

Abstract
Background.
Alcohol use disorders (AUDs) and cigarette smoking both increase risk for development of community-acquired pneumonia (CAP), likely through adverse effects on proximal airway mucociliary clearance and pathogen recognition. Smoking-related alterations on airways gene expression are well-described, but little is known about the impact of AUDs. We measured gene expression in human airways epithelial cells (AECs), hypothesizing that AUDs would be associated with novel differences in gene expression that could alter risk for CAP.
Methods.
Bronchoscopy with airway brushings were performed in participants with AUDs and controls to obtain AECs. An Alcohol Use Disorders Identification Test was used to define AUD. RNA was extracted from AECs, and mRNA expression data were collected on an Agilent micro-array. Differential expression analyses were performed on the filtered and normalized data with correction for multiple testing. Enrichment analyses were performed using clusterProfiler.
Results.
Expression data from 19 control and 18 AUD participants were evaluated. After adjustment for smoking, AUDs were associated with significant differential expression of 520 AEC genes, including genes for ribosomal proteins and genes involved in protein folding. Enrichment analyses indicated significant differential expression of 24 pathways in AUDs, including those implicated in protein targeting to membrane and viral gene expression. Smoking-associated AEC gene expression differences mirrored previous reports, but differed from those associated with AUDs.
Conclusions.
AUDs have a distinct impact on AEC gene expression that may influence proximal airway function independent of smoking. Alcohol-associated alterations may influence risk for CAP through modifying key mechanisms important in protecting proximal airways integrity.
Keywords: alcoholism, smoking, microarray, community-acquired pneumonia, bronchoscopy
Introduction
Alcohol use disorders (AUDs) adversely impact pulmonary immune function, and have been associated with increased risk and severity of both acute and chronic lung diseases. Moreover, AUDs often are present among individuals who smoke cigarettes, which can also impair pulmonary immune function. Importantly, both AUDs (de Roux et al., 2006, Grau et al., 2014) and cigarette smoking (Grau et al., 2014, Gupta et al., 2019, Braeken et al., 2017) are modifiable factors that independently increase risk and severity of community-acquired pneumonia (CAP), a major contributor to death and the most common cause of mortality in the US due to infectious disease (Ramirez et al., 2017, Kochanek, 2019). A classic smoking-related airways disease, chronic obstructive pulmonary disease (COPD), substantially increases risk for CAP (Braeken et al., 2017), as well as lung cancer (Mouronte-Roibas et al., 2016). Notably, AUDs have been associated with unfavorable COPD outcomes as indicated by higher 30-day readmission rates (Singh et al., 2016), suggesting a potential relationship between AUDs and COPD-associated morbidity. Collectively, these epidemiologic insights suggest that AUDs have a measurable impact on proximal airway immune function that may be particularly important to delineate in the context of smoking.
The proximal airways are among the first lung regions exposed to environmental insults, and are therefore critical in governing the lung’s immune response. Airway epithelial cells (AECs) line the proximal airways, where they maintain a conduit for air to enter and exit the distal airways and alveoli. Ciliated and secretory epithelial cells located within the mainstem bronchi support mucociliary clearance, thereby protecting the distal lung from inhaled environmental insults. Dysfunction of AECs is central to the pathogenesis of lung disorders including CAP and COPD (Crystal et al., 2008). Although the influence of cigarette smoke on airways gene expression has been examined in detail (Gower et al., 2011, Hackett et al., 2003, Spira et al., 2004), the impact of AUDs in this context is relatively unexplored. This is a critical oversight as individuals with AUDs frequently smoke, and are often tobacco dependent (Falk et al., 2006). Moreover, after drinking, alcohol contacts the bronchial epithelium via the bronchial circulation, where it may either be metabolized, or excreted unchanged via exhaled breath (Kaphalia and Calhoun, 2013, Manautou and Carlson, 1992), suggesting the potential for a potent biological effect on proximal airways.
We previously reported that alterations in AEC phenotype are associated with alcohol exposure in pre-clinical models and in human participants. Combined alcohol consumption and cigarette smoke exposure have been associated with decrements in normal ciliary beat frequency that may underlie impaired mucociliary clearance (Elliott et al., 2007, Wyatt et al., 2012). Further, aberrant Toll-like receptor (TLR) expression by AECs, and increased inflammatory mediator production, have been reported in the setting of chronic alcohol exposure (Bailey et al., 2015, Bailey et al., 2009). AUD-associated functional alterations in AECs would be expected to adversely affect pathogen recognition and response, thereby contributing to infections common in those with AUDs and smoking, including CAP.
Given the previously demonstrated alterations in AEC phenotype due to AUDs and smoking that could contribute to infectious susceptibility, we sought to use a more unbiased, systematic approach to comprehensively examine differential gene expression in AECs from human participants with AUDs who had no overt lung disease, adjusting for smoking history. We hypothesized that unique genes would be differentially expressed by human AECs in the context of AUDs that would differ from those differentially expressed in the setting of smoking. Further, we postulated that certain genes and enriched gene pathways would be relevant to the development of acute and chronic infectious lung diseases.
Materials and Methods
Screening, recruitment, and enrollment of participants.
Recruitment for the study was conducted between December 2012 and October 2016. Participants with likely AUDs were recruited from the Denver Comprehensive Addictions Rehabilitation and Evaluation Services center (Denver CARES), a non-medical inpatient alcohol and substance detoxification center affiliated with Denver Health and Hospital System in Denver, Colorado. Healthy participants (without AUDs) were recruited during this period via advertisements in the Denver metropolitan area. The Alcohol Use Disorders Identification Test (AUDIT) questionnaire (Reinert and Allen, 2002) was used to characterize alcohol use in all potential participants. The AUDIT has been validated in numerous clinical settings to identify harmful alcohol consumption and likely alcohol dependence, with scores of ≥ 8 in men, or ≥ 5 in women, indicating harmful alcohol use (Reinert and Allen, 2002). All participants provided written informed consent for participation in the study, which was approved by the Colorado Multiple Institutional Review Board.
Participants from Denver CARES with likely AUDs were eligible to participate in the study if the following criteria were present: (1) AUDIT score of ≥ 8 in men or ≥ 5 in women, (2) reported alcohol use within the previous seven days, and (3) age of participant ≥21 years. Since screening was conducted in a detoxification facility, potentially eligible participants were approached for consent after breathalyzer values indicated sobriety. Control participants were eligible to participate if AUDIT scores were < 8 in men or < 5 in women; other inclusion criteria were identical as for AUD participants.
After written informed consent, additional screening measures to establish overall health status were completed in all participants, including a full history and physical exam by a licensed physician; baseline laboratory testing (e.g., complete metabolic profile and complete blood count); urine screening for drugs of abuse; spirometry; and chest radiograph. Abnormalities detected in these additional tests resulted in the participants’ exclusion from participation. They included: (1) significant liver disease by history, serum total bilirubin > 2 mg/dL or albumin < 3.0 mg/dL; (2) history of gastroesophageal bleeding; (3) history of myocardial infarction, severe valvular dysfunction or ejection fraction of < 50%; (4) renal disease, defined as need for dialysis or serum creatinine > 2.0mg/dL; (5) history of lung disease, abnormal chest radiograph, or abnormal spirometry; (6) history of insulin-dependent or non-insulin requiring diabetes mellitus; (7) history of human immunodeficiency virus; (8) current pregnancy; (9) age > 55 (due to increased likelihood of comorbid conditions), and (10) concurrent use of cocaine, opiates, or methamphetamines by self-report or toxicology screen. Information regarding cigarette smoking was obtained from all participants, including current and past smoking habits, number of years smoked, and number of cigarettes smoked per day.
Biological sample collection and processing.
All participants had research specimens collected in the inpatient Clinical and Translational Research Center (CTRC) at the University of Colorado Hospital-Anschutz Medical Campus using previously established protocols (Burnham et al., 2012). All participants had fasted at least six hours prior to bronchoscopy. The average time elapsed since the last alcohol-containing beverage was three days.
Two trained physicians performed bronchoscopy to obtain AEC samples using an identical protocol. For purposes of this investigation, a total of 20 control and 18 AUD participants underwent bronchoscopy. For AEC collection, a sterile cytology brush (ConMed Disposable Bronchial Cytology Brush, Utica, NY) was extended through the bronchoscope into the right or left mainstem bronchus, and then 10 brush passes on the bronchial epithelium were performed in a circumferential fashion around the bronchus in an average of six unique areas. The brush was retracted into its sheath, and removed through the bronchoscope. After removal, the entire brush was immediately placed into a microcentrifuge tube containing 700 μL RNAlater (Qiagen #76104) or Buffer RLT from the RNeasy Plus Mini Kit (Qiagen #74134 with 10uL/mL of Beta-mercaptoethanol added) and stored at −80 °C.
The microcentrifuge tubes containing the brushes were thawed on ice. RNAlater tubes were vortexed vigorously to remove cells from the brushes, the brush was removed, and the cells were pelleted by centrifugation at 13,000 X g for 2 minutes. All of the RNAlater was aspirated off and the cell pellet was lysed with 700μl of Buffer RLT from the RNeasy Plus Mini Kit with 10uL/mL of Beta-mercaptoethanol. Buffer RLT tubes were vortexed vigorously after thawing and the cytology brush removed. At this point, all samples were homogenized using QIAshredder columns (Qiagen #79654, per manufacturer’s protocol). RNA and DNA were then extracted using an Allprep DNA/RNA Mini Kit (Qiagen #80204), per product protocol. The RNA integrity number (RIN) and concentration were examined on an Agilent 4150 TapeStation; samples were determined to have an average RIN of 7.67. RNA expression data were collected on the Agilent SurePrint G3 Gene Expression Microarray for humans, version 3 (Agilent Technologies #G4851C).
