

Abstract
Molecular variant interpretation lacks disease gene-specific cohorts for determining variant enrichment in disease versus healthy populations. To address the molecular etiology of retinal degeneration, specifically the PRPH2-related retinopathies, we reviewed genotype and phenotype information obtained from 187 eyeGENE® participants from 161 families. Clinical details were provided by referring clinicians participating in the eyeGENE® Network. The cohort was sequenced for variants in PRPH2. Variant cDNA clusters and cohort frequency were compared to variants in public databases to help determine pathogenicity by current ACMG/AMP interpretation criteria. The most frequent variant was c.828+3A>T, which affected 28 families (17.4%), and 25 of 79 (31.64%) variants were novel. The majority of missense variants clustered in the D2 intracellular loop of the Peripherin-2 protein, constituting a hotspot. Disease-enrichment was noted for 23 (29.1%) of the variants. Hotspot and disease-enrichment evidence modified variant classification for 16.5% of variants. The missense allele p.Arg172Trp was associated with a younger age of onset. To our knowledge, this is the largest patient cohort review of PRPH2-related retinopathy. Large disease gene-specific cohorts permit gene modeling for hotspot and disease enrichment analysis, providing novel variant classification evidence, including for novel missense variants.
Keywords: eyeGENE, PRPH2, retinopathy, phenotype, genotype, clinical variant interpretation
Graphical Abstarct
INTRODUCTION
Peripherin-2 (NP_000313.2), encoded by the PRPH2 gene (MIM# 179605), is a member of the tetraspanin family, the largest family of transmembrane proteins expressed in mammals that is comprised of 33 proteins (Charrin, Jouannet, Boucheix, & Rubinstein, 2014; Hemler, 2014). Each protein has four transmembrane domains and an intracellular domain (Charrin et al., 2014; Zimmerman et al., 2016). Peripherin-2 localizes to the rims of cone lamellae and the rod disk of the photoreceptor outer segment in the adult retina (Chakraborty, Conley, Fliesler, & Naash, 2010; Conley, Stricker, & Naash, 2010; Loewen & Molday, 2000; Poetsch, Molday, & Molday, 2001; Tam, Moritz, & Papermaster, 2004). Peripherin-2 forms intramolecular disulfide bonds at seven highly conserved cysteine residues within the D2 loop, forming covalently bonded homomultimeric complexes (Boon et al., 2008; Conley et al., 2010; Ding, Stricker, & Naash, 2005; Duncan et al., 2011; Loewen & Molday, 2000; van Lith-Verhoeven et al., 2003). These molecular interactions are essential to preserve structure and function of photoreceptor outer segments (Boon et al., 2008; Chakraborty, Conley, Fliesler, et al., 2010; Chakraborty, Conley, Stuck, & Naash, 2010; Ding et al., 2004; Ding et al., 2005; Duncan et al., 2011; Loewen & Molday, 2000; Tam et al., 2004; van Lith-Verhoeven et al., 2003).
Disease-causing variants in the PRPH2 gene, formerly known as RDS, are associated with several clinical phenotypes including pattern dystrophy (PD), cone rod dystrophy (CRD), Stargardt disease (SD), retinitis pigmentosa (RP), and others (Boon et al., 2008). Pathogenic variants in PRPH2 have previously been detected in 11% of patients with autosomal dominant cone dystrophy (CD) or CRD, and in 8–9% of cases of autosomal dominant RP (Daiger et al., 2008; Kohl et al., 2012). It is estimated that 60–80% of normal Peripherin-2 protein must be present for normal outer segment scaffolding, and it has been hypothesized that earlier onset and a more severe presentation are related to lower protein levels (Kedzierski et al., 2001; Nour, Ding, Stricker, Fliesler, & Naash, 2004; Stuck, Conley, & Naash, 2016). In mice, loss of Prph2 results in the retinal degeneration slow (rds) mouse, which exhibits rod and cone degeneration inherited in a semi-dominant manner (Ma et al., 1995). Misexpression of the human-specific mutation p.Arg172Trp results in rescue of rod but not cone phenotypes, suggesting that multiple mutation-specific mechanisms exist (Ding et al., 2004). Thus far, human genotype-phenotype correlations are poorly understood, in part because of the limited sizes of patient cohorts with PRPH2-associated diseases that have been reported.
Investigations of disease-gene associations have informed treatment plans to slow degeneration and restore vision in clinical trials, yet large cohorts of patient and genetic data for rare retinal diseases are scarce (Cideciyan et al., 2019; Lorenz, Preising, & Stieger, 2010; McIntyre, Williams, & Martens, 2013; Zaneveld, Wang, Wang, & Chen, 2013). Further, variant interpretation evidence is limited to meta-analysis of published evidence and public datasets. Disease population datasets allow for demonstrating variant enrichment in affected individuals and within protein functional domains. Here, we investigated genotype-phenotype relationships among 189 participants with PRPH2-related retinopathy in the National Ophthalmic Genotyping and Phenotyping Network (eyeGENE®), a multicenter genomic medicine initiative of the National Eye Institute, National Institutes of Health (NIH). This permitted a systematic analysis of the contribution of regional protein and patient population enrichment of PRPH2 disease variation by a current variant classification system (Richards et al., 2015).
MATERIALS AND METHODS
Consent, sample collection, and testing
eyeGENE® comprises a registry and DNA repository for individuals with rare inherited ophthalmic diseases and their family members (Blain, Goetz, Ayyagari, & Tumminia, 2013; Brooks et al., 2008; Goetz, Reeves, Tumminia, & Brooks, 2012; Sieving & Collins, 2007). All participants in eyeGENE® were consented to participate in the program through an informed consent process approved by the NIH Combined NeuroScience Institutional Review Board (IRB), commercial IRBs, or the collaborating institution’s IRB, and all research adhered to the tenets of the Declaration of Helsinki. Clinical diagnosis and phenotypic data were reported by the referring clinicians. A blood sample was obtained from each participant and shipped to the eyeGENE® Coordinating Center at the NIH main campus in Bethesda, Maryland. Participant DNA samples were screened for genes associated with their submitted clinically diagnosed ophthalmic disease. DNA from whole blood was extracted using either manual or automated (Autopure LS) methods using the Gentra PureGene chemistry (Qiagen, USA). A de-identified DNA aliquot for each participant was shipped to an eyeGENE® Network clinical laboratory and screened through Sanger or Next generation sequencing. Sanger sequencing included the three coding exons (exons 1–3) and the flanking intronic regions of the PRPH2 gene (RefSeq NM_000322.4) (Sohocki et al., 2001). Next generation sequencing was performed, followed by sequence analysis (Tang, Shi, Li, & Lu, 2008). Targeted Sanger sequencing was performed on affected family members when available. Though gene panel testing became available after eyeGENE® was launched, most participants described in this cohort were screened through single gene testing. Phenotype and genotype characteristics were reviewed to improve variant pathogenicity interpretation and determine if correlations were present. Data are available through controlled access in the NEI Data Commons (“NEI BRICS,” 2018).
