocytes (~2.5pA/pF vs. ~1.0pA/pF) [13]. Hwang et al also compared the caffeine induced Ca2+ release of hiPSC-CMs with those of adult ventricular myocytes, but found significantly lower INCX values of 0.6 pA/pF in hiPSC-CM as compared to 2.5 pA/pF in our hiPSC-CMs [129]. The discrepancy between the two reported values is not readily apparent, but it might be related to developmental stage of hiPSC-CMs used or concentrations of intracellular Ca2+ buffers introduced via the patch pipette in the two studies.
Fractional Ca2+ release (ratio of ICa to caffeine triggered Ca2+ release), appears to vary significantly among hiPSC-CMs, ranging from 0.3–0.6, depending in part on the age of cells in culture and the procedures used to differentiate them. The smaller values of fractional release in hiPSC-CMs as compared to adult cardiomyocytes (0.8–0.9) may reflect the development of subcellular couplons of SR and their proximity to calcium channels that gate them, rather than differences in the mode or mechanisms of their activation, as also reflected in the studies comparing calcium channel inactivation kinetics in 3–5 day versus the 6–8day hiPSC-CM cultures [142].
2.1d. Ca2+ sparks.
Ca2+ release from ryanodine receptor clustered in a dyadic junction has been shown to generate localized rise of intracellular Ca2+ called Ca2+ sparks. In rat ventricular myocytes, Ca2+ sparks represent the release of ~100,000 calcium ions. The morphology of Ca2+ sparks in the subsarcolemmal space of rat ventricular cells, imaged with high resolution TIRF microscopy, compares well to sparks recorded with confocal microscopy [150]. Using TIRF imaging with higher spatial resolution (0.1 μm), we found that the majority of spontaneously occurring sparks in hiPSC-CMs were of brief duration, had spatiotemporal properties analogous to those of adult rat cardiomyocytes, ranged between 40–60ms in duration, had occurrence frequency of 10–40 sparks/s in different cells, see fig 4 [4, 151]. In sharp contrast, TIRF-imaging of Ca2+ sparks in adult rat ventricular cells showed lower occurrence frequencies (7.1 sparks/cell/s) and shorter durations (~15ms) [152]. Confocal line scan recording of sparks in hiPSC-CMs from different groups show occurrence frequencies ranging from 0.007 sparks/μm2/s, [138], to ~4sparks/100μm/s [133]. Other groups, have identified stochastic and repetitively occurring spontaneous Ca2+ sparks at the same cellular sites with duration at half maximum amplitude of 30.9±0.6ms vs. 26.1±1.7ms in adult rat CMs [11]. Ca2+ sparks in hiPSC-CMs appear to be predominately triggered by activation of L-type Ca2+ channels, blocked by nifedipine and enhanced in frequency by elevation of extracellular Ca2+ or 50nM ryanodine [11].
Fig.4.
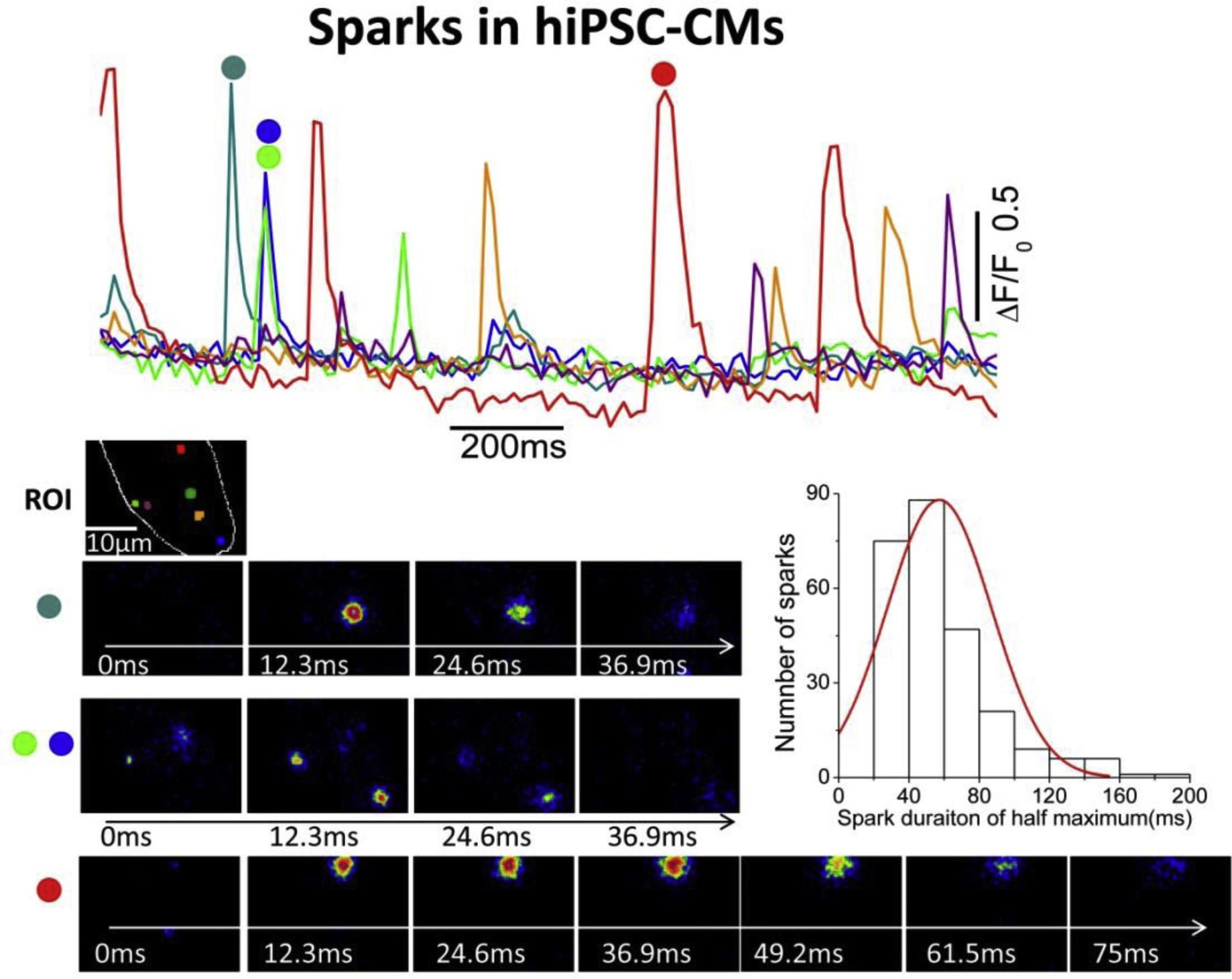
Ca+ Sparks in hiPSC-CMs recorded with TIRF microscopy. The color traces show the time course of normalized Ca2+-dependent fluorescence changes at the selected color-coded regions (ROI) in a single hiPSC-CM. The images show the example of evolution of sparks at different color-coded regions. The sparks were recorded at 12.3ms/frame, ~80Hz. The teal, green and blue sparks are developed and decayed much faster as compared to the red sparks. The histogram shows the distribution of spark durations of half maximum.
2.2. IP3-gated Ca2+ pools
IP3-gated Ca2+ signaling pathway is thought to have an important role in immature developing hearts, playing a critical role in nuclear signaling during growth and development [125, 153]. Although the primary Ca2+-signaling pathway in adult mammalian cardiomyocytes occurs through activation of RyR2-gated store, in some myocytes and at different stages of their development, or humoral regulation, there may be significant contribution from the IP3-gated calcium stores [125, 153]. IP3R-gated Ca2+ release is expressed both in endoplasmic reticulum and in the nuclear envelop [154]. While functional role of IP3R in beat-to-beat regulation of cardiomyocyte contractility remains clouded, an early study shows that IP3R mRNA levels were about 50-fold lower than those of RyR2 in adult cardiac myocytes [155]. Nevertheless, abundant levels of IP3R protein has been reported in embryonic, neonatal, and adult atrial myocytes [123, 125, 156, 157]. This has led to the suggestion that the spontaneous pacing activity of embryonic myocytes and stem cell-derived cardiomyocyte may depend on IP3-mediated Ca2+ signaling [158, 159]. In this respect, since IP3-gated Ca2+ signaling is reported to trigger both sub-sarcolemmal and perinuclear Ca2+ releases in developing cardiomyocytes [125], it is possible that the sub-sarcolemmal Ca2+ release would activate the Na+/Ca2+ exchanger causing the depolarization of the surface membrane and setting off pacing, while Ca2+ released at the nuclear envelop may serve to replenish the SR stores or have a transcriptional cellular role [160]. Cytoplasmic Immuno-staining studies in newborn mice also show that IP3Rs are mostly distributed around the nucleus consistent with the findings in hiPSC-CMs and their possible involvement in modulation of Ca2+-signaling [137, 161]. Consistent with this idea, IP3R antagonist 2-APB or phospholipase C inhibitor U73122 have been reported to slow significantly the kinetics and decrease the amplitude of spontaneously activated Ca2+ transients in hiPSC-CMs [136]. We have failed to confirm these findings in hiPSC-CMs using U73122 at 5μM concentrations, which only slightly affected the beating frequency of hiPSC-CM, suggesting that IP3R Ca2+ signaling pathway does not play a major role in generation of Ca2+ transients and regulation of pacing. In sharp contrast, our studies show that interactions between Ca2+ release from the SR and mitochondria contribute significantly to the mechanism of spontaneous pacing in hiPSC-CMs and rat neonatal cardiomyocytes [13].