Real time PCR experiments were performed for specific genes determined to be differentially expressed in the microarray, using methods previously reported (Fini et al., 2017, Bailey et al., 2015). These experiments used remaining RNA previously extracted from AECs from the participants that had been stored at −80 °C. Since quantity of RNA was limited, a small number of genes were selected for examination. The gene expression assays (primer and probe sets) selected were, if possible, those that correlated to the probes on the Agilent microarray that were found to be differentially expressed, or were the Applied Biosystems TaqMan assays that were deemed by the manufacturer as the best coverage. Inventoried TaqMan Gene Expression Assays (Applied Biosystems) were used to measure mRNA expression levels for CCT7 (Hs00362446_m1), PTMA (Hs02339492_g1), PBX3 (Hs00608415_m1), and BLCAP (Hs00705669_s1). TATA box-binding protein (TBP, assay ID Hs00427620_m1) was used as the endogenous control. For all experiments, 50 ng of RNA was used per cDNA reaction (iScript cDNA Synthesis Kit, BioRad #1708890). For the qRT-PCR, 2 μL of cDNA was brought up to a volume of 9 μl with nuclease-free water, then combined with 1 μl TaqMan Gene Expression Assay and 10 μl TaqMan Gene Expression Master Mix (Applied Biosystems #4369514) in a 20 μl reaction. In order to maximize the limited cDNA, experiments were performed using a 384 well plate (Applied Biosystems #4309849, plate seal Applied Biosystems #4360954). Samples (20 μL reaction for each replicate) were run in triplicate per manufacturer protocol; hold at 50 °C for 2 min, 95 °C for 10 min, followed by PCR (95 °C 19 sec, 60 °C 1 min), for 60 cycles. An Applied Biosystems QuantStudio 6 Flex Real-Time PCR System with sequence-detection software was used for the experiments. The delta-delta-Ct or ddCt algorithm was used to analyze the relative changes in gene expression (Schmittgen and Livak, 2008). This requires the assignment of one or more housekeeping genes (in the case of this experiment, TBP), which are assumed to be uniformly and constantly expressed in all samples, as well as one or more reference samples.
Statistical analyses.
AUDIT scores from all participants were used to define the presence of an AUD. Additional covariates in the analyses included age in years, sex (male or female), and race (white or non-white). Smoking history was also included as a covariate, either as a categorical predictor, defined as current use of cigarettes (yes or no), or as a continuous predictor using pack-year smoking history. A separate model was created for each smoking definition. In the model adjusting for smoking as a categorical variable, we sought to determine the impact of active (current) smoking on gene expression. In the adjusting for smoking as a continuous variable using pack-year history, we could determine the impact of smoking intensity on gene expression. In this way, we could more explore the common association between AUDs and smoking (Grant et al., 2004) in a clinically relevant fashion. Further, given that the smoking history was quite variable, a third analysis was performed including only control (n=11) and AUD (n=11) participants with < 10 pack-years smoking history (Pletcher et al., 2012, Tan et al., 2009), adjusting for covariates including age, sex, and race.
For the microarray analyses, raw expression data were log2 transformed, and normalized using the quantile method in the normalizeBetweenArrays function from the limma package (3.38.3) (Ritchie et al., 2015). Additionally, per the recommendation of the limma vignette for Agilent array data, probes were retained if they were annotated with an Entrez Gene ID or Symbol and were expressed, where expression was defined as probes that were above background on at least 20 arrays (corresponding to the number of samples profiled in the control group). Following normalization, principal component analysis (PCA) plots were used to identify outliers to remove from the expression data, as well as expression box plots, and participant-level summary statistics. The data were re-normalized after outlier removal (Supplemental Figures 1-5).
Differential expression analysis was performed on the filtered and normalized data using the lmFit and eBayes functions in the limma package. Fold-changes and average expression across all samples were log2 transformed. Both raw p-values as well as p-values corrected for multiple testing were calculated. False Discovery Rate (FDR) control was performed using the Benjamini & Hochberg method to correct for multiple testing (Benjamini, 1995).
Enrichment analyses were performed using the clusterProfiler package (v 3.12.0). Only significantly up or down regulated genes (FDR < 0.05) were included in the gene list. All other genes tested were included as the background for the enrichment analyses. Gene sets were identified if they were significantly enriched in either the Gene Ontology (GO) groups 1) Biological Processes (BP), 2) Cellular Components (CC), or 3) Molecular Function (MF), as well as human (hsa) pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG). Enriched ontologies were corrected for multiple testing using the Benjamini-Hochberg method, and the False Discovery Rate was controlled at 0.05.
For qPCR data, differences in relative gene expression in the genes described above between the AUD and control groups were assessed using Wilcoxon/Kruskal-Wallis testing.
Results
Expression data processing.
Exploratory data analysis did not reveal significant clustering for the participants among technical variables such as date labeled, barcode, or position on the array (Supplemental Figure 1). However, as expected, there was clear clustering for both AUD status and smoking status. Following quality control, one control sample was removed as an outlier (Supplemental Figures 2-5), and overall, 15,282 probes (24%) were retained. The outlier identified in the expression data set was also removed from the clinical data set; therefore, the final analyses consisted of 19 control participants and 18 AUD participants with AEC data. AUDIT scores were substantially higher among the AUD participants (Table 1). The two participant groups were otherwise similar in terms of age, sex, and race (white versus non-white). Current smoking was also more prevalent among the AUD participants (83% versus 42%), although pack-years of smoking were similar between the two groups. Notably, approximately 60% of participants in both the control and AUD groups were either not active smokers, or had a very low pack-year history (average 3.3 pack-years). Approximately 40% of participants in both the control and AUD groups had a substantial, 20 pack-year smoking history.
Table 1.
Demographic features of participants
| Control Participants, n=19 |
AUD Participants, n=18 |
P value | |
|---|---|---|---|
| Age in Years (mean ± SD) | 40 ± 7 | 42 ± 6 | 0.26 |
| Male sex (n, %) | 14/19 (74%) | 15/18 (83%) | 0.69 |
| Race/ethnicity (%) | 0.40 | ||
| white | 89% | 78% | |
| non-white | 11% | 22% | |
| AUDIT Score (mean ± SD) | 2 ± 2 | 30 ± 8 | <0.0001 |
| Smoking habits | 0.001 | ||
| (1) Non-smokers | 11/19 (58%) | 3/18 (17%) | |
| (2) Current cigarette smokers with <10 pack-years (Pack-year in this group, mean± SD) | 0/19 (0%) (0 ± 0) | 8/18 (44%) (3 ± 3) | (0.87) |
| (3) Current cigarette smokers with ≥10 pack-years (Pack-year in this group, mean± SD) | 8/19 (42%) (21 ± 7) | 7/18 (40%) (19 ± 5) | (0.44) |
| Pack-years of smoking (among ALL smokers, mean ± SD) | 9 ± 11 | 9 ± 9 | 0.87 |
AUDIT=alcohol use disorders identification test. Fisher’s Exact test used to compare categorical variables. Student’s t test used to compare continuous variables.
Differential expression and Enrichment Analyses based on AUD.
After adjusting for age, sex, and current smoking (yes or no, as a categorical variable), 133 genes were differentially expressed between AUD and control participants’ AECs (FDR < 0.05). Of these, 95 were downregulated in participants with AUDs compared to controls, while 38 were upregulated in participants with AUDs compared to controls. Table 2a summarizes the top 20 most significant differentially expressed genes; the full gene list may be found in Supplemental Table 1. Given the prevalence of smoking among the AUD participants, in a separate model, these data were re-analyzed, adjusting for age, sex, and pack-years smoking as a continuous variable. In this second analysis, 520 genes were differentially expressed between AUD and control participants (FDR <0.05). Of these, 377 were downregulated in AUD participants compared to controls, while 143 were upregulated in AUD participants compared to controls. Table 2b highlights the top 20 differentially expressed genes; the full gene list may be found in Supplemental Table 2. The two models were remarkable for a number of differentially expressed genes in common, including ribosomal proteins (RPLs) L5, L10, and L30; zinc finger (ZNF) 426; chaperonin-containing TCP1, subunit 7 (CCT7), prothymosin-alpha (PTMA); bladder cancer apoptosis inducing factor (BLCAP); and pre-B cell leukemia homeobox 3 (PBX3). In the third model excluding participants with ≥10 pack-years smoking, certain genes remained differentially expressed in the context of AUDs, including RPLs L5, L10, and L30; PTMA; CCT7 and BLCAP (Supplemental Table 3). Representative differentially expressed genes are illustrated in Figure 1. Notably, for certain differentially expressed genes, multiple probes demonstrated differential expression (e.g. CCT7). Representative examples of log2 expression data for probes with most significantly different expression in the context of AUDs (lowest p values) may be found in Figure 2.
Table 2a.