Variant classification
Variant pathogenicity was reported by the CLIA-approved clinical diagnostic laboratories at issuance of the molecular diagnostic report. For this study, variants were re-classified as benign, likely benign, variant of unknown significance (VOUS), likely pathogenic, and pathogenic according to the 2015 ACMG/AMP guidelines (Richards et al., 2015). In silico and conservation score predictions were obtained from an amalgam of tools available through Varsome (https://varsome.com/) and Franklin (https://franklin.genoox.com/). Healthy population frequency data were obtained from gnomAD (v2.1) (“gnomAD browser, PRPH2,” 2018). Variant classification in case of conflicting evidence was cross validated on a Bayesian classification framework (Tavtigian et al., 2018). All indel variants in gnomAD were visually inspected on the Integrative Genomics Viewer (IGV) and four indels (c.1013_1014insGG, c.1012_1013insGCGC, c.910_911insG, c.909_910insGAGAG) were considered to be likely variant-calling artifacts and were discarded from the analysis. ClinVar data were downloaded on Sept. 03, 2018.
Statistics
Statistical analysis was performed using the R system for statistical computing (v. 3.5.1) or IBM® SPSS® Statistics (v. 1.0.0.1298) (“IBM SPSS Software,” 2019; “The R Project for Statistical Computing,” 2018).
RESULTS
PRPH2-related retinopathy cohort characteristics
Through the eyeGENE® program, 1,242 participants were screened for variants in the PRPH2 gene. After removing participant samples with variants which were classified as benign or likely benign in ClinVar, 187 participants from 161 families were included in this study. Those variants classified as benign or likely benign variants were either those with gnomAD allele frequencies over 0.50 or rare variants that are synonymous, intronic, or in the UTR.
The 187 participants harbored 79 reported or suspected PRPH2 disease-associated variants by clinical testing. Variants were analyzed by clinician-provided information, including clinical diagnosis, familial inheritance pattern, sex, electroretinography status (normal/abnormal), and visual acuity (Supp. Tables S1 & S2). For clinical diagnosis indications, 87 (46.5%) individuals were submitted as SD, 45 (24.1%) as PD, 26 (13.9%) as RP, 18 (9.6%) as CRD, and 11 (5.9%) as “Other” [9 Best disease (BD), 1 Choroideremia (CHM) and 1 Doyne honeycomb dystrophy (DHD)].
A similar number of males (91, 48.7%) and females (96, 51.3%) were included in the cohort. Age of onset range was 0 (at birth) to 85 years. Visual acuity range was −0.10 to +3.00 logMAR. Out of 187 participants, 87 had undergone a full field electroretinogram (ffERG) (46.5%). Nineteen (21.8%) were reported to have ERG results within normal range, and 68 (78.2%) were reported abnormal for their age group. Supp. Tables S1 to S4 provide a comprehensive summary of provider-submitted data and associated PRPH2 genotypes, descriptive statistics for the analyzed cohort, PRPH2 variant summary, ERG data, and phenotype data summary, respectively.
Visual acuity (VA) measures were converted to VA Scores: Group 0 (20/12–20/25), Group 0.5 (20/32–20/63), Group 1 (20/80–20/160), Group 2 (20/200–20/400), Group 3 (20/500–20/1000), Group 4 (less than 20/1000), and Group 5 (no light perception, NLP) (Ophthalmology, 2002). VA score (or LogMAR) was compared between clinical diagnoses, which showed similar VA score range between clinical phenotypes, though SD showed the tightest range for average VA score (Supp. Figure S1). To note, most participants were documented to be emmetropic or hyperopic and were rarely myopic. Supp. Figure S2 shows the number of participants with a normal or abnormal ERG as designated by diagnosis category. Among the different diagnoses, provider-reported age of first symptoms (here defined as age of onset; AOO) and number of years with disease (YWD) between AAO and age at exam (AAE) were reviewed (Supp. Table S4). Regression analysis showed no significant association between ffERG normal/abnormal status and AAE (p = 0.304, R=0.111) or reported YWD (p = 0.101, R=0.177). The Pearson Chi-Square test for association was performed and showed a strong association (p = 0.008, Phi and Cramer’s V = 0.446), between ERG status and diagnosis categories.
Intrafamilial and variant-specific variability in several clinical presentations was observed, similar to previous reports (Daiger et al., 2008; Khani et al., 2003). Provider-submitted clinical diagnoses differed between family members in 8 of 15 families with two or more members enrolled in eyeGENE®. For example, 2 members in Family 4 were confirmed to carry the c.828+3A>T variant, the most commonly reported PRPH2-associated allele (Figures 1A – 1C) (Shankar et al., 2015). Participant 4.1 was enrolled at 62 years with a diagnosis of Choroideremia, and participant 4.2 was enrolled at 39 years with a diagnosis of Stargardt disease. Their fundus photos showed distinct phenotypic patterns associated with their respective diagnoses (Figure 1A & 1B). We also observed variability in the clinical diagnosis of unrelated participants with the same variant. Participants 44 (A), 39 (B), and 79 (C) all carry the c.612C>A p.Tyr204* variant (Fundus images in Supp. Figure S3). Participants 44 and 79 were diagnosed with PD and participant 39 was diagnosed with SD. The autofluorescence images in Supp. Figure S4 are from participants 59 (A), 61 (B), and 62 (C) who all carry the c.422A>G p.Tyr141Cys variant. Participants 59 and 62 were diagnosed with SD and participant 61 was diagnosed with PD. These examples highlight the phenotypic diversity seen in this cohort.
Figure 1. Variable Expressions within eyeGENE® Family 4 and Participant Diagnoses:

A: Participant 4.1 was enrolled under the preliminary diagnosis of Choroideremia at age 62 years. Age of onset was at 52 years. The participant’s visual acuity was 20/20 OU, with an abnormal, but recordable full-field ERG. B: Participant 4.2 was enrolled under the preliminary diagnosis of Stargardt disease at age 39 years. Age of onset was at 38 years. The participant’s visual acuity was 20/20 OD and 20/40 OS. An ERG was not performed. C: Pedigree of Family 4. Participant 4.3 was enrolled under the preliminary diagnosis of Stargardt disease but did not carry the familial PRPH2 variant c.828+3A>T. D: Percentage of participants by diagnosis category and variant exon location.