2.3. Mitochondrial Ca2+ signaling
The possible interaction between mitochondrial and SR Ca2+ signaling in cardiac myocytes has been a topic of great interest recently. We have examined the contribution of mitochondrial Ca2+ signaling to spontaneous beating activity of hiPSC-CM using genetically encoded probes targeted to mitochondrial subunit VIII of cytochrome C. These studies showed both release and uptake of Ca2+ by different mitochondrial populations in hiPSC-CM and rat neonatal cardiomyocytes either during spontaneous beating or on rapid application of caffeine [13]. Generally, perinuclear mitochondrial population released Ca2+, though not always, while the peripheral mitochondria took-up Ca2+. FCCP, a mitochondrial uncoupler, at 50nM abolished the spontaneous beating of hiPSC-CM and prolonged the relaxation time of caffeine-induced Ca2+ transients from the SR, suggesting a direct role for mitochondria in the cycling of cytosolic Ca2+ uptake and regulation of pacing [13, 162]. These findings support the idea that in developing myocytes (rat neonatal and hiPSC-CMs) Ca2+ released from mitochondria may contribute to generation and regulation of spontaneous pacing activity. Consist with our finding, another study reports that spontaneously triggered cytoplasmic Ca2+ transients were abolished by 300nM FCCP and 1 μM oligomycin and mitochondrial NCX blocker, CGP-37157, suggesting that mitochondrial Ca2+ flux contributes to the spontaneous beating in hiPSC-derived ventricular-like CMs [163]. Irrespective of possible role of mitochondria in spontaneous pacing of hiPSC-CMs, the targeted mitochondrial Ca2+ probes when used in combination with cytosolic Ca2+ dyes, suggests that mitochondria may play a critical role in uptake of Ca2+ under cellular Ca2+ overload conditions caused either by caffeine-triggered Ca2+ release or sodium withdrawal mediated Ca2+ influx on NCX1. Fig.5 shows simultaneous recording of rise and fall of Ca2+ in a mitochondrion and in the cytosol of a patch-clamped hiPSC-CM. Note that rapid puffs of caffeine or withdrawal of Na+ activate the exchanger current in response to rapid rise and fall of global cytosolic Ca2+ followed by rise of mitochondrial Ca2+ as recorded by GCamP3 probe targeted to mitochondrial matrix. The mitochondrial Ca2+ uptake, though activating with a delay and slower kinetics, outlasts the rise and fall of cytosolic Ca2+ back to baseline levels. Given the differences in Ca2+ uptake functions of mitochondria based on their proximity to Ca2+ release dyadic structures or plasma membrane Ca2+ transporters, it is likely that there will be wide distribution in activation kinetics of Ca2+ uptake or its release in different populations of mitochondria that cannot be easily detected by imaging techniques that fail to differentiate their ultrastructural cellular locations. Thus, the detailed role of mitochondria in cardiac Ca2+ signaling must await the availability of higher resolution imaging technics that use targeted probes to various proteins of Ca2+ signaling pathway.
Fig.5.
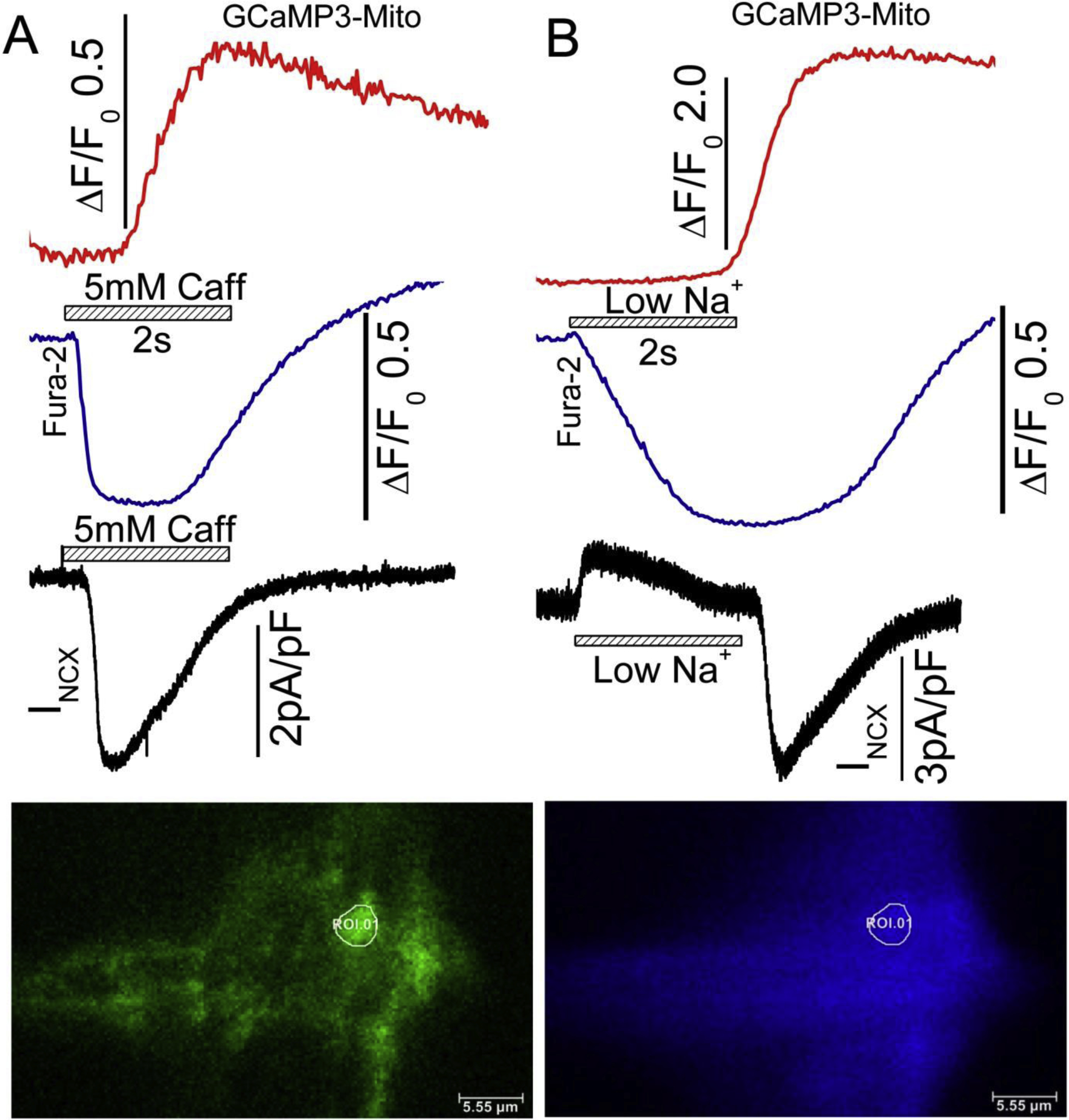
Simultaneously recorded TIRF images of mitochondrial and cytosolic Ca2+ signals in a single patch-clamped hiPSC-CM, in response to Na+ withdrawal (Na+ replaced with TEA) and 5mM caffeine applications. hiPSC-CMs were infected with GCaMP3-mito probe for 72 hours and dialyzed with 0.1mM Fura-2 salt through patch pipet during the experiment. Withdrawal of Na+ activates an outward INCX and its replacement generates a large slowly decaying inward current, that accompanies the rise and fall of cytosolic calcium. The mitochondrial Ca2+ signals recorded from a single mitochondrion (white circle ROI.01 show significant delays when compared to the activation of INCX or rise of cytosolic Ca2+. The blue traces represent cytosolic Fura-2 Ca2+ signal from the same cellular locations. Note that the rise of mitochondrial Ca2+ lags significantly behind the rise in mitochondrial Ca2+. The delays in mitochondrial Ca2+ uptake is shorter on release of SR Ca2+ compared to withdrawal of Na+. The black traces represent the INCX currents activated by Na+ withdraw and caffeine application. GCaMP3-mito probe was excited at 488nm, where increasing fluorescence signal indicates increases in mitochondrial Ca2+. Fura2 was emission signals measured at 405 nm (decreasing fluorescence indicates increase in cytosolic Ca2+). Temperature 35 degrees C.