Summary of top 20 most significant differentially expressed genes, based on alcohol use disorder, adjusted for current smoking as a categorical variable (yes/no). The full gene list may be found in Supplemental Table 1.
| ProbeName | Symbol | EntrezID | Log fold- change |
Average expression |
t | p value | Adjusted p value |
|---|---|---|---|---|---|---|---|
| A_23_P101351 | ZNF426* | 79088 | −1.6139492 | 5.345018 | −6.021046 | 8.00e-07 | 0.0091115 |
| A_33_P3348887 | PBX3* | 5090 | 2.2587736 | 3.696095 | 5.877560 | 1.20e-06 | 0.0091115 |
| A_33_P3353552 | SLC48A1 | 55652 | −0.8148278 | 7.009872 | −5.755836 | 1.80e-06 | 0.0091115 |
| A_23_P120710 | TTC3# | 7267 | −0.6627020 | 7.820126 | −5.645005 | 2.50e-06 | 0.0095240 |
| A_23_P419624 | BLCAP*# | 10904 | −0.7082976 | 8.201873 | −5.496883 | 3.90e-06 | 0.0115758 |
| A_23_P129221 | FAH | 2184 | −1.6225324 | 4.880897 | −5.444574 | 4.50e-06 | 0.0115758 |
| A_23_P217666 | RPL10*# | 6134 | −0.4143518 | 14.179896 | −5.352965 | 6.00e-06 | 0.0130555 |
| A_33_P3219803 | PTMA*# | 5757 | 1.0305163 | 13.786352 | 5.306091 | 6.90e-06 | 0.0131453 |
| A_23_P102404 | CCT7*# | 10574 | −0.4937125 | 10.698977 | −5.209633 | 9.20e-06 | 0.0155959 |
| A_23_P77415 | OSGIN1 | 29948 | −2.6576696 | 3.763180 | −5.110453 | 1.24e-05 | 0.0168854 |
| A_23_P66719 | DHRS13*# | 147015 | −1.6403834 | 4.935906 | −5.098374 | 1.28e-05 | 0.0168854 |
| A_23_P102404 | CCT7*# | 10574 | −0.5118417 | 10.945969 | −5.086847 | 1.33e-05 | 0.0168854 |
| A_23_P309803 | ZNF777 | 27153 | 0.4841887 | 7.937413 | 5.027023 | 1.59e-05 | 0.0186353 |
| A_32_P225604 | RPL5*# | 6125 | −0.4978655 | 13.559090 | −4.936844 | 2.07e-05 | 0.0222872 |
| A_24_P844984 | PIGR | 5284 | 0.6227903 | 13.951730 | 4.907237 | 2.27e-05 | 0.0222872 |
| A_23_P122007 | C5orf30 | 90355 | −1.2693265 | 6.869129 | −4.875647 | 2.49e-05 | 0.0222872 |
| A_23_P115215 | VPS72 | 6944 | −0.4665451 | 7.830694 | −4.852218 | 2.67e-05 | 0.0222872 |
| A_33_P3213419 | LOC100129447 | 100129447 | 1.9628734 | 5.617893 | 4.844685 | 2.73e-05 | 0.0222872 |
| A_23_P105571 | CHPT1 | 56994 | −1.7743049 | 3.603385 | −4.815341 | 2.98e-05 | 0.0222872 |
| A_23_P123330 | RPL30*# | 6156 | −0.4240935 | 13.358958 | −4.812120 | 3.01e-05 | 0.0222872 |
denotes genes that were also differentially expressed in model adjusting for pack-years smoking as a continuous variable (as described in figure 2b).
denotes genes that were also differentially expressed in model that included control (n=11) and AUD (n=11) participants with <10 pack-year smoking history.
Table 2b.
Summary of top 20 most significant differentially expressed genes, based on AUD, adjusted for pack-years smoking as a continuous variable. The full gene list may be found in Supplemental Table 2.
| ProbeName | Symbol | EntrezID | Log fold- change |
Average expression |
t | p value | Adjusted p value |
|---|---|---|---|---|---|---|---|
| A_32_P225604 | RPL5*# | 6125 | −0.6207947 | 13.559090 | −6.684604 | 1.0e-07 | 0.0011786 |
| A_23_P123330 | RPL30*# | 6156 | −0.5323540 | 13.358958 | −6.498355 | 2.0e-07 | 0.0011786 |
| A_23_P101351 | ZNF426* | 79088 | −1.5577416 | 5.345018 | −6.338923 | 3.0e-07 | 0.0011786 |
| A_32_P104432 | SMIM10L2B | 644596 | −1.6107530 | 3.660787 | −6.249990 | 4.0e-07 | 0.0011786 |
| A_23_P217666 | RPL10*# | 6134 | −0.4405652 | 14.179896 | −6.242387 | 4.0e-07 | 0.0011786 |
| A_23_P102404 | CCT7* | 10574 | −0.5501780 | 10.698977 | −6.184088 | 5.0e-07 | 0.0011786 |
| A_33_P3219803 | PTMA*# | 5757 | 1.1026929 | 13.786352 | 6.156888 | 5.0e-07 | 0.0011786 |
| A_23_P123330 | RPL30*# | 6156 | −0.5010585 | 13.564073 | −6.055543 | 7.0e-07 | 0.0012622 |
| A_23_P102404 | CCT7*# | 10574 | −0.5667809 | 10.945969 | −6.049635 | 7.0e-07 | 0.0012622 |
| A_23_P66719 | DHRS13*# | 147015 | −1.7148302 | 4.935906 | −5.880515 | 1.2e-06 | 0.0015521 |
| A_23_P419624 | BLCAP*# | 10904 | −0.6874833 | 8.201873 | −5.878118 | 1.2e-06 | 0.0015521 |
| A_23_P123330 | RPL30*# | 6156 | −0.4944068 | 13.412823 | −5.871070 | 1.3e-06 | 0.0015521 |
| A_23_P123330 | RPL30*# | 6156 | −0.4932770 | 13.263023 | −5.857418 | 1.3e-06 | 0.0015521 |
| A_23_P125519 | RPS4X | 6191 | −0.4533311 | 13.686688 | −5.830215 | 1.4e-06 | 0.0015634 |
| A_23_P123330 | RPL30*# | 6156 | −0.4787982 | 13.288719 | −5.802078 | 1.6e-06 | 0.0015874 |
| A_23_P258002 | CDKN2AIP | 55602 | −1.4786530 | 4.875930 | −5.721616 | 2.0e-06 | 0.0018936 |
| A_23_P123330 | RPL30*# | 6156 | −0.4733975 | 13.372103 | −5.691457 | 2.2e-06 | 0.0019506 |
| A_33_P3348887 | PBX3* | 5090 | 1.7849662 | 3.696095 | 5.633107 | 2.6e-06 | 0.0021941 |
| A_33_P3241582 | RPS15A | 6210 | −0.4600710 | 13.536785 | −5.593442 | 2.9e-06 | 0.0023410 |
| A_23_P102404 | CCT7*# | 10574 | −0.5674271 | 10.999433 | −5.560850 | 3.2e-06 | 0.0023510 |
denotes genes that were also differentially expressed in model adjusting for current smoking as a categorical variable (as described in figure 2a).
denotes genes that were also differentially expressed in model that included control (n=11) and AUD (n=11) participants with <10 pack-year smoking history.
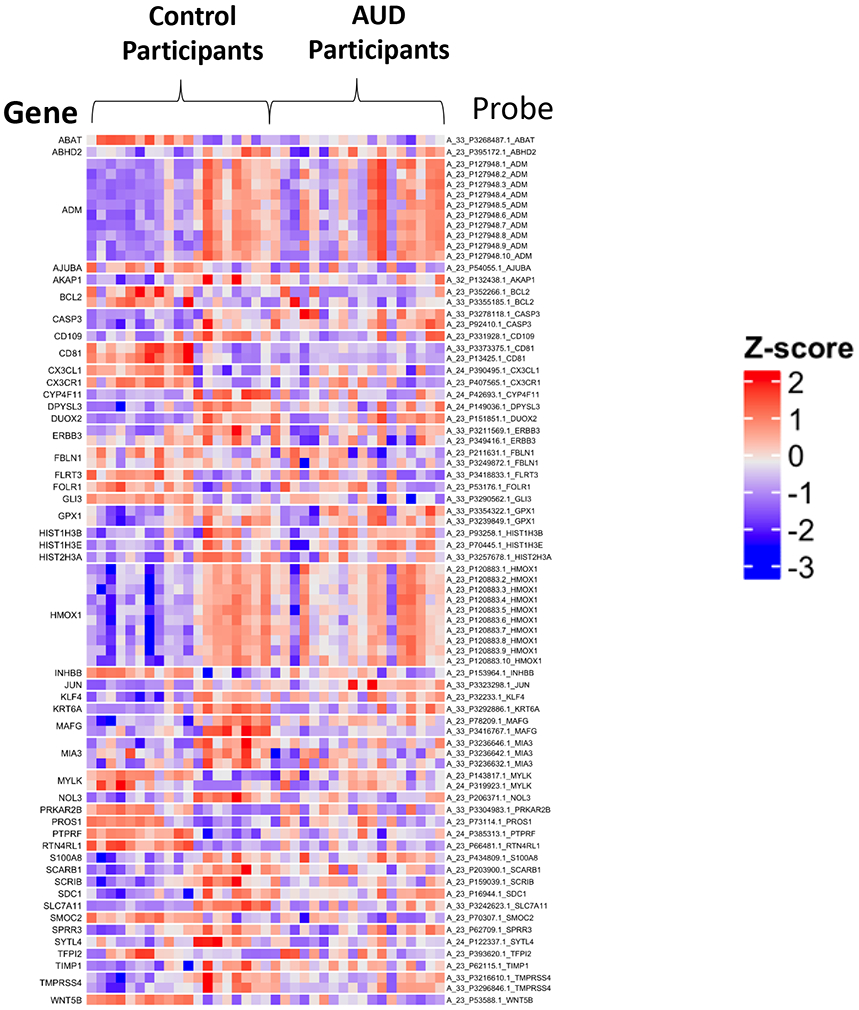
Figure 1.