Genotype-phenotype correlates
Variant characteristics were assessed to explore potential genotype-phenotype correlations. Of the 79 variants, there were 47 (59.5%) previously published variants. Of the disease-associated variants, missense variants accounted for the highest prevalent variant type (50, 63.3%), and there were 24 (30.4%) nonsense or frameshift variants. The majority of CRD, RP, and SD participants had variants in exon 1 (Figure 1D, 77.8%, 50.0%, and 43.8% respectively), while those diagnosed with BD and PD had most of their variants located in exon 2 (Figure 1D, 55.6% and 51.1% respectively). Thirty families (20.3%) had an intronic variant, and most of those families (28, 18.9%) specifically had the intron 2, c.828+3A>T change that is expected to affect splicing of the mRNA and lead to an abnormal protein.(Shankar et al., 2015; Sohocki et al., 2001) The top 7 most recurrent variants were: (1) c.828+3A>T (28/161, 17.4%), (2) c.422A>G p.Tyr141Cys (10/161, 6.2%), (3) c.514C>T p.Arg172Trp (9/161, 5.6%), (4) c.629C>G p.Pro210Arg (7/161, 4.3%), (5) c.424C>T p.Arg142Trp (5/161, 3.1%), (6) c.715C>T p.Gln239* (4/161, 2.5%), and (7) c.612C>A p.Tyr204* (4/161, 2.5%). Table 1 shows the top 10 most common pathogenic or likely pathogenic PRPH2 variants, with 3 or more probands. Across the 28 families with the c.828+3A>T variant (36 affected participants), the reported age of onset ranged from age 12 to 64, with the average age of 44.1 years (median 40 years) (Figure 2A). Diagnoses included SD (22, 61.1%), PD (7, 19.4%), and RP (3, 8.3%). Four participants were diagnosed with BD, CRD, CHM, and DHD, respectively.
Table 1:
Most Common Pathogenic or Likely Pathogenic PRPH2 Variants (≥ 3 Probands)
| cDNA position* | Protein change | Number of probands | gnomAD frequency | References |
|---|---|---|---|---|
| c.828+3A>T | abnormal splicing | 28 | 0.000004288 | Sohocki 2001 Sears 2001 |
| c.389T>C | p.Leu130Pro | 3 | Not Present | Ba-Abbad 2014 |
| c.394delC | p.Gln132Lysfs*7 | 3 | 0.000004063 | Ba-Abbad 2014 |
| c.422A>G | p.Tyr141Cys | 10 | Not Present | Yang 2001, Khani 2003 |
| c.424C>T | p.Arg142Trp | 5 | 0.00001625 | Keilhauer 2006, Anand 2009 |
| c.514C>T | p.Arg172Trp | 9 | Not Present | Anand 2009, Duncan 2011 |
| c.612C>A | p.Tyr204* | 4 | Not Present | Birtel 2018 |
| c.629C>G | p.Pro210Arg | 7 | Not Present | Feist 1994, Duncan 2011 |
| c.647C>T | p.Pro216Leu | 3 | 0.000004061 | Kajiwara 1991, Van Cauwenbergh 2017 |
| c.715C>T | p.Gln239* | 4 | Not Present | Kohl 1997 |
RefSeq: NM_000322.4
Figure 2. Age of Disease Onset of eyeGENE® PRPH2 Variants:

A: Age of onsets were plotted against variant types. Median, 10%, 25%, 75%, 90% of quartiles were displayed in the box plots. The individual data points were also plotted. B: Age of onset for seven of the most recurrent eyeGENE® variants with cohort AF > 0.01. The lowest median age of onset was for the p.Arg172Trp variant at 26 years of age. The p.Arg142Trp variant had the highest median age of onset at 57 years of age.
We performed an in-depth analysis of disease categories, protein location of the variant and functionality, visual acuity score, electroretinogram, and age of onset. Reported AOO of symptoms was compared across variant types and recurrent variants (Figures 2A & 2B). The mean AOO were different but not statistically significant among missense/in-frame deletion (mean = 37.4, SD = 17.2), truncating (nonsense/frameshift) (mean = 42.4, SD = 11.0), or splice-altering alleles (mean = 40.5, SD = 12.4) (one-way ANOVA, p = 0.064). Interestingly, the inter-quartile AOO range in the truncating group (12 years) was significantly smaller than those of the other 2 groups (missense/in-frame deletion: 27 years, splice-altering: 17 years) as demonstrated by Levene’s test for homogeneity of variance (p = 0.00026). This may suggest a more consistent and restricted AOO for alleles leading to PRPH2 haploinsufficiency.
Next, we evaluated AOO among 7 recurrent alleles with allele frequencies in probands greater than 0.01. One-way ANOVA showed that the means of AOO among recurrent PRPH2 alleles were different (p = 0.0027). Welch 2 sample t-test showed that a lower AOO was associated with c.514C>T p.Arg172Trp (mean = 27.7 years, SD = 12.7 years, p = 0.0036) as compared to other variants (mean = 40.5 years, SD = 15.1 years). This trend holds true when considering the proband alone vs. others (AOO mean of c.514C>T: 25.6 years (SD = 14.7.): 40.4 years (SD = 15.0), p = 0.016, Welch two sample t-test). Table 2 lists the participants with the p.Arg172Trp change along with their phenotypic description. Notably, p.Arg172Trp has been associated with both earlier cone dysfunction in patients and a dominant negative mechanism in cone photoreceptors of mouse models (Conley, Stuck, et al., 2014; Ding et al., 2004; Gill, Georgiou, Kalitzeos, Moore, & Michaelides, 2019; Nakazawa, Wada, & Tamai, 1995). Thus, this observation of a distinctly earlier onset with p.Arg172Trp can be explained by cone dysfunction leading to earlier central vision loss and dominant negative mechanism impairing function of the normal PRPH2 allele.