2.4. Maturity of Ca2+ signaling in hiPSC-CMs
2.4.a. Spontaneous beating and intracellular rhythmic Calcium Oscillations
The spontaneously pacing properties of hiPSC-CMs is similar to that observed in rat neonatal cardiomyocytes and is considered to reflect the immature state of the hiPSC-CMs. Supporting this idea spontaneous rhythmic beating is absent or rare in freshly isolated adult ventricular myocytes. Not generally considered, however, are the findings that adult ventricular myocytes when maintained in culture conditions for a week or longer, lose their brick-like shapes, become flat, develop protruding cellular processes, generate repetitive calcium oscillations, and beat rhythmically. It is therefore reasonable to ask whether rhythmic beating is a consequence of myocyte shape changes or whether it represents regression of myocyte to an immature state? We propose the former possibility because Ca2+ signaling, as discussed in previous sections of this review, does not show significant differences in its biophysical or pharmacological parameters from those of adult cardiomyocytes to suggest possible regression of myocytes into an immature state. It also remains unrecognized that the spontaneous rhythmic beating, in rat neonatal cardiomyocytes and hiPSC-CMs, is initiated by regular and rhythmic rises and falls of cytosolic Ca2+ that persists even when the myocyte membrane potentials are voltage-clamped at a constant holding potentials of-50 or −70mVs, and that the rhythmic Ca2+ oscillations are insensitive to applications of nifedipine (1.0uM) or Cd2+(1.0mM), that prevent activation of any plasma-membrane ionic channels [13]. Since these spontaneous Ca2+ oscillations in hiPSC-CMs or neonatal rat cardiomyocytes were blocked by very low concentrations (50nM) of mitochondrial uncoupler, FCCP [13], we considered the possibility that they may originate directly from mitochondria or indirectly through mitochondrial modulation of SR stores [13]. What indeed triggers this rhythmic Ca2+ oscillation remains somewhat unclear. Perhaps the mechanisms responsible for cyclic generation ATP initiate directly or indirectly the release of Ca2+ from the mitochondria. Considering the large surface area of hiPSC-CMs compared to their small cell volumes, and their robust Ca2+ release mechanism, it is likely that the Na/Ca exchanger is effectively activated to depolarize the membrane to trigger spontaneous trains of action potentials. The robust expression of Na/Ca exchanger in hiPSC-CMs, and the paucity of t-tubules in them would further enhance the effectiveness of released calcium to depolarize the membrane and initiate spontaneous rhythmic beating. We propose therefore that spontaneous beating of hiPSC-CMs or neonatal rat cardiomyocytes are caused by changes in myocyte shape that creates large surface areas with smaller cellular volumes, allowing the robust cytosolic rise of Ca2+ to depolarize the cell and set off rhythmic spontaneous beating, rather than a genetic consequence of regression to an immature cellular state. It is intriguing to consider whether a similar mechanism may also be active in cardiac pacemaker cells of the mammalian SA-node, where the myocytes are small with surface to volume ratios exceeding unity and have robust Ca2+ releases [162].
2.4.b. Cellular maturity and ASIC family of Channels
HiPSC-CMs unexpectedly express acid sensitive ion channels (ASIC), which are absent in adult rat ventricular or neonatal rat cardiomyocytes. ASIC currents in hiPS-CMs show the same biophysical and pharmacological properties as those reported for ASIC currents in neuronal tissues [164, 165]. Significant expressions of proton-gated ASIC currents, ASIC1 mRNA, and ASIC1 protein was reported in 15–60 days old hiPSC-CMs, but only small or undetectable levels in hiPSC-CM cultures older than 100-days post differentiation, suggesting that younger myocytes may have remnants of neuronal signaling pathways [166]. The presence of ASIC family of proteins in hiPSC-CM lines may be a consequence of ancestral dermal-fibroblasts origins of these cells, but only low or undetectable levels of ASIC mRNA or proton-gated currents was found in dermal fibroblast [166]. Measurements of ASIC currents in cardiomyocytes derived from hematopoietic cells may be helpful in probing this possibility, but remain to be carried out.
2.4.c. Hormonal and metabolic procedures to promote maturity
To promote maturation of hiPSC-derived cardiomyocytes, multiple approaches have been attempted that include long term maintenance of cells in culture, hormone treatments, 3D culture conditions, mechanical stretching, electrical stimulation, and addition of fatty acids to the culture media [133, 167–170]. It appears that maintaining hiPSC-CMs in culture for prolonged periods (80–120days) can promote some degree of maturation in cellular ultrastructure and Ca2+ handling properties, such as enhanced rate of Ca2+ release and uptake, but with no significant effects on the amplitude of Ca2+ transients [131]. In another study, maturation in culture was accompanied by tight packing of myofibrils forming parallel arrays of sarcomeres, accompanied by the appearance of mature Z-, A-, H-, and I-bands, but not M-bands in 180-days hiPSC-CMs cultures, where M-bands became detectable in 360 day [169]. In sharp contrast, Hwang et al report that although 15 day hiPSC-CMs exhibit lower twitch tensions, Ca2+ transients and SR Ca2+ stores compared to 21 day old hiPSC-CMs, the Ca2+ handling properties did not continue to mature further in 21 to 30 day myocytes [129].
Tri-iodo-l-thyronine (T3) is reported to increase the hiPSC-CM myocyte size, sarcomere length, mitochondrial respiratory capacity, where the enhancement of contractile force is accompanied by increases in rates of calcium release and reuptake and SR-ATPase expression [168]. Similarly, fatty acid-treatment of hiPSC-CMs appears to also improve contractile force and kinetics of calcium transients as well as action potential upstroke velocity [167]. Combined use of thyroid hormones and dexamethasone appear also to induce t-tubular development, enhance the CICR, and EC-coupling of hiPSC-CMs grown on matrigel [133]. A combination of biochemical factors, thyroid hormone, dexamethasone, and insulin-like growth factor-1 (TDI) are also reported to enhance hiPSC-CMs maturation in 3D cardiac micro-tissues. In such cultures, the levels of t-tubule-associated genes junctophilin2 and RyR2, increased significantly when cardiomyocytes were treated with TDI. Similarly, the mRNA and protein levels of sarcoplasmic reticular Ca2+ ATPase and the Na/Ca exchanger were also significantly increased in the treated myocytes [171]. Thus, it appears that manipulating hiPSC-CM culture environment can lead to more robust EC-coupling parameters, but it is unclear whether these procedures alter the fundamental or qualitative nature of gating of Ca2+ release by ICa, the sensitivity of RyR2 to calcium, or pharmacology of EC-coupling. Although great emphasis has been placed on development of t-tubular network as an indicator of cardiomyocyte maturity, it must be noted that the mammalian atrial myocytes, with their paucity of t-tubules, have only minor Ca2+ signaling differences as compared to ventricular cells that have fully developed t-tubular network. It is likely that Ca2+ signaling and its regulation evolves and matures early in myocyte development, long before the myocyte attains its adult structural form. Nevertheless, maturation studies are likely to provide procedures to create metabolically and structurally more robust myocytes that could better survive the challenging micro-environments of infarcted tissue when transplanted into the diseased heart.
III. Conclusion and future trends and directions
In this review, we have attempted to compare Ca2+ signaling parameters of hiPSC-CMs with those of native mammalian cardiomyocytes. The comparative studies suggest that qualitatively hiPSC-CMs have similar Ca2+ signaling properties as those of adult cardiomyocytes, but quantitative differences do exist in the magnitude of calcium transients and their kinetics and the ability of ICa to release fully the RyR2 gated stores of SR. These differences may be, in part, related to subcellular geometry dyadic couplons and their proximity to sarcolemmal calcium channels and absence of t-tubular network in developing hiPSC-CMs, and not to a fundamental difference in CICR mechanism.
The flat and often spindle-shaped hiPSC-CMs with disorganized sarcomeric structures beat spontaneously unlike the adult cardiomyocytes, a property, we believe, arising from their large surface to volume ratios, where the robust calcium release effectively depolarizes the cells leading to spontaneous rhythmic pacing, quite similar to neonatal rat cardiomyocytes. The finding that NCX currents were two to 3 folds larger in hiPSC-CMs would further enhance their spontaneous beating rates. Despite these quantitative differences in the Ca2+ signaling parameters, the qualitative aspects of Ca2+ signaling are extremely similar in hiPSC-CMs and adult cardiomyocytes, suggesting that these cells may be used as reliable models for studies of human heart EC-coupling.
It is critical to recognize, that maturity of cardiac myocytes is a multi-dimensional time- and metabolic-dependent process. It is quite likely that some cellular mechanism mature early while others might be slower to reach maturation. In using hiPSC-CMs as a model of cardiac function or pathology, the structural and biophysical maturity of the signaling pathways to be studied must be taken into account. Clearly, cell morphology and development of sub-cellular architecture must be considered in structural or functional studies dependent on the structural development. Nevertheless, on the central issue, Cardiac Ca2+ signaling of hiPSC-CMs of this review, once the Ca2+ signaling processes of these cells are critically evaluated and compared with those of adult cardiomyocytes, and have been found to be similar, conclusions may be drawn as to the physiology or pathophysiology of the signaling pathway, irrespective of structural difference of the two human cell types. Although quantitative differences should be taken into consideration, it is not sufficient to dismiss hiPSC-CM calcium signaling and pathologies imposed on them by the experimenter, simply based on sweeping statements of cells immaturity. There is, for instance, little difference in Ca2+ signaling between atrium and ventricles even though atrium expresses low levels or lacks t-tubular network, as compared to ventricular cells. Structural immaturity does not necessarily indicate functional immaturity of some of the fundamental signaling pathways.
The field of Ca2+ signaling in adult cardiomyocytes still remains an active area of research as state-of-the-art technologies ranging from molecular, genetic, electrophysiological and imaging approaches are making it possible to explore in greater details the molecular mechanism regulating cardiac EC-coupling. Even though it is generally accepted that ICa-gated Ca2+ release is the dominant pathway for activation of contraction in the heart, other pathways that include transport of Ca2+ across the membrane or Ca2+ released from ER and mitochondria are being evaluated for their role in modulation of cardiac Ca2+ signaling under pathophysiological or developmental conditions. There still remain questions as to the microarchitecture of dyadic coupling, the couplons, with other ion transporting proteins, the mechanism of voltage-dependence of CICR gain, and the extent to which development or pathology alters these parameters. Transgenic animals either overexpressing or deleted of specific proteins of Ca2+ signaling, though have provided some clarity to EC-coupling regulation, have generated more questions as to, for instance why deletion of NCX1, or CSQ2 in adult heart fails to create significant changes in Ca2+ release mechanism or induce cardiac pathology?