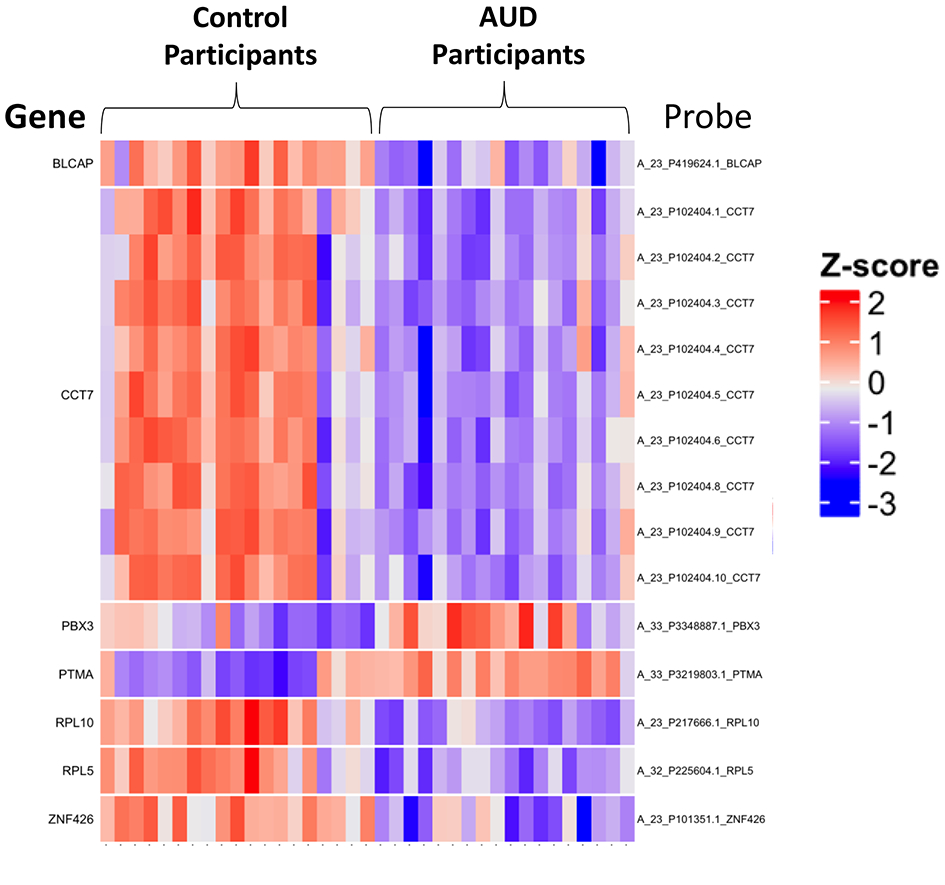
Heatmap of selected airways epithelial cell gene expression in control participants and participants with alcohol use disorders (AUDs). Airways epithelial cell expression among control participants and participants with AUDs was assessed by microarray (Agilent SurePrint G3 for humans, version 3). After filtering, data were log2 transformed. Selected top differentially expressed genes between control and AUD participants included zinc finger 426 (ZNF 426), prothymosin alpha (PTMA), bladder cancer apoptosis inducing factor (BLCAP), ribosomal proteins L5 and L10 (RPL5, RPL10), pre-B cell leukemia homeobox 3 (PBX3), and chaperonin-containing TCP1, subunit 7 (CCT7). For some genes (i.e. CCT7), multiple probes were differentially expressed.
Figure 2.
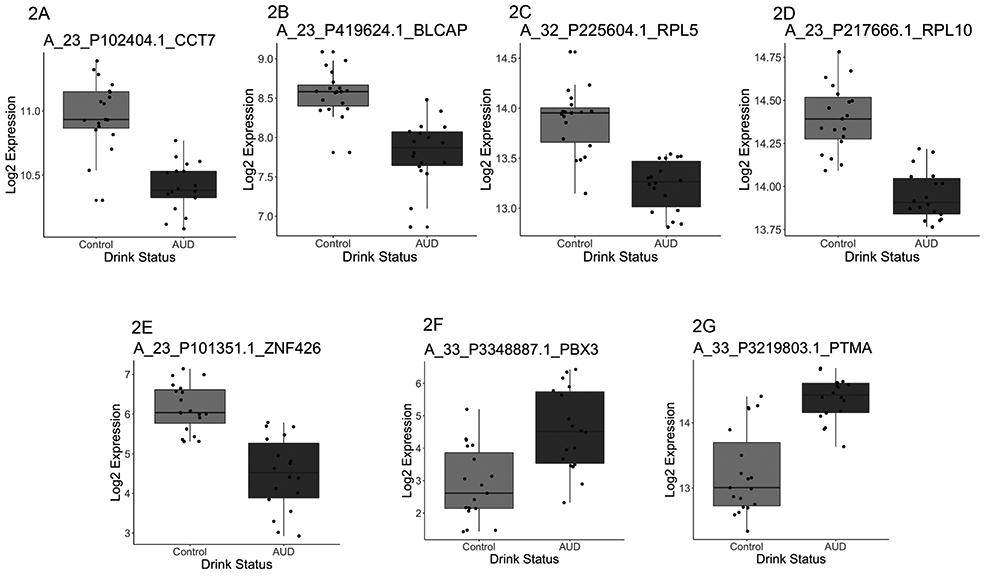
Genes with differential expression in airway epithelial cells in the context of alcohol use disorders (AUDs). Representative log2 expression data are presented for specific genes. In general, AUDs were associated with down-regulation in gene expression for the majority of genes examined, including (2A) chaperonin-containing TCP1, subunit 7 (CCT7), (2B) bladder cancer apoptosis inducing factor (BLCAP), (2C) ribosomal protein L5 (RPL5), (2D) RPL10, and (2E) zinc finger 426 (ZNF 426). However, a relative increase in gene expression was noted for (2F) pre-B cell leukemia homeobox 3 (PBX3), and (2G) prothymosin alpha (PTMA).
Enrichment analysis was performed with clusterProfiler (Yu et al., 2012) using the 520 significantly differentially expressed genes in the context of AUDs adjusted for pack-years, including GO terms from all 3 ontologies. Using this strategy, 24 gene ontologies were enriched. Fifteen were related to biological processes (BP), seven were related to cellular components (CC), and two were related to molecular functions (MF). GO terms relevant to BP included signal recognition protein (SRP)-dependent cotranslational protein targeting (GO:0006605), translation (GO:0006412), and viral gene expression (GO:0019080) were significantly enriched in AUD participants. For ontologies relevant to CC, enrichment of ontologies related to cytosolic ribosomes (GO:0022626) was present. Finally, for MF ontologies, enrichment of structural molecule activity (GO:0005198) was noted. The full list of enriched genome ontologies in the AUD context may be found in Supplemental Table 4. Figure 3 illustrates a heatmap of differentially expressed genes within representative GO term 0006412 (biological process of translation). Supplemental Figures 6 and 7 illustrate heatmaps of significantly enriched GO terms 0006605 (protein targeting), and 0006614 (SRP-dependent cotranslational protein targeting to membrane). Enrichment analyses were also performed using gene expression data examined in the context of AUDs, where current smoking was included as a binary variable (133 differentially expressed genes). Genes were significantly overrepresented in six ontologies, all of which were common to ontologies overrepresented in models accounting for smoking pack-years (data not shown). KEGG analysis demonstrated enrichment for 22 genes involved in ribosome-related pathways (hsa030100).
Figure 3.
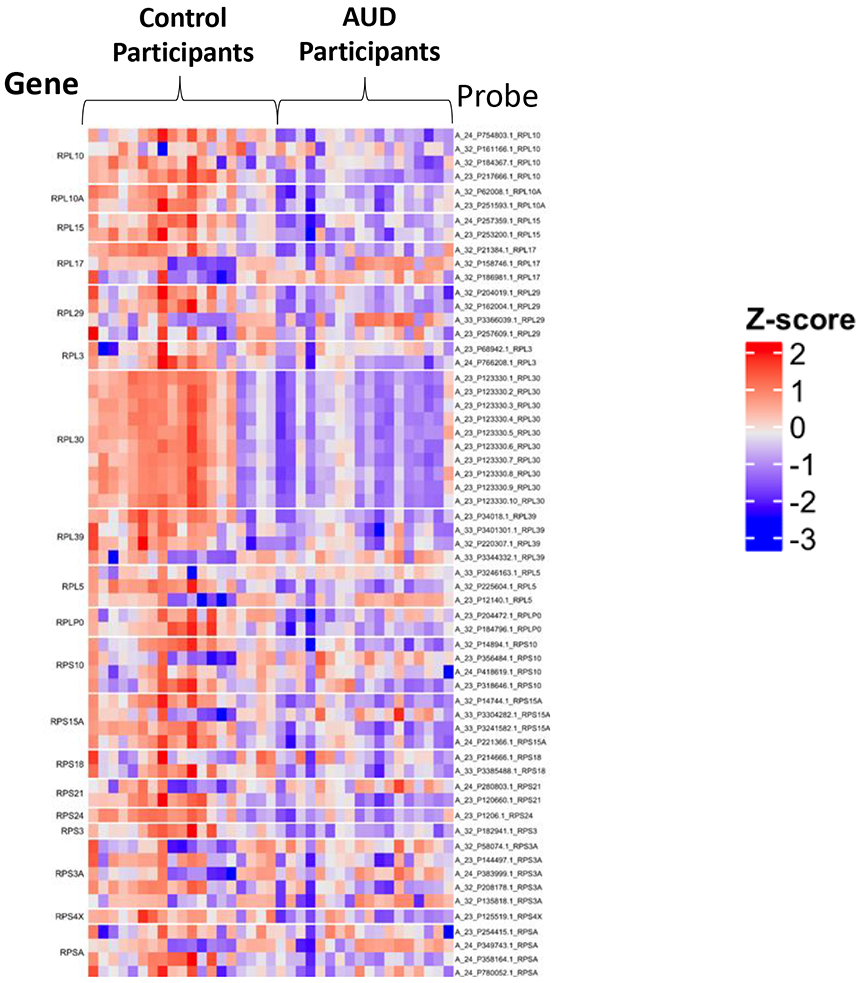
Representative heatmap of genes within gene ontology (GO) term 0006412 that maps to the biological process of translation. Of the 364 genes in the GO term, 39 had significantly different expression (adjusted p=0.008). Additional GO terms that differed on the basis of AUD history may be found in Supplemental Table 4.