Table 2:
Participants with p.Arg172Trp Variant
| Family code | Diagnosis | Age y | Age of onset y | BCVA logMAR OD | BCVA logMAR OS | ERG Response | Fundus Appearance |
|---|---|---|---|---|---|---|---|
| 12.1 | PD | 52 | 32 | +0.60 | +0.18 | N | OU: Depigmented lesion in fovea, central hypofluorescence surrounded by hyperfluorescence by FA, foveal atrophy, ‘questionable dark choroid’ by FA |
| 12.2 | PD | 26 | 26 | −0.12 | 0.00 | ND | OU: Depigmented lesion in fovea, Hyperpigmentation, Central hypofluorescence surrounded by hyperfluorescence by FA |
| 22 | BD | 56 | 20 | +1.30 | +1.30 | ND | OU: Scar/atrophy |
| 72 | CRD | 12 | 12 | −0.10 | 0.00 | ND | OU: Macular degeneration, granular macular pigmentation, Bull’s eye pattern w/hypopigmented parafovea, normal OCT thickness |
| 73.1 | CRD | 56 | 15 | +1.00 | +1.30 | A | OU: Macular degeneration, vascular attenuation |
| 73.2 | CRD | 60 | 36 | +0.30 | +0.30 | A | OU: Macular degeneration |
| 73.3 | CRD | 38 | 38 | +0.30 | +0.10 | A | OU: Macular degeneration, central choroidal show |
| 73.4 | CRD | 63 | 30 | +1.30 | +1.20 | ND | OU: Macular degeneration, vascular attenuation |
| 74 | PD | 56 | 55 | 0.00 | +0.10 | ND | OU: Depigmented lesion in fovea |
| 75 | RP | 46 | 24 | +2.00 | +2.00 | ND | OU: Retinal degeneration, intraretinal pigment spicules, macular degeneration, peripheral retinal degeneration, vascular attenuation |
| 127 | CRD | 49 | 41 | +0.30 | +0.18 | A | OU: Macular degeneration |
| 133 | RP | 58 | 20 | +0.70 | +3.00 | ND | OU: Retinal degeneration, intraretinal pigment spicules, vascular attenuation |
| 144 | SD | 15 | 11 | +0.48 | +1.00 | ND | OU: Macular degeneration, vascular attenuation |
Age at exam in years (y); Reported age of onset in years (y); PD-Pattern dystrophy, BD-Best disease, CRD-Cone rod dystrophy, RP-Retinitis pigmentosa, SD-Stargardt disease; Full-field ERG response N-Normal, A-Abnormal, ND-Not done
PRPH2 gene- and population-level analyses
To investigate whether other variant characteristics might inform phenotypic expression of PRPH2-related retinopathy, we analyzed variants in our disease cohort and the general population (gnomAD v2.1) to compare relative allele frequency and location within the cDNA and protein position. In the gnomAD database, there is no sequence variant present that has an allele frequency (AF) between 0.002 and 0.5 in the PRPH2 coding region and canonical splicing sites; however, there are numerous variants (92 synonymous and 190 missense) with AF less than 0.002 that spread throughout the whole coding region (Figure 3A). As mentioned above, the most recurrent variant in the PRPH2 retinopathy cohort was the splice site variant c.828+3A>T (28 of 161 probands). When examining the coding region only, variants in the PRPH2 patient cohort appeared to be markedly enriched at cDNA positions c.421 – 720 (49 unique nonsynonymous variants found in 91 probands within 300 base pairs). In contrast, there were only 26 unique nonsynonymous variants found in 40 probands within the 741 base pairs outside of c.421 – 720. Thus, 69% or 5.6-fold more disease-associated variants were located within the c.421–721 coding region than the rest of the coding region after normalizing length (Figure 3A and Supp. Table S3). To facilitate region-specific comparisons of our PRPH2 retinopathy cohort to the gnomAD dataset, we introduced sub-gene length-adjusted adj∑AF for each region of interest, which was defined as the sum of AF normalized to 1000 bp of DNA sequence (i.e. ∑AF/DNA size*1000). It is noteworthy that the adj∑AFs of nonsynonymous variants in the PRPH2 patient cohort are expected to be much higher than those in the gnomAD, because the PRPH2 patient cohort was selected based on PRPH2 sequencing. Because protein domain is the basic unit of protein function, we postulated that the c.421 – 720 (a.a. 141 – 240) could correspond to a protein domain(s) critical to PRPH2 function. Indeed, c.421–720 constitutes 3/4 of c.369–762, which encodes the 132-residue D2 intracellular loop (a.a. 123–254, Figure 3B). Thus, we attempted to relax the genetically defined c.421–720 region to the whole region encoding the D2 loop for adj∑AF analysis. The adj∑AF of nonsynonymous variants for the D2 loop in the PRPH2 patient cohort was 0.80, a 5.7-fold increase as compared to the region outside of D2 (0.14). The adj∑AF of nonsynonymous variants for the D2 loop and that for outside of the D2 loop in gnomAD was 0.0053 and 0.0093, respectively, suggesting that the D2 loop is intolerant to variation. Further, when looking at the missense variants only, the 100 a.a. (a.a. 141 – 240) in the D2 loop includes 27 residues altered by one or more missense mutation(s) in PRPH2 cohort, while there were only eleven residues with disease-associated missense alleles in the remaining 246 residues. Thus, this segment of the D2 loop represents a hotspot of pathogenic variation.
Figure 3. Schematic Representations of Peripherin-2 Protein and cDNA Positions of Variants and Allele Frequencies in eyeGENE® Probands and gnomAD:

A: Top, PRPH2 gnomAD allele frequencies by cDNA position and variant type, only variants with gnomAD allele frequencies less than 0.5 are shown. Middle, cDNA variant location and allele frequencies in eyeGENE® probands. Bottom, cDNA variant location and number of submitters in ClinVar. B: Schematic representation of Peripherin-2 protein with the distribution of variants found in eyeGENE® participants by color. D1 and D2 represent the two intradiskal loops. The N and C termini are located in the space between the disk membrane and the rod outer segment plasma membrane, with the fusion peptide region from a.a. 312 to 326. Missense variants are indicated in red, truncating variants in blue, in-frame deletions in orange, and sites with both missense and truncating variants in pink. Figure created and modified from Protter (Omasits, Ahrens, Muller, & Wollscheid, 2014).
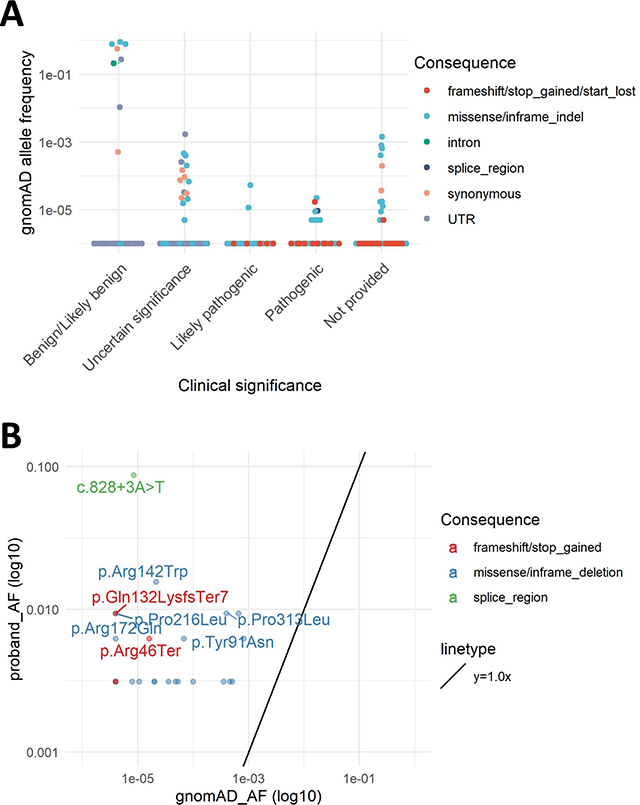
As expected, a significant number of variants were present in our cohort, ClinVar, and gnomAD. Twenty-nine variants were classified as pathogenic in ClinVar, 8 of which were found in gnomAD, with allele frequency range of 4.0e-6 to 2.1e-5. Similarly, 17 variants were classified as likely pathogenic in ClinVar, 2 of which were also found in gnomAD: c.425G>A p.Arg142Gln and c.623G>A p.Gly208Asp. The AFs in gnomAD are 1.1e-05 and 5.3e-05, respectively (Figure 4A). The highest AF in gnomAD for these pathogenic or likely pathogenic variants is 5.3e-05 (c.623G>A p.Gly208Asp). The sum carrier frequency for these pathogenic or likely pathogenic ClinVar variants in gnomAD is approximately 1:3758, which, given an average age of onset of approximately 40 years of age across the cohort (Supp. Table S1), possibly represent presymptomatic adults in the gnomAD general population cohort.