With the emergence of hiPSC-CMs technology, human cells may become a primary source for basic investigation of human cardiac Ca2+ signaling and its regulation, but the issues related to maturity and developmental stages of the cells must be addressed and resolved. Since these cells lend themselves well to genetic engineering, the combined use of hiPSC lines and CRSPR/Cas9 gene editing techniques, that allows introduction of missense mutations causing cardiac pathology, are making it possible to study the pathophysiology of cardiac disease and its pharmacotherapy systematically in the lab before attempting it in patients afflicted with the disease. Perhaps more compelling is the possibility of applying gene editing technology to probe the sites that bind Ca2+ on RyR2 and CSQ2 or are phosphorylated by PKA and CaMKII. We feel that such molecular and genetic engineering techniques will open unlimited vistas in advancing cardiovascular research.
Since the human heart cells are of limited availability, generation of stem-cells derived cardiomyocytes provides a unique source, not only to probe the fundamental processes that underlie Ca2+ signaling in human heart, but also address pathophysiological alterations associated with cardiac disease in in vitro conditions. Possible use of hiPSC-CMs for cardiac regeneration and transplantation therapy also provides an intriguing possibility, but before using such cells clinically, it is paramount that their physiology is fully explored and understood. It is critical not to use cells with improper Ca2+-handling and electrophysiology that would introduce aberrant sources of Ca2+ release and Ca2+-mediated after-depolarizations and/or automaticity resulting in various forms of potentially lethal arrhythmias.
Highlights.
Functional analysis of Ca2+ signaling parameters in human pluripotent stem cell derived cardiomyocytes as compared to adult mammalian cardiomyocytes
Discussion of maturity level of hiPSC-CMs and whether hiPSC-CMs can serve as reliable models for adult human cardiac myocytes
Evaluation of contributions of ICa-gated Ca2+ release, Na/Ca exchanger, and mitochondria to hiPSC-CM Ca2+ signaling
Calcium signaling and pathophysiological consequences of genetic deletion or overexpression of proteins of Ca2+ signaling pathway in mice
Qualitatively, hiPSC-CMs serve as a good model of adult mammalian cardiac Ca2+ signaling despite the quantitative differences between the two cell types
Acknowledgement
We thank Dr. Naohiro Yamaguchi for critical easing of the manuscript. The studies were supported by National Institutes of Health Grants 1R56HL147054-01, 2019.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interests
No conflicts of interest, financial or otherwise, are declared by the authors.
- References
- [1].Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S, Induction of pluripotent stem cells from adult human fibroblasts by defined factors, Cell, 131 (2007) 861–872. [DOI] [PubMed] [Google Scholar]
- [2].Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, Thomson JA, Kamp TJ, Functional cardiomyocytes derived from human induced pluripotent stem cells, Circulation research, 104 (2009) e30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hoekstra M, Mummery CL, Wilde AA, Bezzina CR, Verkerk AO, Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias, Front Physiol, 3 (2012) 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhang XH, Haviland S, Wei H, Saric T, Fatima A, Hescheler J, Cleemann L, Morad M, Ca2+ signaling in human induced pluripotent stem cell-derived cardiomyocytes (iPS-CM) from normal and catecholaminergic polymorphic ventricular tachycardia (CPVT)-afflicted subjects, Cell calcium, 54 (2013) 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, Seyfarth M, Sinnecker D, Schomig A, Laugwitz KL, Patient-specific induced pluripotent stem-cell models for long-QT syndrome, N Engl J Med, 363 (2010) 1397–1409. [DOI] [PubMed] [Google Scholar]
- [6].Fatima A, Xu G, Shao K, Papadopoulos S, Lehmann M, Arnaiz-Cot JJ, Rosa AO, Nguemo F, Matzkies M, Dittmann S, Stone SL, Linke M, Zechner U, Beyer V, Hennies HC, Rosenkranz S, Klauke B, Parwani AS, Haverkamp W, Pfitzer G, Farr M, Cleemann L, Morad M, Milting H, Hescheler J, Saric T, In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells, Cell Physiol Biochem, 28 (2011) 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gunaseeli I, Doss MX, Antzelevitch C, Hescheler J, Sachinidis A, Induced pluripotent stem cells as a model for accelerated patient- and disease-specific drug discovery, Curr Med Chem, 17 (2010) 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Maury Y, Gauthier M, Peschanski M, Martinat C, Human pluripotent stem cells for disease modelling and drug screening, Bioessays, 34 (2012) 61–71. [DOI] [PubMed] [Google Scholar]
- [9].Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, Kolaja KL, Swanson BJ, January CT, High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents, American journal of physiology. Heart and circulatory physiology, 301 (2011) H2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lee YK, Ng KM, Lai WH, Chan YC, Lau YM, Lian Q, Tse HF, Siu CW, Calcium homeostasis in human induced pluripotent stem cell-derived cardiomyocytes, Stem Cell Rev Rep, 7 (2011) 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang GQ, Wei H, Lu J, Wong P, Shim W, Identification and characterization of calcium sparks in cardiomyocytes derived from human induced pluripotent stem cells, PloS one, 8 (2013) e55266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gherghiceanu M, Barad L, Novak A, Reiter I, Itskovitz-Eldor J, Binah O, Popescu LM, Cardiomyocytes derived from human embryonic and induced pluripotent stem cells: comparative ultrastructure, Journal of cellular and molecular medicine, 15 (2011) 2539–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang XH, Wei H, Saric T, Hescheler J, Cleemann L, Morad M, Regionally diverse mitochondrial calcium signaling regulates spontaneous pacing in developing cardiomyocytes, Cell calcium, 57 (2015) 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Morad M, Trautwein W, The effect of the duration of the action potential on contraction in the mammalian heart muscle, Pflugers Archiv fur die gesamte Physiologie des Menschen und der Tiere, 299 (1968) 66–82. [DOI] [PubMed] [Google Scholar]
- [15].Morad M, Orkand RK, Excitation-concentration coupling in frog ventricle: evidence from voltage clamp studies, The Journal of physiology, 219 (1971) 167–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Morad M, Goldman YE, Trentham DR, Rapid photochemical inactivation of Ca2+-antagonists shows that Ca2+ entry directly activates contraction in frog heart, Nature, 304 (1983) 635–638. [DOI] [PubMed] [Google Scholar]
- [17].Beeler GW Jr., Reuter H, The relation between membrane potential, membrane currents and activation of contraction in ventricular myocardial fibres, The Journal of physiology, 207 (1970) 211–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Morad M, Goldman YE, Excitation-contraciton coupling in heart muscle: membrane control of development of tension, Prog Biophysics Mol Biol, 27(1973)259–313. [Google Scholar]
- [19].Schneider MF, Chandler WK, Voltage dependent charge movement of skeletal muscle: a possible step in excitation-contraction coupling, Nature, 242 (1973) 244–246. [DOI] [PubMed] [Google Scholar]
- [20].Fabiato A, Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum, The American journal of physiology, 245 (1983) C1–14. [DOI] [PubMed] [Google Scholar]
- [21].Fabiato A, Fabiato F, Calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cells from adult human, dog, cat, rabbit, rat, and frog hearts and from fetal and new-born rat ventricles, Ann N Y Acad Sci, 307 (1978) 491–522. [DOI] [PubMed] [Google Scholar]
- [22].Fabiato A, Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell, The Journal of general physiology, 85 (1985) 247–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nabauer M, Callewaert G, Cleemann L, Morad M, Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes, Science, 244 (1989) 800–803. [DOI] [PubMed] [Google Scholar]
- [24].Cleemann L, Morad M, Role of Ca2+ channel in cardiac excitation-contraction coupling in the rat: evidence from Ca2+ transients and contraction, The Journal of physiology, 432 (1991) 283–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Beuckelmann DJ, Wier WG, Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells, The Journal of physiology, 405 (1988) 233–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Barcenas-Ruiz L, Wier WG, Voltage dependence of intracellular [Ca2+]i transients in guinea pig ventricular myocytes, Circulation research, 61 (1987) 148–154. [DOI] [PubMed] [Google Scholar]
- [27].Adachi-Akahane S, Cleemann L, Morad M, BAY K 8644 modifies Ca2+ cross signaling between DHP and ryanodine receptors in rat ventricular myocytes, The American journal of physiology, 276 (1999) H1178–1189. [DOI] [PubMed] [Google Scholar]