Gene expression differences were assessed with RT-PCR using the remaining bronchial airway RNA from participants who had been included in the microarray. Specific genes were selected based on differential expression results and for their potential relevance to airways diseases based on literature review. Significant differences in gene expression measured in AECs between control and AUD participants were confirmed for CCT7 (p=0.04) and PBX3 (p=0.01), Figure 4. BLCAP and PTMA expression did not significantly differ between AUD and control participants, but changes in gene expression for these two probes were of the same directionality as in the array.
Figure 4.
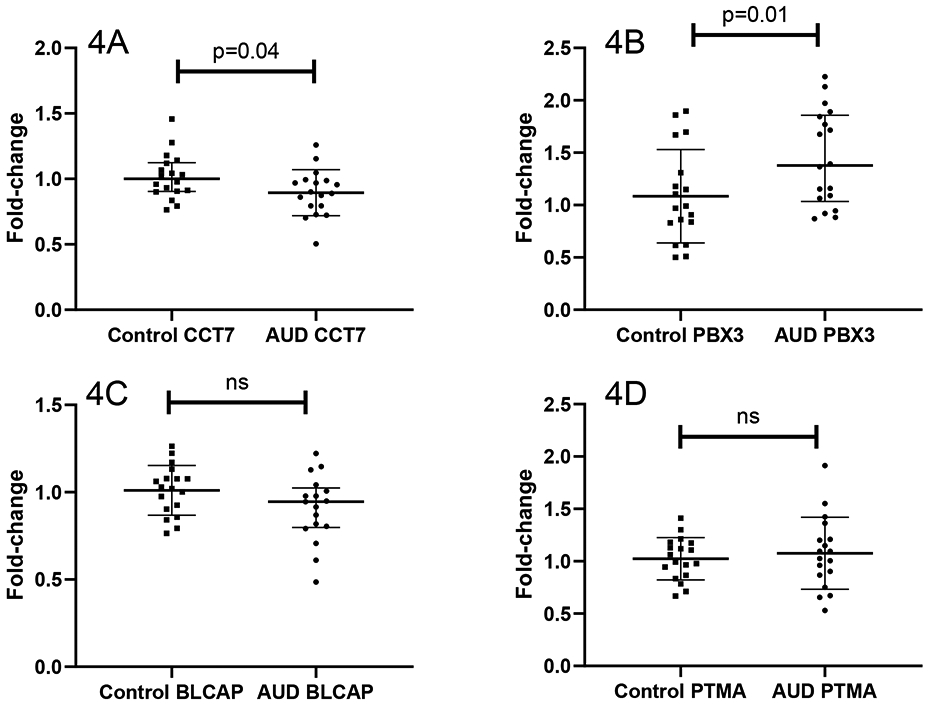
Confirmatory PCR analyses. Real-time PCR was performed using RNA from participants that was available post-array to assess specific genes determined to be differentially expessed in the micro-array. For both chaperonin-containing TCP1, subunit 7 (CCT7) (5A) and pre-B cell leukemia homeobox 3 (PBX3) (5B), significant differences in gene expression were observed, corresponding to microarray findings. Gene expression for (5C) bladder cancer apoptosis inducing factor (BLCAP), and (5D) prothymosin alpha (PTMA) did not differ significantly between control and alcohol use disorder participants, although directionality of gene expression corresponded to that observed in the micro-array analyses.
Differential expression and Enrichment Analyses based on smoking.
Upon examining the data based on current smoking (yes/no), after adjusting for age, sex, and AUD participant status (AUD versus control), 838 genes were differentially expressed (FDR < 0.05). Of these, 404 were downregulated, and 434 were upregulated in current smokers compared to non-smokers. Table 3 summarizes the top 20 differentially expressed genes according to current smoking history; the full gene list may be found in Supplemental Table 5. Notably, none of these 20 genes were among the top 20 differentially expressed in models examining expression differences in the context of AUDs (described in Tables 2a and 2b). Comparing differentially expressed genes in the context of AUDs (to an FDR <0.05) to differentially expressed genes in the context of current smoking (to an FDR <0.05) revealed that twelve genes were differentially expressed in common. Among these twelve genes, oxidative stress induced growth inhibitor-1 (OSGIN1) and ribosomal protein L30 (RPL30) fell among the top twenty.
Table 3.
Summary of top 20 most significant differentially expressed genes, based on current smoking, adjusted for AUD as a categorical variable (yes/no). The full gene list may be found in Supplemental Table 4.
| ProbeName | Symbol | EntrezID | Log fold- change |
Average expression |
t | p value | Adjusted p value |
|---|---|---|---|---|---|---|---|
| A_33_P3233645 | MT1G | 4495 | −4.1143306 | 4.341725 | −8.601759 | 0e+00 | 0.0000073 |
| A_23_P3038 | GPX2 | 2877 | 2.3171582 | 9.580692 | 7.382457 | 0e+00 | 0.0001126 |
| A_23_P55779 | CYP2A13 | 1553 | −3.3948401 | 7.132334 | −7.135260 | 0e+00 | 0.0001536 |
| A_23_P60248 | TXN | 7295 | 1.0313451 | 13.214024 | 6.939364 | 1e-07 | 0.0002040 |
| A_33_P3290343 | CYP1B1 | 1545 | 6.3972981 | 4.686245 | 6.706007 | 1e-07 | 0.0002926 |
| A_23_P101642 | PTPRH | 5794 | 5.3610256 | 5.153402 | 6.667352 | 1e-07 | 0.0002926 |
| A_33_P3238433 | ALDH3A1 | 218 | 2.1892620 | 12.951323 | 6.626265 | 1e-07 | 0.0002926 |
| A_23_P151851 | DUOX2 | 50506 | 5.3899431 | 5.481190 | 6.425314 | 2e-07 | 0.0003830 |
| A_24_P152968 | AKR1C1 | 1645 | 1.9798421 | 9.627513 | 6.418102 | 2e-07 | 0.0003830 |
| A_23_P21990 | SLC23A1 | 9963 | −3.0017571 | 5.582623 | −6.405647 | 3e-07 | 0.0003830 |
| A_23_P150876 | VPS37B | 79720 | 0.9257283 | 9.553437 | 6.361066 | 3e-07 | 0.0003830 |
| A_24_P129341 | AKR1B10 | 57016 | 5.4038808 | 7.263562 | 6.348004 | 3e-07 | 0.0003830 |
| A_23_P142738 | TMEM178A | 130733 | −1.9814992 | 3.091291 | −6.326403 | 3e-07 | 0.0003830 |
| A_33_P3380992 | AKR1B15 | 441282 | 5.4180491 | 6.548961 | 6.164309 | 5e-07 | 0.0005761 |
| A_23_P100632 | JPT1 | 51155 | 0.8700925 | 11.661904 | 6.094492 | 6e-07 | 0.0006519 |
| A_24_P120462 | DNAH5 | 1767 | −2.5727260 | 4.241725 | −6.078069 | 7e-07 | 0.0006519 |
| A_23_P126623 | PGD | 5226 | 1.3527388 | 9.449167 | 6.057662 | 7e-07 | 0.0006521 |
| A_23_P209625 | CYP1B1 | 1545 | 6.5025153 | 5.704521 | 6.034444 | 8e-07 | 0.0006601 |
| A_33_P3395321 | JPT1 | 51155 | 0.9377850 | 9.992488 | 6.007443 | 8e-07 | 0.0006778 |
| A_23_P207213 | ALDH3A1 | 218 | 2.4120472 | 11.328098 | 5.976383 | 9e-07 | 0.0007066 |
Enrichment analyses were also performed using the 838 genes differentially expressed related to smoking, adjusted for AUD status. Sixty-nine gene ontologies were enriched related to BPs, eleven were related to CCs, and 33 were related to MF. GO terms relevant to BPs enriched in the smoking context included epithelial cell differentiation (GO:0030855), response to toxic substance (GO:0009636), response to wounding (GO:0009611), regulation of cell motility (GO:2000145), and regulation of cell migration (GO:0030334). GO terms relevant to CCs that were enriched included intrinsic component of plasma membrane (GO:0031226), apical plasma membrane (GO:0016324), and extracellular matrix (GO:0031012). Finally, enriched ontologies relevant to MF included oxidoreductase activity (GO:0016491), calcium ion binding (GO:0005509), and signaling receptor activity (GO:0038023). Figure 5 illustrates a heatmap of representative differentially expressed genes in for GO term 0009611 that maps to the BP of response to wounding. Supplemental Figures 8 and 9 illustrate representative gene expression in GO 0016491 (oxidoreductase activity) and GO 0031226 (intrinsic component of the plasma membrane). The full list of enriched genome ontologies in the smoking context may be found in Supplemental Table 6. KEGG pathways enriched in the context of cigarette smoking, adjusted for alcohol consumption, included those relevant to ferroptosis (hsa04216, nine genes), mineral absorption (hsa04978), the pentose phosphate pathway (hsa00030), hypoxia-inducible factor 1 pathway (hsa04066, twelve genes), and glycolysis/gluconeogenesis (hsa00010).