Figure 4. ACMG Classification of Variants, ClinVar Concordance, Fisher’s Exact Test of gnomAD Allele Frequency versus Proband Allele Frequency, and eyeGENE® Proband Allele Frequencies versus gnomAD Allele Frequencies:

A: gnomAD allele frequency of ClinVar variants according to ClinVar classification. There were 29 variants classified as pathogenic in ClinVar, of which 2 were also found in gnomAD: c.425G>A p.Arg142Gln and c.623G>A p.Gly208Asp. B: eyeGENE® proband allele frequency and gnomAD allele frequency for the common variants between these two datasets. The variants with p value
< 0.00063 in the Fisher’s Exact test were annotated. C: PM1 and PS4 help determination of pathogenicity by ACMG classification criteria. D: Concordance of classifications between ClinVar and this study.
A hallmark of variant pathogenicity is enrichment in disease populations compared to the general population. The large number of probands in this study provided the first PRPH2-related disease cohort to calculate disease-specific allele frequencies. To further evaluate variant pathogenicity, each variant was tested for significant enrichment in the PRPH2 cohort as compared to variants found in gnomAD using the Fisher’s exact test (Figure 4B and Supp. Table S3). A Bonferroni-corrected p-value of 0.00063 (0.05/79 variants, two variants were in cis) was used as the cut-off to determine significant enrichment. Of the 79 variants, 29.1% were statistically enriched in the PRPH2 cohort.
Finally, we performed a systematic variant classification of the PRPH2 eyeGENE® cohort utilizing hotspot and disease population enrichment using current clinical variant interpretation guidelines (Richards et al., 2015). A stepwise inclusion of PM1 and PS4 in baseline ACMG evidence (all evidence except PM1 and PS4) showed a substantial elevation in number of pathogenic/likely pathogenic variants (51.9 % to 68.4%), whereas a decrease in variants of uncertain significance from 48.1 % to 31.6 % (Figure 4C). Addition of hotspot (PM1) and disease status enrichment (PS4) each reduced the burden of variants of uncertain significance, and together shifted 16.5% (13 of 79) variants from uncertain significance to pathogenic or likely pathogenic (Figure 4C). Notably, improvement in classification certainty included 4 of 25 (16%) of novel missense variants. Forty-five (27 pathogenic/likely pathogenic and 18 variants of uncertain significance) of the 79 variants in our cohort were not reported in ClinVar (Figure 4D).
Variant interpretations from our PRPH2 cohort modeling of protein hotspot and disease population enrichment were compared to current clinical variant interpretations. The ClinVar database is an established depot of expert-curated human variation classification with clinical significance and phenotype relationship (Landrum & Kattman, 2018). Of the 79 variants in our cohort, 34 are present in ClinVar at the time of manuscript preparation. Variant interpretation was not provided for 8 variants present in ClinVar, and 45 variants were not listed previously in ClinVar.
DISCUSSION
To our knowledge, this is the largest patient cohort studied with molecularly confirmed PRPH2-related eye disease. Though marked phenotypic diversity is exhibited (Supp. Figures S3 & S4), we identified novel genotype-phenotype associations with age of onset, including a significantly narrow range for truncating mutations, and an earlier onset for patients with p.Arg172Trp compared to other alleles. This cohort represents a wide range of ages of onset, clinical diagnoses, and intrafamilial variability. No association was found between clinical diagnosis and years with disease or visual outcome as determined by ANOVA. There was an association found between clinical diagnosis and electroretinography findings.
Another benefit of this large cohort was our ability to evaluate variants on a disease-population level. We were able to assess variation of PRPH2 to add important evidence to variant classification, including the D2 loop as a hotspot, and expected frequency of recurrent causal PRPH2 variants. The information in our cohort such as co-segregation, mutation-hotspot domain, and significant enrichment in the disease cohort would also help provide additional criteria for variant entries already present in ClinVar. Overall, this cohort is a powerful tool allowing for disease-specific allele frequency calculations for the first time, and through comparison to publicly available general population frequencies, we are able to calculate for statistical enrichment of disease-association for each variant, and globally map disease location hotspots.
This study highlights the variability seen across provider-submitted information for patient phenotypes, and thus emphasizes the importance of consistent and structured data collection. The lack of uniformity across collected diagnosis elements led to a challenging review of results to link genotype and phenotype. For example, raw values for ERG components (A wave, B wave, etc.) were collected for some diagnoses while an inferential categorization of ERG results (normal, abnormal, not recordable or not done) was collected for others. Recognizing the effort required on the part of clinical staff enrolling participants, the original eyeGENE® questionnaires were primarily designed to be completed with minimal burden, which contributed to some of the variability in the data collected. In addition, family members may have been enrolled by different clinicians at different clinics, which may have confounded the disparity between diagnoses within a family. We were unable to review longitudinal data because only single visit data were collected. Variability in testing performed by multiple testing sites is another limitation of this study. Though testing was indicated based on participant symptoms, we cannot rule out alternate or additional causes of disease phenotype, which could also explicate the variability in phenotypic expression.
While it is difficult to do a comparison and trend analysis in a gene that shows such phenotypic heterogeneity, even within the same family, there are some generalizations that may be made for these patients as seen in this cohort. Overall, the average reported age of onset is 40 years of age. However, as age of onset is related to visual acuity loss based on disease involvement of the fovea, the concept may be interpreted inconsistently and may account for some of the early ages that were reported (‘childhood’, ages 5, 7, 10, or 12), or there may be other unknown factors involved that account for the earlier than expected onset. Surprisingly, the majority of participants were emmetropic or hyperopic, but were rarely myopic.
Like prior studies, the phenotype-genotype correlations in this cohort are also difficult to determine (Khani et al., 2003; Meunier et al., 2011; Roosing et al., 2014; van Lith-Verhoeven et al., 2003; Yang et al., 2003). Shankar et al. (2015) reviewed the c.828+3A>T variant and found that 97 patients of 19 different families had diagnoses of RP, macular dystrophy, and PD. The c.828+3A>T variant was classified as a founder mutation, so as one might expect, this variant was at the highest frequency in this cohort (Shankar et al., 2015). Within the families with c.422A>G p.Tyr141Cys (14 affected patients), the symptoms and diagnoses included SD, PD, and CRD (Khani et al., 2003). Though lipofuscin deposits or “flecks” are commonly seen with ABCA4-related Stargardt disease, flecks were noted in 53 out of 78 PRPH2-positive Stargardt cases (67.9%) (Duncker et al., 2015). As seen by this cohort, molecular confirmation can help to confirm the initial clinical diagnosis.