- [28].Morad M, Chau M, Learning about cardiac calcium signaling from genetic engineering, Ann N Y Acad Sci, 1015 (2004) 1–15. [DOI] [PubMed] [Google Scholar]
- [29].Altamirano J, Bers DM, Voltage Dependence of Cardiac Excitation-Contraction Coupling: Unitary Ca2+ Current Amplitude and Open Channel Probability, Circulation Research,101(2007), 590–597. [DOI] [PubMed] [Google Scholar]
- [30].Xu L, Meissner G, Vang J, Abernethy D, Soldatov N, Morad M, The Carboxyl-terminal tail of α1C L-type Ca2+ channel directly modulates ryanodine receptor incorporated in lipid bilayer. FASEB Journal, 13(1999) A1033. [Google Scholar]
- [31].Kurokawa J, Soldatov N, Adachi-Akahane S, Cleemann L, Morad M, Regulation of Ca2+ Sparks by Carboxy-Terminal Tail of Cardiac Ca2+ Channel, Biophysical Jorunal, 78(2000) 149A. [Google Scholar]
- [32].Woo SH, Soldatov NM, Morad M, Modulation of Ca2+ signalling in rat atrial myocytes: possible role of the alpha1C carboxyl terminal, The Journal of physiology, 552 (2003) 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bers DM, Cardiac excitation-contraction coupling, Nature, 415 (2002) 198–205. [DOI] [PubMed] [Google Scholar]
- [34].Fleischer S, Inui M, Biochemistry and biophysics of excitation-contraction coupling, Annu Rev Biophys Biophys Chem, 18 (1989) 333–364. [DOI] [PubMed] [Google Scholar]
- [35].Kirchberber MA, Tada M, Katz AM, Phospholamban: a regulatory protein of the cardiac sarcoplasmic reticulum, Recent Adv Stud Cardiac Struct Metab, 5 (1975) 103–115. [PubMed] [Google Scholar]
- [36].Verboomen H, Wuytack F, De Smedt H, Himpens B, Casteels R, Functional difference between SERCA2a and SERCA2b Ca2+ pumps and their modulation by phospholamban, Biochem J, 286 (Pt 2) (1992) 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kimura Y, Kurzydlowski K, Tada M, MacLennan DH, Phospholamban regulates the Ca2+-ATPase through intramembrane interactions, The Journal of biological chemistry, 271 (1996) 21726–21731. [DOI] [PubMed] [Google Scholar]
- [38].Sham JS, Jones LR, Morad M, Phospholamban mediates the beta-adrenergic-enhanced Ca2+ uptake in mammalian ventricular myocytes, The American journal of physiology, 261 (1991) H1344–1349. [DOI] [PubMed] [Google Scholar]
- [39].Karczewski P, Kuschel M, Baltas LG, Bartel S, Krause EG, Site-specific phosphorylation of a phospholamban peptide by cyclic nucleotide- and Ca2+/calmodulin-dependent protein kinases of cardiac sarcoplasmic reticulum, Basic research in cardiology, 92 Suppl 1 (1997) 37–43. [DOI] [PubMed] [Google Scholar]
- [40].Nabauer M, Morad M, Ca2(+)-induced Ca2+ release as examined by photolysis of caged Ca2+ in single ventricular myocytes, The American journal of physiology, 258 (1990) C189–193. [DOI] [PubMed] [Google Scholar]
- [41].Meissner G, Rios E, Tripathy A, Pasek DA, Regulation of skeletal muscle Ca2+ release channel (ryanodine receptor) by Ca2+ and monovalent cations and anions, The Journal of biological chemistry, 272 (1997) 1628–1638. [DOI] [PubMed] [Google Scholar]
- [42].Du GG, MacLennan DH, Ca(2+) inactivation sites are located in the COOH-terminal quarter of recombinant rabbit skeletal muscle Ca(2+) release channels (ryanodine receptors), The Journal of biological chemistry, 274 (1999) 26120–26126. [DOI] [PubMed] [Google Scholar]
- [43].Meissner G, Henderson JS, Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin, The Journal of biological chemistry, 262 (1987) 3065–3073. [PubMed] [Google Scholar]
- [44].Balshaw DM, Xu L, Yamaguchi N, Pasek DA, Meissner G, Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor), The Journal of biological chemistry, 276 (2001) 20144–20153. [DOI] [PubMed] [Google Scholar]
- [45].Xu L, Meissner G, Mechanism of calmodulin inhibition of cardiac sarcoplasmic reticulum Ca2+ release channel (ryanodine receptor), Biophysical journal, 86 (2004) 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gyorke I, Hester N, Jones LR, Gyorke S, The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium, Biophysical journal, 86 (2004) 2121–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sham JS, Song LS, Chen Y, Deng LH, Stern MD, Lakatta EG, Cheng H, Termination of Ca2+ release by a local inactivation of ryanodine receptors in cardiac myocytes, Proceedings of the National Academy of Sciences of the United States of America, 95 (1998) 15096–15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cleemann L, Wang W, Morad M, Two-dimensional confocal images of organization, density, and gating of focal Ca2+ release sites in rat cardiac myocytes, Proceedings of the National Academy of Sciences of the United States of America, 95 (1998) 10984–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Adachi-Akahane S, Cleemann L, Morad M, Cross-signaling between L-type Ca2+ channels and ryanodine receptors in rat ventricular myocytes, The Journal of general physiology, 108 (1996) 435–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sham JS, Cleemann L, Morad M, Functional coupling of Ca2+ channels and ryanodine receptors in cardiac myocytes, Proceedings of the National Academy of Sciences of the United States of America, 92 (1995) 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cheng H, Lederer WJ, Cannell MB, Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle, Science, 262 (1993) 740–744. [DOI] [PubMed] [Google Scholar]
- [52].Meissner G, The structural basis of ryanodine receptor ion channel function, The Journal of general physiology, 149 (2017) 1065–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen SR, MacLennan DH, Identification of calmodulin-, Ca(2+)-, and ruthenium red-binding domains in the Ca2+ release channel (ryanodine receptor) of rabbit skeletal muscle sarcoplasmic reticulum, The Journal of biological chemistry, 269 (1994) 22698–22704. [PubMed] [Google Scholar]
- [54].Chen SR, Zhang L, MacLennan DH, Characterization of a Ca2+ binding and regulatory site in the Ca2+ release channel (ryanodine receptor) of rabbit skeletal muscle sarcoplasmic reticulum, The Journal of biological chemistry, 267 (1992) 23318–23326. [PubMed] [Google Scholar]
- [55].Chen SR, Zhang L, MacLennan DH, Antibodies as probes for Ca2+ activation sites in the Ca2+ release channel (ryanodine receptor) of rabbit skeletal muscle sarcoplasmic reticulum, The Journal of biological chemistry, 268 (1993) 13414–13421. [PubMed] [Google Scholar]
- [56].Chen SR, Ebisawa K, Li X, Zhang L, Molecular identification of the ryanodine receptor Ca2+ sensor, The Journal of biological chemistry, 273 (1998) 14675–14678. [DOI] [PubMed] [Google Scholar]
- [57].Li P, Chen SR, Molecular basis of Ca(2)+ activation of the mouse cardiac Ca(2)+ release channel (ryanodine receptor), The Journal of general physiology, 118 (2001) 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].des Georges A, Clarke OB, Zalk R, Yuan Q, Condon KJ, Grassucci RA, Hendrickson WA, Marks AR, Frank J, Structural Basis for Gating and Activation of RyR1, Cell, 167 (2016) 145–157 e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Murayama T, Ogawa H, Kurebayashi N, Ohno S, Horie M, Sakurai T, A tryptophan residue in the caffeine-binding site of the ryanodine receptor regulates Ca(2+) sensitivity, Communications biology, 1 (2018) 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang XH, Wei H, Yamaguchi N, Morad M, CRISPR/Cas9 engineered Q3925E-RyR2 mutation in human induced pluripotent stem cells impairs caffeine triggered Ca2+ release, Biophysical Jorunal, 116(2019) 237a. [Google Scholar]
- [61].Xia YL, Zhang XH, Yamaguchi N, Morad M, Probing the RyR2 Ca2+ and caffeine binding sites by mutagenesis in human stem-cell derived cardiomyocytes by CRISPR/Cas9 gene editing, Biophysical journal, 116(2020) 255a [Google Scholar]
- [62].Du GG, Khanna VK, MacLennan DH, Mutation of divergent region 1 alters caffeine and Ca(2+) sensitivity of the skeletal muscle Ca(2+) release channel (ryanodine receptor), The Journal of biological chemistry, 275 (2000) 11778–11783. [DOI] [PubMed] [Google Scholar]
- [63].Xiong H, Feng X, Gao L, Xu L, Pasek DA, Seok JH, Meissner G, Identification of a two EF-hand Ca2+ binding domain in lobster skeletal muscle ryanodine receptor/Ca2+ release channel, Biochemistry, 37 (1998) 4804–4814. [DOI] [PubMed] [Google Scholar]
- [64].Gomez AC, Yamaguchi N, Two regions of the ryanodine receptor calcium channel are involved in Ca(2+)-dependent inactivation, Biochemistry, 53 (2014) 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zalk R, Clarke OB, des Georges A, Grassucci RA, Reiken S, Mancia F, Hendrickson WA, Frank J, Marks AR, Structure of a mammalian ryanodine receptor, Nature, 517 (2015) 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Callewaert G, Cleemann L, Morad M, Caffeine-induced Ca2+ release activates Ca2+ extrusion via Na+-Ca2+ exchanger in cardiac myocytes, The American journal of physiology, 257 (1989) C147–152. [DOI] [PubMed] [Google Scholar]
- [67].Reeves JP, Hale CC, The stoichiometry of the cardiac sodium-calcium exchange system, The Journal of biological chemistry, 259 (1984) 7733–7739. [PubMed] [Google Scholar]