Figure 5.
Representative heatmap of genes within gene ontology (GO) term 0009611 that maps to the biological process of response to wounding. Of the 590 genes in the GO term, 48 had significantly different expression (adjusted p=0.0000007). Additional GO terms that differed on the basis of smoking history may be found in Supplemental Table 6.
Discussion
In our investigations, we found AUDs to be independently associated with differential expression of 520 genes in human AECs. In participants with AUDs, approximately 70% of differentially expressed AEC genes were downregulated, while 30% were upregulated. Top differentially expressed genes associated with AUDs differed from those differentially expressed in association with cigarette smoking, and included genes fundamentally involved in protein processing. Enrichment analyses suggested that post-translational protein processing and protein targeting, along with pathways involved in viral transcription were significantly influenced by AUDs. By delineating the potential impact of AUDs on AEC gene expression, previously unrecognized mechanisms contributing to the predisposition for CAP among those with AUDs may be identified. Further, our findings also extend current knowledge regarding the superimposed impact of AUDs and smoking on development of chronic lung conditions, such as COPD. Collectively, these data suggest a measurable impact of alcohol consumption on AEC gene expression independent of smoking. Our findings pave the way for additional mechanistic studies to examine candidate genes relevant to lung infections in AUDs that will promote a more comprehensive understanding of disease pathogenesis. Further, our observations support the concept that alcohol and its metabolites can measurably influence gene expression, and potentially cell function, in tissue types other than liver and brain.
In order to protect the host from inhaled pathogens, chemicals, and other toxicants in the environment, mucociliary clearance serves as an upper airway defense mechanism where it is further supported by innate immune effectors in this lung region. Prior investigations examining the impact of AUDs on lung have focused on mucociliary function. After ethanol ingestion, up to 10% may be excreted unchanged in urine, sweat, and breath. Importantly, unmetabolized ethanol reaches the breath via the upper airways through the bronchial circulation, exposing AECs to substantial concentrations of ethanol en route (reviewed in (Sapkota and Wyatt, 2015)). Using in vitro models of chronic alcohol exposure, an uncoupling of cilio-stimulatory pathways is suggested, leading to impaired mucociliary function (Wyatt et al., 2018, Wyatt et al., 2012). Additionally, in human AECs, AUDs have been associated with altered Toll-like receptor (TLR) expression that is critical for recognition of bacterial patterns (i.e. lipopolysaccharides on Gram (−) organisms). Further, the airways of participants with AUDs are characterized as having enhanced quantity of pro-inflammatory cytokines interleukin-6 and −8 (Bailey et al., 2015, Bailey et al., 2009). Collectively, these investigations suggest that chronic alcohol consumption may impair pathogen recognition and clearance at the level of the upper airway, paving the way for the development of lower respiratory tract infections. Notably, in our current investigation, differentially expressed genes did not correspond to ciliary function or pathogen recognition. Reasons for these differences are unclear, but notably, the most substantial differences in TLR expression from prior investigations existed in participants with dual alcohol and tobacco exposure. It is therefore possible that participants in our current investigation had quantitatively different alcohol and/or tobacco exposure, or that we were underpowered to detect these difference. Nevertheless, the unbiased approach to characterize AUD-associated AEC gene expression we employed will provide novel information to support identification of candidate genes with roles in pulmonary infection.
We did observe specific genes with differential expression in the context of AUDs that may influence the host’s response to pathogens, including CCT7 and PBX3. CCT7 demonstrated down-regulation in the AUD context. CCT7 encodes a member of the chaperonin-containing tailless complex polypeptide-1 complex, also known as TRiC. TRiC plays a role in protein folding and proteostasis (Hartl et al., 2011), making it is possible the diminished expression of CCT7 could contribute to an alcohol-associated “senescent” phenotype that has been reported in studies of the liver (Ji, 2015, Nagy et al., 2016). AECs unable to appropriately fold critical polypeptides might be expected to respond inadequately to infectious insults. Expression of PBX3 was up-regulated in AECs from AUD participants. PBX3 is a proto-oncogene, and member of the PBX family of 3-amino-acid loop extension homeobox (HOX) proteins, ubiquitously expressed in a number of tissue types including lung (Milech et al., 2001). PBX3 protein has been implicated in solid malignancies involving epithelial cells, including prostate and colorectal tumors, where it is believed to contribute to the invasive and metastatic nature of these diseases (Wang et al., 2016). PBX proteins can interact with HOX proteins which increases DNA-binding affinity, thereby promoting transcription of downstream target genes (Monica et al., 1991). One in vitro study using A549 cells, a human epithelial cell line commonly used in lung cancer investigations, was conducted to understand mechanisms underlying proliferation and invasion of human lung tumors. A549 cell viability, migration, and invasion were all increased when PBX3 expression was upregulated, suggesting that PBX3 may have a potential regulatory role in growth and metastasis of lung cancer (Li et al., 2018). The heightened expression of PBX3 we observed in the context of AUDs may therefore disrupt airway cell homeostasis, promoting impaired response to infection and potentially cancer.
We observed enriched gene ontologies in the AUD context relevant to the biological process of translation and co-translational protein targeting to endoplasmic reticulum (ER). Enriched GO terms featured overlap in a number of ribosomal protein genes, including RPL5 and RPL10. Given prior reports and our data, we speculate that the relationship between AUDs and development of CAP may be linked to aberrant ribosomal protein structure, leading to augmented ER stress in AECs that in turn impairs the host’s response to pathogens. ER stress is implicated in the pathogenesis of a variety of alcohol-related organ disorders, including alcohol-associated neurotoxicity (George et al., 2017), hepatotoxicity (Rodriguez et al., 2019), and potentially cardiomyopathy (Li et al., 2019). Proposed mechanisms include disruption of intracellular calcium homeostasis, resulting in abnormal ER protein folding and modification (Yang and Luo, 2015). Importantly, ribosomal proteins are particularly critical in initiating and sustaining viral infections, since viruses have a limited genomic repertoire and must use host cellular factors in order to propagate (Li, 2019). Viral infections are common etiologic agents in CAP, and can predispose patients to develop secondary bacterial pneumonias (Prasso and Deng, 2017). A relationship between alcohol exposure and respiratory syncytial virus infection has been previously demonstrated, with pathogenesis involving ciliated proximal epithelial cells (Wyatt et al., 2018). AUD-associated alterations in ribosomal gene expression could additionally influence the cell’s ability to appropriately localize normal membrane and secretory proteins required to maintain airways homeostasis (Saraogi et al., 2014, Akopian et al., 2013). In the setting of AUDs, baseline disruption in the highly orchestrated series of events necessary for fundamental protein processing, coupled with viral exposure, could conceivably heighten the risk for pulmonary infections. A schematic illustrating the potential impact of AUDs on pulmonary immunity involving the airways is depicted in Figure 6.
Figure 6.
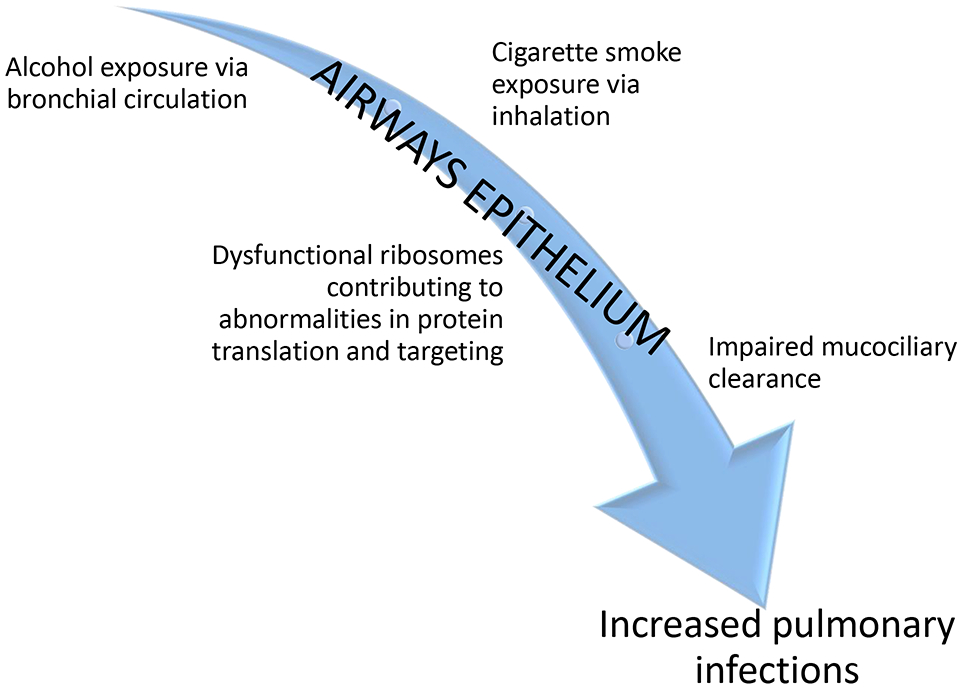
Schematic depicting the downstream chain of events initiated by chronic exposure to alcohol and cigarette smoking on the airways epithelium.