In this study, we systematically included moderate level ACMG evidence for pathogenicity (PM1: Located in a mutational hot spot and/or critical and well-established functional domain (e.g. active site of an enzyme) without benign variation) and strong ACMG evidence (PS4: The prevalence of the variant in affected individuals is significantly increased compared to the prevalence in controls) (“NEI BRICS,” 2018). A stepwise inclusion of PM1 and PS4 in baseline ACMG evidence (all evidence except PM1 and PS4) showed a substantial elevation in number of pathogenic/likely pathogenic variants (51.9 % to 68.4%), whereas a decrease in variants of uncertain significance from 48.1 % to 31.6 %. (Figure 4C). This analysis showed concordance between ClinVar and our variant analysis for 22 pathogenic/likely pathogenic and 4 variants of uncertain significance which validates our approach. Our analyses also classified 5 variants as pathogenic/likely pathogenic and 3 as variants of uncertain significance for which ACMG class was not provided in ClinVar. Reported variants in this study will greatly expand the ClinVar PRPH2 entries as 45 (27 pathogenic/likely pathogenic and 18 variants of uncertain significance) of the 79 variants in our cohort were not reported in the ClinVar (Figure 4D).
Most variants were found in the D2 loop of the protein, which is critical for protein-protein interactions. This loop appears less tolerant to variation compared to the cytoplasmic tail of Peripherin-2. If the mutation occurs at or near one of the conserved cysteine residues, it may interfere with disulfide bond formation between molecules, which would then cause improper protein folding and assembly within the outer segment disks (Boon et al., 2008). The C-terminal domain has been shown to contain an amphiphatic helix which is thought to have a direct role in creating the high curvature bend of the photoreceptor disk rim (Charrin et al., 2014). Additionally, the C-terminus is required for initiation of outer segment morphogenesis and disc formation, while outer segment maturation requires full length Peripherin-2 protein structure (Conley, Al-Ubaidi, Han, & Naash, 2014). The requirement for the C-terminus in pathogenesis of disease, and the implication of the apparent tolerance to mutation remains to be seen. Other potential factors that may affect phenotypic variability include levels of mutant versus normal allele gene expression, gene to gene interactions, cumulative effects of variation in other genes, and environmental effects (Boon et al., 2008; Khani et al., 2003; Poloschek et al., 2010; Tam et al., 2004). Having large patient cohorts in which to study these multiple factors, along with their individual phenotypes and assorted variants, would be useful to better characterize the highly complex Peripherin-2 protein.
Conclusion
Peripherin-2 forms oligomeric complexes with other proteins, including the rod outer segment membrane protein-1 (ROM-1) and LRRC32/GARP (leucine-rich repeat-containing protein-32) expressed in rods, and is critical to the proper formation of the outer segment (Boon et al., 2008; Chakraborty, Conley, Fliesler, et al., 2010; Chakraborty, Conley, Stuck, et al., 2010; Conley et al., 2010; Ding et al., 2004; Ding et al., 2005; Duncan et al., 2011; Loewen & Molday, 2000; Poetsch et al., 2001; Poloschek et al., 2010; Tam et al., 2004; van Lith-Verhoeven et al., 2003). The ability to review phenotype-genotype associations within a single rare disorder is advantageous for understanding phenotypic range and properly categorizing causal genetic variation. Though it is still difficult to determine whether phenotypic trends exist for the Peripherin-2 protein due to variable phenotypic expression, variant- and gene-level information is improved on cohort analysis, and this model can be applied to other disease genes. Continued review of reported variants is essential to understanding the etiology of disease as we relate genomic variations with phenotypic outcome. Functional studies will continue to be important in determining the biochemical relationships between Peripherin-2 and other proteins and their effects on phenotypes and disease progression.
Supplementary Material
SUPP. TABLE S1: Variant Table w/Phenotypic Summary
SUPP. TABLE S3: Variant Summary Table
SUPP. FIGURE S1: Visual Acuity Scoring and Diagnosis
eyeGENE® Participants Average Visual Acuity Score: Individual participants (indicated by dot) were plotted against average visual acuity score (OD/OS) and color-coded by diagnosis category (Red = Cone rod dystrophy, Gold = Other, Green = Pattern dystrophy, Blue = Retinitis pigmentosa, Purple = Stargardt disease). Figure shows similar visual acuity score range between clinical phenotypes.
SUPP. FIGURE S2: eyeGENE® Participant Breakdown of ERG Status and Diagnosis Category
eyeGENE® Participant Breakdown of ERG Status and Diagnosis Category: Chart showing the number of participants with a normal or abnormal ERG as designated by diagnosis category.
SUPP. FIGURE S3: PRPH2-positive Fundus Images of Participants 44 (A), 39 (B), and 79 (C)
These fundus images are from participants 44 (A), 39 (B), and 79 (C) who all carry the c.612C>A p.Tyr204* variant. Participants 44 and 79 were diagnosed with Pattern dystrophy and participant 39 was diagnosed with Stargardt disease.
SUPP. FIGURE S4: PRPH2-positive Autofluorescence Images of Participants 59 (A), 61 (B), and 62 (C)
These autofluorescence images are from participants 59 (A), 61 (B), and 62 (C) who all carry the c.422A>G p.Tyr141Cys variant. Participants 59 and 62 were diagnosed with Stargardt disease and participant 61 was diagnosed with Pattern dystrophy.
SUPP. TABLE S2: Descriptive Statistics Table
SUPP. TABLE S4: ERG Data with Diagnosis, Years with Disease, and Age at Exam
ACKNOWLEDGEMENTS
The authors would like to thank the eyeGENE® Network participants, clinicians, and eyeGENE® Working Group for their valuable contributions to this research.
Funding Information: The DNA samples used for the analyses described in this study were obtained from the National Eye Institute – National Ophthalmic Genotyping and Phenotyping Network (ClinicalTrials.gov Identifier NCT00378742), which has been funded in part from the National Institutes of Health/National Eye Institute Intramural Research Program and under Contract No. HHS-N-260-2007-00001-C.
Footnotes
Disclosure: None.