- [68].Bers DM, Bridge JH, Relaxation of rabbit ventricular muscle by Na-Ca exchange and sarcoplasmic reticulum calcium pump. Ryanodine and voltage sensitivity, Circulation research, 65 (1989) 334–342. [DOI] [PubMed] [Google Scholar]
- [69].Leblanc N, Hume JR, Sodium current-induced release of calcium from cardiac sarcoplasmic reticulum, Science, 248 (1990) 372–376. [DOI] [PubMed] [Google Scholar]
- [70].Lederer WJ, Niggli E, Hadley RW, Sodium-calcium exchange in excitable cells: fuzzy space, Science, 248 (1990) 283. [DOI] [PubMed] [Google Scholar]
- [71].Sham JS, Cleemann L, Morad M, Gating of the cardiac Ca2+ release channel: the role of Na+ current and Na(+)-Ca2+ exchange, Science, 255 (1992) 850–853. [DOI] [PubMed] [Google Scholar]
- [72].Scriven DR, Asghari P, Schulson MN, Moore ED, Analysis of Cav1.2 and ryanodine receptor clusters in rat ventricular myocytes, Biophysical journal, 99 (2010) 3923–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Scriven DR, Moore ED, Ca(2)(+) channel and Na(+)/Ca(2)(+) exchange localization in cardiac myocytes, Journal of molecular and cellular cardiology, 58 (2013) 22–31. [DOI] [PubMed] [Google Scholar]
- [74].Maylie J, Nunzi MG, Morad M, Excitation-contraction coupling in ventricular muscle of dogfish (squalus acanthias). Bull. Mt. Desert Island Biol. Lab 19 (1979) 84–87. [Google Scholar]
- [75].Page SG, Niedergerke R, Structures of physiological interest in the frog heart ventricle, Journal of cell science, 11 (1972) 179–203. [DOI] [PubMed] [Google Scholar]
- [76].Fan J, Shuba YM, Morad M, Regulation of cardiac sodium-calcium exchanger by beta-adrenergic agonists, Proceedings of the National Academy of Sciences of the United States of America, 93 (1996) 5527–5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Nabauer M, Morad M, Modulation of contraction by intracellular Na+ via Na(+)-Ca2+ exchange in single shark (Squalus acanthias) ventricular myocytes, The Journal of physiology, 457 (1992) 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Vilsen B, Andersen JP, Deduced amino acid sequence and E1-E2 equilibrium of the sarcoplasmic reticulum Ca(2+)-ATPase of frog skeletal muscle. Comparison with the Ca(2+)-ATPase of rabbit fast twitch muscle, FEBS letters, 306 (1992) 213–218. [DOI] [PubMed] [Google Scholar]
- [79].Morad M, Sanders C, Weiss J, The Inotropic Actions of Adrenaline on Frog Ventricular Muscle: Relaxing Versus Potentiating Effects, The journal of physiology, 311(1981) 585–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Klitzner T, Morad M, Excitation-contraction coupling in frog ventricle. Possible Ca2+ transport mechanisms, Pflugers Arch, 398 (1983) 274–283. [DOI] [PubMed] [Google Scholar]
- [81].Shuba YM, Iwata T, Naidenov VG, Oz M, Sandberg K, Kraev A, Carafoli E, Morad M, A novel molecular determinant for cAMP-dependent regulation of the frog heart Na+-Ca2+ exchanger, The Journal of biological chemistry, 273 (1998) 18819–18825. [DOI] [PubMed] [Google Scholar]
- [82].He LP, Cleemann L, Soldatov NM, Morad M, Molecular determinants of cAMP-mediated regulation of the Na+-Ca2+ exchanger expressed in human cell lines, The Journal of physiology, 548 (2003) 677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Woo SH, Morad M, Bimodal regulation of Na(+)--Ca(2+) exchanger by beta-adrenergic signaling pathway in shark ventricular myocytes, Proceedings of the National Academy of Sciences of the United States of America, 98 (2001) 2023–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Janowski E, Day R, Kraev A, Roder JC, Cleemann L, Morad M, beta-adrenergic regulation of a novel isoform of NCX: sequence and expression of shark heart NCX in human kidney cells, American journal of physiology. Heart and circulatory physiology, 296 (2009) H1994–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Adachi-Akahane S, Lu L, Li Z, Frank JS, Philipson KD, Morad M, Calcium signaling in transgenic mice overexpressing cardiac Na(+)-Ca2+ exchanger, The Journal of general physiology, 109 (1997) 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Baumer AT, Flesch M, Kilter H, Philipson KD, Bohm M, Overexpression of the Na(+)-Ca2+ exchanger leads to enhanced inotropic responsiveness to Na(+)-channel agonist without sarcoplasmic reticulum protein changes in transgenic mice, Biochemical and biophysical research communications, 249 (1998) 786–790. [DOI] [PubMed] [Google Scholar]
- [87].Terracciano CM, Souza AI, Philipson KD, MacLeod KT, Na+-Ca2+ exchange and sarcoplasmic reticular Ca2+ regulation in ventricular myocytes from transgenic mice overexpressing the Na+-Ca2+ exchanger, The Journal of physiology, 512 (Pt 3) (1998) 651–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wang J, Chan TO, Zhang XQ, Gao E, Song J, Koch WJ, Feldman AM, Cheung JY, Induced overexpression of Na+/Ca2+ exchanger transgene: altered myocyte contractility, [Ca2+]i transients, SR Ca2+ contents, and action potential duration, American journal of physiology. Heart and circulatory physiology, 297 (2009) H590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Roos KP, Jordan MC, Fishbein MC, Ritter MR, Friedlander M, Chang HC, Rahgozar P, Han T, Garcia AJ, Maclellan WR, Ross RS, Philipson KD, Hypertrophy and heart failure in mice overexpressing the cardiac sodium-calcium exchanger, J Card Fail, 13 (2007) 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Reuter H, Henderson SA, Han T, Mottino GA, Frank JS, Ross RS, Goldhaber JI, Philipson KD, Cardiac excitation-contraction coupling in the absence of Na(+) - Ca2+ exchange, Cell calcium, 34 (2003) 19–26. [DOI] [PubMed] [Google Scholar]
- [91].Goldhaber JI, Henderson SA, Reuter H, Pott C, Philipson KD, Effects of Na+-Ca2+ exchange expression on excitation-contraction coupling in genetically modified mice, Ann N Y Acad Sci, 1047 (2005) 122–126. [DOI] [PubMed] [Google Scholar]
- [92].Muth JN, Bodi I, Lewis W, Varadi G, Schwartz A, A Ca(2+)-dependent transgenic model of cardiac hypertrophy: A role for protein kinase Calpha, Circulation, 103 (2001) 140–147. [DOI] [PubMed] [Google Scholar]
- [93].Song LS, Guia A, Muth JN, Rubio M, Wang SQ, Xiao RP, Josephson IR, Lakatta EG, Schwartz A, Cheng H, Ca(2+) signaling in cardiac myocytes overexpressing the alpha(1) subunit of L-type Ca(2+) channel, Circulation research, 90 (2002) 174–181. [DOI] [PubMed] [Google Scholar]
- [94].Muth JN, Yamaguchi H, Mikala G, Grupp IL, Lewis W, Cheng H, Song LS, Lakatta EG, Varadi G, Schwartz A, Cardiac-specific overexpression of the alpha(1) subunit of the L-type voltage-dependent Ca(2+) channel in transgenic mice. Loss of isoproterenol-induced contraction, The Journal of biological chemistry, 274 (1999) 21503–21506. [DOI] [PubMed] [Google Scholar]
- [95].Knollmann BC, Knollmann-Ritschel BE, Weissman NJ, Jones LR, Morad M, Remodelling of ionic currents in hypertrophied and failing hearts of transgenic mice overexpressing calsequestrin, The Journal of physiology, 525 Pt 2 (2000) 483–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Jones LR, Suzuki YJ, Wang W, Kobayashi YM, Ramesh V, Franzini-Armstrong C, Cleemann L, Morad M, Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin, J Clin Invest, 101 (1998) 1385–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K, Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia, J Clin Invest, 116 (2006) 2510–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Kornyeyev D, Petrosky AD, Zepeda B, Ferreiro M, Knollmann B, Escobar AL, Calsequestrin 2 deletion shortens the refractoriness of Ca(2)(+) release and reduces rate-dependent Ca(2)(+)-alternans in intact mouse hearts, Journal of molecular and cellular cardiology, 52 (2012) 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Kryshtal DO, Gryshchenko O, Gomez-Hurtado N, Knollmann BC, Impaired calcium-calmodulin-dependent inactivation of Cav1.2 contributes to loss of sarcoplasmic reticulum calcium release refractoriness in mice lacking calsequestrin 2, Journal of molecular and cellular cardiology, 82 (2015) 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Chopra N, Kannankeril PJ, Yang T, Hlaing T, Holinstat I, Ettensohn K, Pfeifer K, Akin B, Jones LR, Franzini-Armstrong C, Knollmann BC, Modest reductions of cardiac calsequestrin increase sarcoplasmic reticulum Ca2+ leak independent of luminal Ca2+ and trigger ventricular arrhythmias in mice, Circulation research, 101 (2007) 617–626. [DOI] [PubMed] [Google Scholar]
- [101].Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S, Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: Mechanism for hereditary arrhythmia, Proceedings of the National Academy of Sciences of the United States of America, 100 (2003) 11759–11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Masaki H, Sako H, Kadambi VJ, Sato Y, Kranias EG, Yatani A, Overexpression of phospholamban alters inactivation kinetics of L-type Ca2+ channel currents in mouse atrial myocytes, Journal of molecular and cellular cardiology, 30 (1998) 317–325. [DOI] [PubMed] [Google Scholar]