A number of prior investigations examining association of tobacco smoking and gene expression in human airways have been conducted, but none explicitly considered alcohol consumption in their analyses (Ammous et al., 2008, Gower et al., 2011, Spira et al., 2004). Nevertheless, epidemiologic studies suggest that tobacco-dependent individuals are substantially more likely to have an AUD (Grant et al., 2004). Concomitant use of alcohol and tobacco have been linked to development of malignancy, and among individuals with AUDs, smoking cigarettes enhances risk for tobacco-associated diseases such as heart disease (reviewed in (Adams, 2017)). In this investigation, we sought to clarify differences between AUD- and previously described smoking-related alterations in AEC gene expression. In one early study using an Affymetrix-based array, 152 uniquely named genes were differentially expressed in the small airways in the context of smoking, with relatively equal numbers up- and down-regulated (Ammous et al., 2008). In our current investigation, AEC gene expression referable to smoking was remarkable for genes involved in oxidative stress and signal transduction. Differentially expressed gene in the smoking context common across the prior study and our current work included glucose-6-phosphate dehydrogenase (G6PD), aldo-ketoreductase family 1, member B10 (AKR1B10), and glutathione peroxidase-2 (GPX2). Another study compiled data from four sets of gene expression profiles (Gower et al., 2011). Focusing on genes expressed in mainstem bronchi from current smokers compared to non-smokers, differential expression for MT1G, GPX2, CYP1B1, ALDH3A1, AKR1C1, AKR1B10, and PGD were consistent with gene expression in our current data. In contrast, none of the AUD-associated genes differentially expressed in our data were significantly expressed in the context of smoking in these earlier data. Similarities in smokers’ AEC gene expression between our current study and prior investigations supports to the validity of our observations, and helps confirm the novelty of gene expression differences we report in the context of AUDs.
Although we believe that our work highlights unique differences in AEC gene expression associated with AUDs, it is not without limitations. First, the participant numbers were relatively modest. However, to our knowledge, our study has the largest sample size to date delineating the impact of AUDs on AEC gene expression. Moreover, the sample size in our study is comparable to prior published investigations exploring alterations in gene expression in the context of tobacco smoking (Ammous et al., 2008, Carolan et al., 2006, Hackett et al., 2003, Spira et al., 2004). Additionally, we controlled for demographic variables such as age and sex in our analyses to more specifically examine the impact of AUDs and smoking on gene expression. We acknowledge that cigarette use was present in a substantial number of participants and may have impacted our observations, and considered this variable in our analyses. However, exclusion of cigarette smoking would have substantially increased the time required to recruit participants for this investigation, given the common use of cigarettes among participants with AUDs. Further, we believe that including participants with cigarette smoking enhances the clinical relevance of our results. In the future, expanding this cohort to include additional participants of non-white race, non-smokers, and an increased number of women will be important to establish specific racial, smoking, and sex influences on gene expression. Second, although we utilized specific probes indicated by the array results in confirmatory qPCR experiments, we were not able to replicate all gene expression differences demonstrated in the array. Although it remains possible that differences in gene expression determined by array are false positives, it is also possible that the probe selected was not sufficient to detect gene expression in the qPCR experiments. Our qPCR experiments were limited by the available specimen quantities that precluded additional experiments substituting different probes. Regardless, replication of our observations in a secondary cohort of participants with AUDs and matched control individuals will be important to validate our findings. Third, it should be acknowledged that gene expression differences we observed may not correlate with protein expression due to post-translational modifications and degradation of newly manufactured proteins. Therefore, extending inferences between our transcriptomic data and protein quantity or function are speculative. An assessment of protein content or other functional assays will be critical to establish the mechanistic relevance of this line of investigation. Lastly, the use of micro-array to examine the transcriptome is only one type of assay that can help establish associations between environmental exposures and tissue function. Certainly, emerging technologies (for example, single cell RNA-Seq) will be increasingly useful to further define alterations in gene expression. We expect that our preliminary observations will require replication in additional participants to further advance insights in the area of airways epithelial biology in the context of alcohol exposure.
In conclusion, our work highlights unique alterations in AEC gene expression related to chronic, heavy alcohol consumption that may be relevant to the pathophysiology of CAP as well as chronic airways diseases. Given the fact that unhealthy alcohol consumption is frequently present among those who smoke, identifying novel genes and gene pathways implicated in lung diseases that are influenced by alcohol may provide new investigative avenues to delineate mechanisms underlying alcohol- and smoking-related lung disorders, including CAP and COPD. Improved understanding of alcohol’s impact on airways has the potential to inform new interventions that could attenuate disease development.
Supplementary Material
Acknowledgements
The authors would like to thank the clients and staff of Denver CARES for their participation in this project.
Funding information: NIH-NHLBI: K23HL136785 (SKM), Research Career Scientist Award (IK6 BX003781) from the Department of Veterans Affairs (TAW), NIH-NHLBI: R21HL121572 (IVY), NIH-NIAAA: R24AA019661 (ELB), and NIH-NCATS: UL1TR001082 (ELB)
References
- ADAMS S 2017. Psychopharmacology of Tobacco and Alcohol Comorbidity: a Review of Current Evidence. Current addiction reports, 4, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AKOPIAN D, SHEN K, ZHANG X & SHAN S-O 2013. Signal recognition particle: an essential protein-targeting machine. Annual review of biochemistry, 82, 693–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AMMOUS Z, HACKETT NR, BUTLER MW, RAMAN T, DOLGALEV I, O'CONNOR TP, HARVEY B-G & CRYSTAL RG 2008. Variability in small airway epithelial gene expression among normal smokers. Chest, 133, 1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAILEY KL, ROMBERGER DJ, KATAFIASZ DM, HEIRES AJ, SISSON JH, WYATT TA & BURNHAM EL 2015. TLR2 and TLR4 Expression and Inflammatory Cytokines are Altered in the Airway Epithelium of Those with Alcohol Use Disorders. Alcohol Clin Exp Res, 39, 1691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAILEY KL, WYATT TA, ROMBERGER DJ & SISSON JH 2009. Alcohol functionally upregulates Toll-like receptor 2 in airway epithelial cells. Alcoholism, clinical and experimental research, 33, 499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENJAMINI Y, HOCHBERG Y 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B, 57, 289–300. [Google Scholar]
- BRAEKEN DC, ROHDE GG, FRANSSEN FM, DRIESSEN JH, VAN STAA TP, SOUVEREIN PC, WOUTERS EF & DE VRIES F 2017. Risk of community-acquired pneumonia in chronic obstructive pulmonary disease stratified by smoking status: a population-based cohort study in the United Kingdom. Int J Chron Obstruct Pulmon Dis, 12, 2425–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURNHAM EL, MCCORD JM, BOSE S, BROWN LA, HOUSE R, MOSS M & GAYDOS J 2012. Protandim does not influence alveolar epithelial permeability or intrapulmonary oxidative stress in human subjects with alcohol use disorders. Am. J. Physiol Lung Cell Mol. Physiol, 302, L688–L699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAROLAN BJ, HEGUY A, HARVEY B-G, LEOPOLD PL, FERRIS B & CRYSTAL RG 2006. Up-regulation of Expression of the <em>Ubiquitin Carboxyl-Terminal Hydrolase L1</em> Gene in Human Airway Epithelium of Cigarette Smokers. Cancer Research, 66, 10729. [DOI] [PubMed] [Google Scholar]
- CRYSTAL RG, RANDELL SH, ENGELHARDT JF, VOYNOW J & SUNDAY ME 2008. Airway epithelial cells: current concepts and challenges. Proceedings of the American Thoracic Society, 5, 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE ROUX A, CAVALCANTI M, MARCOS MA, GARCIA E, EWIG S, MENSA J & TORRES A 2006. Impact of alcohol abuse in the etiology and severity of community-acquired pneumonia. Chest, 129, 1219–25. [DOI] [PubMed] [Google Scholar]
- ELLIOTT MK, SISSON JH & WYATT TA 2007. Effects of cigarette smoke and alcohol on ciliated tracheal epithelium and inflammatory cell recruitment. Am J Respir Cell Mol Biol, 36, 452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FALK DE, YI HY & HILLER-STURMHOFEL S 2006. An epidemiologic analysis of co-occurring alcohol and tobacco use and disorders: findings from the National Epidemiologic Survey on Alcohol and Related Conditions. Alcohol Res Health, 29, 162–71. [PMC free article] [PubMed] [Google Scholar]