DATA AVAILABILITY STATEMENT
Data are available upon request and have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
REFERENCES
- Blain D, Goetz KE, Ayyagari R, & Tumminia SJ (2013). eyeGENE(R): a vision community resource facilitating patient care and paving the path for research through molecular diagnostic testing. Clin Genet, 84(2), 190–197. doi: 10.1111/cge.12193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon CJ, den Hollander AI, Hoyng CB, Cremers FP, Klevering BJ, & Keunen JE (2008). The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog Retin Eye Res, 27(2), 213–235. doi: 10.1016/j.preteyeres.2008.01.002 [DOI] [PubMed] [Google Scholar]
- Brooks BP, Macdonald IM, Tumminia SJ, Smaoui N, Blain D, Nezhuvingal AA, … National Ophthalmic Disease Genotyping, N. (2008). Genomics in the era of molecular ophthalmology: reflections on the National Ophthalmic Disease Genotyping Network (eyeGENE). Arch Ophthalmol, 126(3), 424–425. doi: 10.1001/archopht.126.3.424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty D, Conley SM, Fliesler SJ, & Naash MI (2010). The function of oligomerization-incompetent RDS in rods. Adv Exp Med Biol, 664, 39–46. doi: 10.1007/978-1-4419-1399-9_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty D, Conley SM, Stuck MW, & Naash MI (2010). Differences in RDS trafficking, assembly and function in cones versus rods: insights from studies of C150S-RDS. Hum Mol Genet, 19(24), 4799–4812. doi: 10.1093/hmg/ddq410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrin S, Jouannet S, Boucheix C, & Rubinstein E (2014). Tetraspanins at a glance. J Cell Sci, 127(Pt 17), 3641–3648. doi: 10.1242/jcs.154906 [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, … Russell SR (2019). Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med, 25(2), 225–228. doi: 10.1038/s41591-018-0295-0 [DOI] [PubMed] [Google Scholar]
- Conley SM, Al-Ubaidi MR, Han Z, & Naash MI (2014). Rim formation is not a prerequisite for distribution of cone photoreceptor outer segment proteins. FASEB J, 28(8), 3468–3479. doi: 10.1096/fj.14-251397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley SM, Stricker HM, & Naash MI (2010). Biochemical analysis of phenotypic diversity associated with mutations in codon 244 of the retinal degeneration slow gene. Biochemistry, 49(5), 905–911. doi: 10.1021/bi901622w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley SM, Stuck MW, Burnett JL, Chakraborty D, Azadi S, Fliesler SJ, & Naash MI (2014). Insights into the mechanisms of macular degeneration associated with the R172W mutation in RDS. Hum Mol Genet, 23(12), 3102–3114. doi: 10.1093/hmg/ddu014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiger SP, Sullivan LS, Gire AI, Birch DG, Heckenlively JR, & Bowne SJ (2008). Mutations in known genes account for 58% of autosomal dominant retinitis pigmentosa (adRP). Adv Exp Med Biol, 613, 203–209. doi: 10.1007/978-0-387-74904-4_23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding XQ, Nour M, Ritter LM, Goldberg AF, Fliesler SJ, & Naash MI (2004). The R172W mutation in peripherin/rds causes a cone-rod dystrophy in transgenic mice. Hum Mol Genet, 13(18), 2075–2087. doi: 10.1093/hmg/ddh211 [DOI] [PubMed] [Google Scholar]
- Ding XQ, Stricker HM, & Naash MI (2005). Role of the second intradiscal loop of peripherin/rds in homo and hetero associations. Biochemistry, 44(12), 4897–4904. doi: 10.1021/bi048414i [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JL, Talcott KE, Ratnam K, Sundquist SM, Lucero AS, Day S, … Roorda A (2011). Cone structure in retinal degeneration associated with mutations in the peripherin/RDS gene. Invest Ophthalmol Vis Sci, 52(3), 1557–1566. doi: 10.1167/iovs.10-6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncker T, Tsang SH, Woods RL, Lee W, Zernant J, Allikmets R, … Sparrow JR (2015). Quantitative Fundus Autofluorescence and Optical Coherence Tomography in PRPH2/RDS- and ABCA4-Associated Disease Exhibiting Phenotypic Overlap. Invest Ophthalmol Vis Sci, 56(5), 3159–3170. doi: 10.1167/iovs.14-16343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill JS, Georgiou M, Kalitzeos A, Moore AT, & Michaelides M (2019). Progressive cone and cone-rod dystrophies: clinical features, molecular genetics and prospects for therapy. Br J Ophthalmol. doi: 10.1136/bjophthalmol-2018-313278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- gnomAD browser, PRPH2. (2018). Retrieved from http://gnomad.broadinstitute.org/gene/ENSG00000112619
- Goetz KE, Reeves MJ, Tumminia SJ, & Brooks BP (2012). eyeGENE(R): a novel approach to combine clinical testing and researching genetic ocular disease. Curr Opin Ophthalmol, 23(5), 355–363. doi: 10.1097/ICU.0b013e32835715c9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemler ME (2014). Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer, 14(1), 49–60. [DOI] [PubMed] [Google Scholar]
- IBM SPSS Software. (2019). Retrieved from https://www.ibm.com/analytics/spss-statistics-software
- Kedzierski W, Nusinowitz S, Birch D, Clarke G, McInnes RR, Bok D, & Travis GH (2001). Deficiency of rds/peripherin causes photoreceptor death in mouse models of digenic and dominant retinitis pigmentosa. Proc Natl Acad Sci U S A, 98(14), 7718–7723. doi: 10.1073/pnas.141124198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khani SC, Karoukis AJ, Young JE, Ambasudhan R, Burch T, Stockton R, … Ayyagari R (2003). Late-onset autosomal dominant macular dystrophy with choroidal neovascularization and nonexudative maculopathy associated with mutation in the RDS gene. Invest Ophthalmol Vis Sci, 44(8), 3570–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl S, Kitiratschky V, Papke M, Schaich S, Sauer A, & Wissinger B (2012). Genes and mutations in autosomal dominant cone and cone-rod dystrophy. Adv Exp Med Biol, 723, 337–343. doi: 10.1007/978-1-4614-0631-0_44 [DOI] [PubMed] [Google Scholar]
- Landrum MJ, & Kattman BL (2018). ClinVar at five years: Delivering on the promise. Hum Mutat, 39(11), 1623–1630. doi: 10.1002/humu.23641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewen CJ, & Molday RS (2000). Disulfide-mediated oligomerization of Peripherin/Rds and Rom-1 in photoreceptor disk membranes. Implications for photoreceptor outer segment morphogenesis and degeneration. J Biol Chem, 275(8), 5370–5378. [DOI] [PubMed] [Google Scholar]
- Lorenz B, Preising M, & Stieger K (2010). Retinal blinding disorders and gene therapy: implications for molecular and clinical aspects. Curr Gene Ther, 10, 350–370. [DOI] [PubMed] [Google Scholar]
- Ma J, Norton JC, Allen AC, Burns JB, Hasel KW, Burns JL, … Travis GH (1995). Retinal degeneration slow (rds) in mouse results from simple insertion of a t haplotype-specific element into protein-coding exon II. Genomics, 28(2), 212–219. doi: 10.1006/geno.1995.1133 [DOI] [PubMed] [Google Scholar]
- McIntyre JC, Williams CL, & Martens JR (2013). Smelling the roses and seeing the light: gene therapy for ciliopathies. Trends Biotechnol, 31(6), 355–363. doi: 10.1016/j.tibtech.2013.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier I, Senechal A, Dhaenens CM, Arndt C, Puech B, Defoort-Dhellemmes S, … Hamel CP (2011). Systematic screening of BEST1 and PRPH2 in juvenile and adult vitelliform macular dystrophies: a rationale for molecular analysis. Ophthalmology, 118(6), 1130–1136. doi: 10.1016/j.ophtha.2010.10.010 [DOI] [PubMed] [Google Scholar]
- Nakazawa M, Wada Y, & Tamai M (1995). Macular dystrophy associated with monogenic Arg172Trp mutation of the peripherin/RDS gene in a Japanese family. Retina, 15(6), 518–523. [DOI] [PubMed] [Google Scholar]
- NEI Data Commons. (2018). Retrieved from https://neidatacommons.nei.nih.gov/
- Nour M, Ding XQ, Stricker H, Fliesler SJ, & Naash MI (2004). Modulating expression of peripherin/rds in transgenic mice: critical levels and the effect of overexpression. Invest Ophthalmol Vis Sci, 45(8), 2514–2521. doi: 10.1167/iovs.04-0065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omasits U, Ahrens C, Muller S, & Wollscheid B (2014). Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics(30), 884–886. [DOI] [PubMed] [Google Scholar]
- Ophthalmology I. C. o. (2002). Visual Standards: Aspects and Ranges of Vision Loss with Emphasis on Population Surveys. 35.