- [103].Kadambi VJ, Ponniah S, Harrer JM, Hoit BD, Dorn GW 2nd, Walsh RA, Kranias EG, Cardiac-specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice, J Clin Invest, 97 (1996) 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Hoit BD, Tramuta DA, Kadambi VJ, Dash R, Ball N, Kranias EG, Walsh RA, Influence of transgenic overexpression of phospholamban on postextrasystolic potentiation, Journal of molecular and cellular cardiology, 31 (1999) 2007–2015. [DOI] [PubMed] [Google Scholar]
- [105].Chu G, Luo W, Slack JP, Tilgmann C, Sweet WE, Spindler M, Saupe KW, Boivin GP, Moravec CS, Matlib MA, Grupp IL, Ingwall JS, Kranias EG, Compensatory mechanisms associated with the hyperdynamic function of phospholamban-deficient mouse hearts, Circulation research, 79 (1996) 1064–1076. [DOI] [PubMed] [Google Scholar]
- [106].Wolska BM, Stojanovic MO, Luo W, Kranias EG, Solaro RJ, Effect of ablation of phospholamban on dynamics of cardiac myocyte contraction and intracellular Ca2+, The American journal of physiology, 271 (1996) C391–397. [DOI] [PubMed] [Google Scholar]
- [107].Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, Kranias EG, Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation, Circulation research, 75 (1994) 401–409. [DOI] [PubMed] [Google Scholar]
- [108].Slack JP, Grupp IL, Dash R, Holder D, Schmidt A, Gerst MJ, Tamura T, Tilgmann C, James PF, Johnson R, Gerdes AM, Kranias EG, The enhanced contractility of the phospholamban-deficient mouse heart persists with aging, Journal of molecular and cellular cardiology, 33 (2001) 1031–1040. [DOI] [PubMed] [Google Scholar]
- [109].Kaneko M, Hashikami K, Yamamoto S, Matsumoto H, Nishimoto T, Phospholamban Ablation Using CRISPR/Cas9 System Improves Mortality in a Murine Heart Failure Model, PloS one, 11 (2016) e0168486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chen M, Xu D, Wu AZ, Kranias E, Lin SF, Chen PS, Chen Z, Phospholamban regulates nuclear Ca(2+) stores and inositol 1,4,5-trisphosphate mediated nuclear Ca(2+) cycling in cardiomyocytes, Journal of molecular and cellular cardiology, 123 (2018) 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Woo SH, Cleemann L, Morad M, Ca2+ current-gated focal and local Ca2+ release in rat atrial myocytes: evidence from rapid 2-D confocal imaging, The Journal of physiology, 543 (2002) 439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].McNutt NS, Fawcett DW, The ultrastructure of the cat myocardium. II. Atrial muscle, The Journal of cell biology, 42 (1969) 46–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Carl SL, Felix K, Caswell AH, Brandt NR, Ball WJ Jr., Vaghy PL, Meissner G, Ferguson DG, Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium, The Journal of cell biology, 129 (1995) 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Berlin JR, Spatiotemporal changes of Ca2+ during electrically evoked contractions in atrial and ventricular cells, The American journal of physiology, 269 (1995) H1165–1170. [DOI] [PubMed] [Google Scholar]
- [115].Huser J, Lipsius SL, Blatter LA, Calcium gradients during excitation-contraction coupling in cat atrial myocytes, The Journal of physiology, 494 (Pt 3) (1996) 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kockskamper J, Sheehan KA, Bare DJ, Lipsius SL, Mignery GA, Blatter LA, Activation and propagation of Ca(2+) release during excitation-contraction coupling in atrial myocytes, Biophysical journal, 81 (2001) 2590–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Woo SH, Cleemann L, Morad M, Spatiotemporal characteristics of junctional and nonjunctional focal Ca2+ release in rat atrial myocytes, Circulation research, 92 (2003) e1–11. [DOI] [PubMed] [Google Scholar]
- [118].Arora R, Aistrup GL, Supple S, Frank C, Singh J, Tai S, Zhao A, Chicos L, Marszalec W, Guo A, Song LS, Wasserstrom JA, Regional distribution of T-tubule density in left and right atria in dogs, Heart rhythm, 14 (2017) 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Luss I, Boknik P, Jones LR, Kirchhefer U, Knapp J, Linck B, Luss H, Meissner A, Muller FU, Schmitz W, Vahlensieck U, Neumann J, Expression of cardiac calcium regulatory proteins in atrium v ventricle in different species, Journal of molecular and cellular cardiology, 31 (1999) 1299–1314. [DOI] [PubMed] [Google Scholar]
- [120].Gadeberg HC, Bond RC, Kong CH, Chanoit GP, Ascione R, Cannell MB, James AF, Heterogeneity of T-Tubules in Pig Hearts, PloS one, 11 (2016) e0156862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Richards MA, Clarke JD, Saravanan P, Voigt N, Dobrev D, Eisner DA, Trafford AW, Dibb KM, Transverse tubules are a common feature in large mammalian atrial myocytes including human, American journal of physiology. Heart and circulatory physiology, 301 (2011) H1996–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Dibb KM, Clarke JD, Horn MA, Richards MA, Graham HK, Eisner DA, Trafford AW, Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure, Circulation. Heart failure, 2 (2009) 482–489. [DOI] [PubMed] [Google Scholar]
- [123].Kim JC, Son MJ, Subedi KP, Li Y, Ahn JR, Woo SH, Atrial local Ca2+ signaling and inositol 1,4,5-trisphosphate receptors, Progress in biophysics and molecular biology, 103 (2010) 59–70. [DOI] [PubMed] [Google Scholar]
- [124].Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W, Bootman MD, Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart, Curr Biol, 10 (2000) 939–942. [DOI] [PubMed] [Google Scholar]
- [125].Janowski E, Berrios M, Cleemann L, Morad M, Developmental aspects of cardiac Ca(2+) signaling: interplay between RyR- and IP(3)R-gated Ca(2+) stores, American journal of physiology. Heart and circulatory physiology, 298 (2010) H1939–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Bassani JW, Yuan W, Bers DM, Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes, The American journal of physiology, 268 (1995) C1313–1319. [DOI] [PubMed] [Google Scholar]
- [127].Wier WG, Egan TM, Lopez-Lopez JR, Balke CW, Local control of excitation-contraction coupling in rat heart cells, The Journal of physiology, 474 (1994) 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kim JJ, Yang L, Lin B, Zhu X, Sun B, Kaplan AD, Bett GC, Rasmusson RL, London B, Salama G, Mechanism of automaticity in cardiomyocytes derived from human induced pluripotent stem cells, Journal of molecular and cellular cardiology, 81 (2015) 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Hwang HS, Kryshtal DO, Feaster TK, Sanchez-Freire V, Zhang J, Kamp TJ, Hong CC, Wu JC, Knollmann BC, Comparable calcium handling of human iPSC-derived cardiomyocytes generated by multiple laboratories, Journal of molecular and cellular cardiology, 85 (2015) 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Rao C, Prodromakis T, Kolker L, Chaudhry UA, Trantidou T, Sridhar A, Weekes C, Camelliti P, Harding SE, Darzi A, Yacoub MH, Athanasiou T, Terracciano CM, The effect of microgrooved culture substrates on calcium cycling of cardiac myocytes derived from human induced pluripotent stem cells, Biomaterials, 34 (2013) 2399–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Lundy SD, Zhu WZ, Regnier M, Laflamme MA, Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells, Stem cells and development, 22 (2013) 1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Li S, Chen G, Li RA, Calcium signalling of human pluripotent stem cell-derived cardiomyocytes, The Journal of physiology, 591 (2013) 5279–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Parikh SS, Blackwell DJ, Gomez-Hurtado N, Frisk M, Wang L, Kim K, Dahl CP, Fiane A, Tonnessen T, Kryshtal DO, Louch WE, Knollmann BC, Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes, Circulation research, 121 (2017) 1323–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Kane C, Couch L, Terracciano CM, Excitation-contraction coupling of human induced pluripotent stem cell-derived cardiomyocytes, Front Cell Dev Biol, 3 (2015) 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Ivashchenko CY, Pipes GC, Lozinskaya IM, Lin Z, Xiaoping X, Needle S, Grygielko ET, Hu E, Toomey JR, Lepore JJ, Willette RN, Human-induced pluripotent stem cell-derived cardiomyocytes exhibit temporal changes in phenotype, American journal of physiology. Heart and circulatory physiology, 305 (2013) H913–922. [DOI] [PubMed] [Google Scholar]
- [136].Itzhaki I, Rapoport S, Huber I, Mizrahi I, Zwi-Dantsis L, Arbel G, Schiller J, Gepstein L, Calcium handling in human induced pluripotent stem cell derived cardiomyocytes, PloS one, 6 (2011) e18037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Germanguz I, Sedan O, Zeevi-Levin N, Shtrichman R, Barak E, Ziskind A, Eliyahu S, Meiry G, Amit M, Itskovitz-Eldor J, Binah O, Molecular characterization and functional properties of cardiomyocytes derived from human inducible pluripotent stem cells, Journal of cellular and molecular medicine, 15 (2011) 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Jung CB, Moretti A, Mederos y Schnitzler M, Iop L, Storch U, Bellin M, Dorn T, Ruppenthal S, Pfeiffer S, Goedel A, Dirschinger RJ, Seyfarth M, Lam JT, Sinnecker D, Gudermann T, Lipp P, Laugwitz KL, Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia, EMBO molecular medicine, 4 (2012) 180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Fong AH, Romero-Lopez M, Heylman CM, Keating M, Tran D, Sobrino A, Tran AQ, Pham HH, Fimbres C, Gershon PD, Botvinick EL, George SC, Hughes CC, Three-Dimensional Adult Cardiac Extracellular Matrix Promotes Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes, Tissue engineering. Part A, 22 (2016) 1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Uzun AU, Mannhardt I, Breckwoldt K, Horvath A, Johannsen SS, Hansen A, Eschenhagen T, Christ T, Ca(2+)-Currents in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Effects of Two Different Culture Conditions, Front Pharmacol, 7 (2016) 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Tanaka T, Tohyama S, Murata M, Nomura F, Kaneko T, Chen H, Hattori F, Egashira T, Seki T, Ohno Y, Koshimizu U, Yuasa S, Ogawa S, Yamanaka S, Yasuda K, Fukuda K, In vitro pharmacologic testing using human induced pluripotent stem cell-derived cardiomyocytes, Biochemical and biophysical research communications, 385 (2009) 497–502. [DOI] [PubMed] [Google Scholar]