- FINI MA, GAYDOS J, MCNALLY A, KAROOR V & BURNHAM EL 2017. Alcohol abuse is associated with enhanced pulmonary and systemic xanthine oxidoreductase activity. Am J Physiol Lung Cell Mol Physiol, 313, L1047–l1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GÉNIER S, DEGRANDMAISON J, MOREAU P, LABRECQUE P, HÉBERT TE & PARENT J-L 2016. Regulation of GPCR expression through an interaction with CCT7, a subunit of the CCT/TRiC complex. Molecular biology of the cell, 27, 3800–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEORGE AK, BEHERA J, KELLY KE, ZHAI Y & TYAGI N 2017. Hydrogen sulfide, endoplasmic reticulum stress and alcohol mediated neurotoxicity. Brain Res Bull, 130, 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOWER AC, STEILING K, BROTHERS JF 2ND, LENBURG ME & SPIRA A 2011. Transcriptomic studies of the airway field of injury associated with smoking-related lung disease. Proc Am Thorac Soc, 8, 173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRANT BF, HASIN DS, CHOU SP, STINSON FS & DAWSON DA 2004. Nicotine dependence and psychiatric disorders in the United States: results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry, 61, 1107–15. [DOI] [PubMed] [Google Scholar]
- GRAU I, ARDANUY C, CALATAYUD L, SCHULZE MH, LINARES J & PALLARES R 2014. Smoking and alcohol abuse are the most preventable risk factors for invasive pneumonia and other pneumococcal infections. Int J Infect Dis, 25, 59–64. [DOI] [PubMed] [Google Scholar]
- GUPTA NM, LINDENAUER PK, YU PC, IMREY PB, HAESSLER S, DESHPANDE A, HIGGINS TL & ROTHBERG MB 2019. Association Between Alcohol Use Disorders and Outcomes of Patients Hospitalized With Community-Acquired Pneumonia. JAMA Netw Open, 2, e195172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HACKETT NR, HEGUY A, HARVEY B-G, O'CONNOR TP, LUETTICH K, FLIEDER DB, KAPLAN R & CRYSTAL RG 2003. Variability of Antioxidant-Related Gene Expression in the Airway Epithelium of Cigarette Smokers. American Journal of Respiratory Cell and Molecular Biology, 29, 331–343. [DOI] [PubMed] [Google Scholar]
- HARTL FU, BRACHER A & HAYER-HARTL M 2011. Molecular chaperones in protein folding and proteostasis. Nature, 475, 324–32. [DOI] [PubMed] [Google Scholar]
- JI C 2015. Advances and New Concepts in Alcohol-Induced Organelle Stress, Unfolded Protein Responses and Organ Damage. Biomolecules, 5, 1099–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAPHALIA L & CALHOUN WJ 2013. Alcoholic lung injury: Metabolic, biochemical and immunological aspects. Toxicology Letters, 222, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOCHANEK KD M. SL; XU J; ARIAS E 2019. Deaths: Final Data for 2017. National Vital Statistics Reports. June 24, 2019 ed. [PubMed] [Google Scholar]
- LABISSO WL, RAULIN AC, NWIDU LL, KOCON A, WAYNE D, ERDOZAIN AM, MORENTIN B, SCHWENDENER D, ALLEN G, ENTICOTT J, GERDES HK, JOHNSON L, GRZESKOWIAK J, DRIZOU F, TARBOX R, OSNA NA, KHARBANDA KK, CALLADO LF & CARTER WG 2018. The Loss of alpha- and beta-Tubulin Proteins Are a Pathological Hallmark of Chronic Alcohol Consumption and Natural Brain Ageing. Brain Sci, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI D, LI H, YANG Y & KANG L 2018. Long Noncoding RNA Urothelial Carcinoma-Associated 1 Promotes the Proliferation and Metastasis of Human Lung Tumor Cells by Regulating MicroRNA-144. Oncol Res, 26, 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- LI N, WEN C, HUANG P, TAO Y, JIN F & LIU X 2019. Atorvastatin reduces alcohol-induced endoplasmic reticulum stress in AC16 cardiomyocytes. Scand Cardiovasc J, 53, 42–47. [DOI] [PubMed] [Google Scholar]
- LI S 2019. Regulation of Ribosomal Proteins on Viral Infection. Cells, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANAUTOU JE & CARLSON GP 1992. Comparison of rat pulmonary and hepatic cytosolic alcohol dehydrogenase activities. J Toxicol Environ Health, 35, 7–18. [DOI] [PubMed] [Google Scholar]
- MILECH N, KEES UR & WATT PM 2001. Novel alternative PBX3 isoforms in leukemia cells with distinct interaction specificities. Genes Chromosomes Cancer, 32, 275–80. [DOI] [PubMed] [Google Scholar]
- MONICA K, GALILI N, NOURSE J, SALTMAN D & CLEARY ML 1991. PBX2 and PBX3, new homeobox genes with extensive homology to the human proto-oncogene PBX1. Mol Cell Biol, 11, 6149–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOURONTE-ROIBAS C, LEIRO-FERNANDEZ V, FERNANDEZ-VILLAR A, BOTANA-RIAL M, RAMOS-HERNANDEZ C & RUANO-RAVINA A 2016. COPD, emphysema and the onset of lung cancer. A systematic review. Cancer Lett, 382, 240–244. [DOI] [PubMed] [Google Scholar]
- NAGY LE, DING W-X, CRESCI G, SAIKIA P & SHAH VH 2016. Linking Pathogenic Mechanisms of Alcoholic Liver Disease With Clinical Phenotypes. Gastroenterology, 150, 1756–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLETCHER MJ, VITTINGHOFF E, KALHAN R, RICHMAN J, SAFFORD M, SIDNEY S, LIN F & KERTESZ S 2012. Association between marijuana exposure and pulmonary function over 20 years. JAMA, 307, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRASSO JE & DENG JC 2017. Postviral Complications: Bacterial Pneumonia. Clin Chest Med, 38, 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMIREZ JA, WIEMKEN TL, PEYRANI P, ARNOLD FW, KELLEY R, MATTINGLY WA, NAKAMATSU R, PENA S, GUINN BE, FURMANEK SP, PERSAUD AK, RAGHURAM A, FERNANDEZ F, BEAVIN L, BOSSON R, FERNANDEZ-BOTRAN R, CAVALLAZZI R, BORDON J, VALDIVIESO C, SCHULTE J & CARRICO RM 2017. Adults Hospitalized With Pneumonia in the United States: Incidence, Epidemiology, and Mortality. Clin Infect Dis, 65, 1806–1812. [DOI] [PubMed] [Google Scholar]
- REINERT DF & ALLEN JP 2002. The Alcohol Use Disorders Identification Test (AUDIT): a review of recent research. Alcohol Clin. Exp. Res, 26, 272–279. [PubMed] [Google Scholar]
- RITCHIE ME, PHIPSON B, WU D, HU Y, LAW CW, SHI W & SMYTH GK 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res, 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RODRIGUEZ WE, WAHLANG B, WANG Y, ZHANG J, VADHANAM MV, JOSHI-BARVE S, BAUER P, CANNON R, AHMADI AR, SUN Z, CAMERON A, BARVE S, MALDONADO C, MCCLAIN C & GOBEJISHVILI L 2019. Phosphodiesterase 4 Inhibition as a Therapeutic Target for Alcoholic Liver Disease: From Bedside to Bench. Hepatology, 70, 1958–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAPKOTA M & WYATT TA 2015. Alcohol, Aldehydes, Adducts and Airways. Biomolecules, 5, 2987–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARAOGI I, AKOPIAN D & SHAN S-O 2014. Regulation of cargo recognition, commitment, and unloading drives cotranslational protein targeting. The Journal of Cell Biology, 205, 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMITTGEN TD & LIVAK KJ 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc, 3, 1101–8. [DOI] [PubMed] [Google Scholar]
- SINGH G, ZHANG W, KUO YF & SHARMA G 2016. Association of Psychological Disorders With 30-Day Readmission Rates in Patients With COPD. Chest, 149, 905–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPIRA A, BEANE J, SHAH V, LIU G, SCHEMBRI F, YANG X, PALMA J & BRODY JS 2004. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proceedings of the National Academy of Sciences of the United States of America, 101, 10143–10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAN WC, LO C, JONG A, XING L, FITZGERALD MJ, VOLLMER WM, BUIST SA & SIN DD 2009. Marijuana and chronic obstructive lung disease: a population-based study. CMAJ, 180, 814–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG S, LI C, WANG W & XING C 2016. PBX3 promotes gastric cancer invasion and metastasis by inducing epithelial-mesenchymal transition. Oncol Lett, 12, 3485–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WYATT TA, BAILEY KL, SIMET SM, WARREN KJ, SWEETER JM, DEVASURE JM, PAVLIK JA & SISSON JH 2018. Alcohol potentiates RSV-mediated injury to ciliated airway epithelium. Alcohol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WYATT TA, SISSON JH, ALLEN-GIPSON DS, MCCASKILL ML, BOTEN JA, DEVASURE JM, BAILEY KL & POOLE JA 2012. Co-exposure to cigarette smoke and alcohol decreases airway epithelial cell cilia beating in a protein kinase Cepsilon-dependent manner. Am J Pathol, 181, 431–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAM AY, XIA Y, LIN HT, BURLINGAME A, GERSTEIN M & FRYDMAN J 2008. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol, 15, 1255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG F & LUO J 2015. Endoplasmic Reticulum Stress and Ethanol Neurotoxicity. Biomolecules, 5, 2538–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG H, WANG H, CZURA CJ & TRACEY KJ 2005. The cytokine activity of HMGB1. J Leukoc Biol, 78, 1–8. [DOI] [PubMed] [Google Scholar]
- YU G, WANG LG, HAN Y & HE QY 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics, 16, 284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Y, FENG J, CUI J, YANG G & ZHU X 2018. Pre-B cell leukemia transcription factor 3 induces inflammatory responses in human umbilical vein endothelial cells and murine sepsis via acting a competing endogenous RNA for high mobility group box 1 protein. Mol Med Rep, 17, 5805–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Z, HUANG A, ZHANG A & ZHOU C 2017. HuR promotes breast cancer cell proliferation and survival via binding to CDK3 mRNA. Biomed Pharmacother, 91, 788–795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.