- Poetsch A, Molday LL, & Molday RS (2001). The cGMP-gated channel and related glutamic acid-rich proteins interact with peripherin-2 at the rim region of rod photoreceptor disc membranes. J Biol Chem, 276(51), 48009–48016. doi: 10.1074/jbc.M108941200 [DOI] [PubMed] [Google Scholar]
- Poloschek CM, Bach M, Lagreze WA, Glaus E, Lemke JR, Berger W, & Neidhardt J (2010). ABCA4 and ROM1: implications for modification of the PRPH2-associated macular dystrophy phenotype. Invest Ophthalmol Vis Sci, 51(8), 4253–4265. doi: 10.1167/iovs.09-4655 [DOI] [PubMed] [Google Scholar]
- The R Project for Statistical Computing. (2018, 7/5/2019). Retrieved from https://www.r-project.org/
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Committee, A. L. Q. A. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 17(5), 405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosing S, Thiadens AA, Hoyng CB, Klaver CC, den Hollander AI, & Cremers FP (2014). Causes and consequences of inherited cone disorders. Prog Retin Eye Res, 42, 1–26. doi: 10.1016/j.preteyeres.2014.05.001 [DOI] [PubMed] [Google Scholar]
- Shankar SP, Birch DG, Ruiz RS, Hughbanks-Wheaton DK, Sullivan LS, Bowne SJ, … Daiger SP (2015). Founder Effect of a c.828+3A>T Splice Site Mutation in Peripherin 2 (PRPH2) Causing Autosomal Dominant Retinal Dystrophies. JAMA Ophthalmol, 133(5), 511–517. doi: 10.1001/jamaophthalmol.2014.6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieving PA, & Collins FS (2007). Genetic ophthalmology and the era of clinical care. JAMA, 297(7), 733–736. [DOI] [PubMed] [Google Scholar]
- Sohocki MM, Daiger SP, Bowne SJ, Rodriquez JA, Northrup H, Heckenlively JR, … Sullivan LS (2001). Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat, 17(1), 42–51. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuck MW, Conley SM, & Naash MI (2016). PRPH2/RDS and ROM-1: Historical context, current views and future considerations. Prog Retin Eye Res, 52, 47–63. doi: 10.1016/j.preteyeres.2015.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Moritz OL, & Papermaster DS (2004). The C terminus of peripherin/rds participates in rod outer segment targeting and alignment of disk incisures. Mol Biol Cell, 15(4), 2027–2037. doi: 10.1091/mbc.e03-09-0650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Shi X, Li X, & Lu Z (2008). DNA sequencing by synthesis with degenerate primers. J Genet Genomics, 35(9), 545–551. doi: 10.1016/S1673-8527(08)60074-0 [DOI] [PubMed] [Google Scholar]
- Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, … ClinGen Sequence Variant Interpretation Working, G. (2018). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med, 20(9), 1054–1060. doi: 10.1038/gim.2017.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lith-Verhoeven JJ, van den Helm B, Deutman AF, Bergen AA, Cremers FP, Hoyng CB, & de Jong PT (2003). A peculiar autosomal dominant macular dystrophy caused by an asparagine deletion at codon 169 in the peripherin/RDS gene. Arch Ophthalmol, 121(10), 1452–1457. doi: 10.1001/archopht.121.10.1452 [DOI] [PubMed] [Google Scholar]
- Yang Z, Lin W, Moshfeghi DM, Thirumalaichary S, Li X, Jiang L, … Zhang K (2003). A novel mutation in the RDS/Peripherin gene causes adult-onset foveomacular dystrophy. Am J Ophthalmol, 135(2), 213–218. [DOI] [PubMed] [Google Scholar]
- Zaneveld J, Wang F, Wang X, & Chen R (2013). Dawn of ocular gene therapy: implications for molecular diagnosis in retinal disease. Sci China Life Sci, 56(2), 125–133. doi: 10.1007/s11427-013-4443-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman B, Kelly B, McMillan BJ, Seegar TCM, Dror RO, Kruse AC, & Blacklow SC (2016). Crystal Structure of a Full-Length Human Tetraspanin Reveals a Cholesterol-Binding Pocket. Cell, 167(4), 1041–1051 e1011. doi: 10.1016/j.cell.2016.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPP. TABLE S1: Variant Table w/Phenotypic Summary
SUPP. TABLE S3: Variant Summary Table
SUPP. FIGURE S1: Visual Acuity Scoring and Diagnosis
eyeGENE® Participants Average Visual Acuity Score: Individual participants (indicated by dot) were plotted against average visual acuity score (OD/OS) and color-coded by diagnosis category (Red = Cone rod dystrophy, Gold = Other, Green = Pattern dystrophy, Blue = Retinitis pigmentosa, Purple = Stargardt disease). Figure shows similar visual acuity score range between clinical phenotypes.
SUPP. FIGURE S2: eyeGENE® Participant Breakdown of ERG Status and Diagnosis Category
eyeGENE® Participant Breakdown of ERG Status and Diagnosis Category: Chart showing the number of participants with a normal or abnormal ERG as designated by diagnosis category.
SUPP. FIGURE S3: PRPH2-positive Fundus Images of Participants 44 (A), 39 (B), and 79 (C)
These fundus images are from participants 44 (A), 39 (B), and 79 (C) who all carry the c.612C>A p.Tyr204* variant. Participants 44 and 79 were diagnosed with Pattern dystrophy and participant 39 was diagnosed with Stargardt disease.
SUPP. FIGURE S4: PRPH2-positive Autofluorescence Images of Participants 59 (A), 61 (B), and 62 (C)
These autofluorescence images are from participants 59 (A), 61 (B), and 62 (C) who all carry the c.422A>G p.Tyr141Cys variant. Participants 59 and 62 were diagnosed with Stargardt disease and participant 61 was diagnosed with Pattern dystrophy.
SUPP. TABLE S2: Descriptive Statistics Table
SUPP. TABLE S4: ERG Data with Diagnosis, Years with Disease, and Age at Exam