- [142].Fernandez-Morales JC, Hua W, Yao Y, Morad M, Regulation of Ca(2+) signaling by acute hypoxia and acidosis in cardiomyocytes derived from human induced pluripotent stem cells, Cell calcium, 78 (2019) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Zhao Z, Lan H, El-Battrawy I, Li X, Buljubasic F, Sattler K, Yucel G, Lang S, Tiburcy M, Zimmermann WH, Cyganek L, Utikal J, Wieland T, Borggrefe M, Zhou XB, Akin I, Ion Channel Expression and Characterization in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes, Stem Cells Int, 2018 (2018) 6067096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Yokoo N, Baba S, Kaichi S, Niwa A, Mima T, Doi H, Yamanaka S, Nakahata T, Heike T, The effects of cardioactive drugs on cardiomyocytes derived from human induced pluripotent stem cells, Biochemical and biophysical research communications, 387 (2009) 482–488. [DOI] [PubMed] [Google Scholar]
- [145].Zwi L, Caspi O, Arbel G, Huber I, Gepstein A, Park IH, Gepstein L, Cardiomyocyte differentiation of human induced pluripotent stem cells, Circulation, 120 (2009) 1513–1523. [DOI] [PubMed] [Google Scholar]
- [146].Jung G, Fajardo G, Ribeiro AJ, Kooiker KB, Coronado M, Zhao M, Hu DQ, Reddy S, Kodo K, Sriram K, Insel PA, Wu JC, Pruitt BL, Bernstein D, Time-dependent evolution of functional vs. remodeling signaling in induced pluripotent stem cell-derived cardiomyocytes and induced maturation with biomechanical stimulation, FASEB journal: official publication of the Federation of American Societies for Experimental Biology, 30 (2016) 1464–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Pioner JM, Santini L, Palandri C, Martella D, Lupi F, Langione M, Querceto S, Grandinetti B, Balducci V, Benzoni P, Landi S, Barbuti A, Ferrarese Lupi F, Boarino L, Sartiani L, Tesi C, Mack DL, Regnier M, Cerbai E, Parmeggiani C, Poggesi C, Ferrantini C, Coppini R, Optical Investigation of Action Potential and Calcium Handling Maturation of hiPSC-Cardiomyocytes on Biomimetic Substrates, International journal of molecular sciences, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Chen G, Li S, Karakikes I, Ren L, Chow MZ, Chopra A, Keung W, Yan B, Chan CW, Costa KD, Kong CW, Hajjar RJ, Chen CS, Li RA, Phospholamban as a crucial determinant of the inotropic response of human pluripotent stem cell-derived ventricular cardiomyocytes and engineered 3-dimensional tissue constructs, Circulation. Arrhythmia and electrophysiology, 8 (2015) 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Pessah IN, Stambuk RA, Casida JE, Ca2+-activated ryanodine binding: mechanisms of sensitivity and intensity modulation by Mg2+, caffeine, and adenine nucleotides, Molecular pharmacology, 31 (1987) 232–238. [PubMed] [Google Scholar]
- [150].Cleemann L, DiMassa G, Morad M, Ca2+ sparks within 200 nm of the sarcolemma of rat ventricular cells: evidence from total internal reflection fluorescence microscopy, Advances in experimental medicine and biology, 430 (1997) 57–65. [DOI] [PubMed] [Google Scholar]
- [151].Wei H, Zhang XH, Clift C, Yamaguchi N, Morad M, CRISPR/Cas9 Gene editing of RyR2 in human stem cell-derived cardiomyocytes provides a novel approach in investigating dysfunctional Ca(2+) signaling, Cell calcium, 73 (2018) 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Pahlavan S, Morad M, Total internal reflectance fluorescence imaging of genetically engineered ryanodine receptor-targeted Ca(2+) probes in rat ventricular myocytes, Cell calcium, 66 (2017) 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Puceat M, Jaconi M, Ca2+ signalling in cardiogenesis, Cell calcium, 38 (2005) 383–389. [DOI] [PubMed] [Google Scholar]
- [154].Rizzuto R, Pozzan T, Microdomains of intracellular Ca2+: molecular determinants and functional consequences, Physiological reviews, 86 (2006) 369–408. [DOI] [PubMed] [Google Scholar]
- [155].Moschella MC, Marks AR, Inositol 1,4,5-trisphosphate receptor expression in cardiac myocytes, The Journal of cell biology, 120 (1993) 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Luo D, Yang D, Lan X, Li K, Li X, Chen J, Zhang Y, Xiao RP, Han Q, Cheng H, Nuclear Ca2+ sparks and waves mediated by inositol 1,4,5-trisphosphate receptors in neonatal rat cardiomyocytes, Cell calcium, 43 (2008) 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Sedan O, Dolnikov K, Zeevi-Levin N, Leibovich N, Amit M, Itskovitz-Eldor J, Binah O, 1,4,5-Inositol trisphosphate-operated intracellular Ca(2+) stores and angiotensin-II/endothelin-1 signaling pathway are functional in human embryonic stem cell-derived cardiomyocytes, Stem cells, 26 (2008) 3130–3138. [DOI] [PubMed] [Google Scholar]
- [158].Sasse P, Zhang J, Cleemann L, Morad M, Hescheler J, Fleischmann BK, Intracellular Ca2+ oscillations, a potential pacemaking mechanism in early embryonic heart cells, The Journal of general physiology, 130 (2007) 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Mery A, Aimond F, Menard C, Mikoshiba K, Michalak M, Puceat M, Initiation of embryonic cardiac pacemaker activity by inositol 1,4,5-trisphosphate-dependent calcium signaling, Molecular biology of the cell, 16 (2005) 2414–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Kapoor N, Maxwell JT, Mignery GA, Will D, Blatter LA, Banach K, Spatially defined InsP3-mediated signaling in embryonic stem cell-derived cardiomyocytes, PloS one, 9 (2014) e83715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Garcia KD, Shah T, Garcia J, Immunolocalization of type 2 inositol 1,4,5-trisphosphate receptors in cardiac myocytes from newborn mice, American journal of physiology. Cell physiology, 287 (2004) C1048–1057. [DOI] [PubMed] [Google Scholar]
- [162].Morad M, Zhang XH, Mechanisms of spontaneous pacing: sinoatrial nodal cells, neonatal cardiomyocytes, and human stem cell derived cardiomyocytes, Canadian journal of physiology and pharmacology, 95 (2017) 1100–1107. [DOI] [PubMed] [Google Scholar]
- [163].Xie A, Zhou A, Liu H, Shi G, Liu M, Boheler KR, Dudley SC Jr., Mitochondrial Ca2+ flux modulates spontaneous electrical activity in ventricular cardiomyocytes, PloS one, 13 (2018) e0200448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Konnerth A, Lux HD, Morad M, Proton-induced transformation of calcium channel in chick dorsal root ganglion cells, The Journal of physiology, 386 (1987) 603–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Osmakov DI, Andreev YA, Kozlov SA, Acid-sensing ion channels and their modulators, Biochemistry. Biokhimiia, 79 (2014) 1528–1545. [DOI] [PubMed] [Google Scholar]
- [166].Zhang XH, Saric T, Mehrjardi NZ, Hamad S, Morad M, Acid-Sensitive Ion Channels Are Expressed in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes, Stem cells and development, 28 (2019) 920–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Yang X, Rodriguez ML, Leonard A, Sun L, Fischer KA, Wang Y, Ritterhoff J, Zhao L, Kolwicz SC Jr., Pabon L, Reinecke H, Sniadecki NJ, Tian R, Ruohola-Baker H, Xu H, Murry CE, Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells, Stem Cell Reports, 13 (2019) 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Yang X, Rodriguez M, Pabon L, Fischer KA, Reinecke H, Regnier M, Sniadecki NJ, Ruohola-Baker H, Murry CE, Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells, Journal of molecular and cellular cardiology, 72 (2014) 296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Kamakura T, Makiyama T, Sasaki K, Yoshida Y, Wuriyanghai Y, Chen J, Hattori T, Ohno S, Kita T, Horie M, Yamanaka S, Kimura T, Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture, Circ J, 77 (2013) 1307–1314. [DOI] [PubMed] [Google Scholar]
- [170].Tu C, Chao BS, Wu JC, Strategies for Improving the Maturity of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes, Circulation research, 123 (2018) 512–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Huang CY, Peres Moreno Maia-Joca R, Ong CS, Wilson I, DiSilvestre D, Tomaselli GF, Reich DH, Enhancement of human iPSC-derived cardiomyocyte maturation by chemical conditioning in a 3D environment, Journal of molecular and cellular cardiology, 138 (2020) 1–11. [DOI] [PubMed] [Google Scholar]