

Abstract
Coronavirus pandemic has caused a vast number of deaths worldwide. Thus creating an urgent need to develop effective counteragents against novel coronavirus disease (COVID-19). Many antiviral drugs have been repurposed for treatment but implicated minimal recovery, which further advanced the need for clearer insights and innovation to derive effective therapeutics. Strategically, Noscapine, an approved antitussive drug with positive effects on lung linings may show favorable outcomes synergistically with antiviral drugs in trials. Hence, we have theoretically examined the combinatorial drug therapy by culminating the existing experimental results with in silico analyses. We employed the antitussive noscapine in conjugation with antiviral drugs (Chloroquine, Umifenovir, Hydroxychloroquine, Favlplravir and Galidesivir). We found that Noscapine-Hydroxychloroquine (Nos-Hcq) conjugate has strong binding affinity for the main protease (Mpro) of SARS-CoV-2, which performs key biological function in virus infection and progression. Nos-Hcq was analyzed through molecular dynamics simulation. The MD simulation for 100 ns affirmed the stable binding of conjugation unprecedentedly through RMSD and radius of gyration plots along with critical reaction coordinate binding free energy profile. Also, dynamical residue cross-correlation map with principal component analysis depicted the stable binding of Nos-Hcq conjugate to Mpro domains with optimal secondary structure statistics of complex dynamics. Also, we reveal the drugs with stable binding to major domains of Mpro can significantly improve the work profile of reaction coordinates, drug accession and inhibitory regulation of Mpro. The designed combinatorial therapy paves way for further prioritized in vitro and in vivo investigations for drug with robust binding against Mpro of SARS-CoV-2.
Keywords: SARS-CoV-2, noscapine conjugates, main protease enzyme coronavirus, molecular dynamics simulation
Graphical Abstract

Communicated by Ramaswamy H. Sarma
1. Introduction
The recent outbreak of the novel Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) is unprecedented. The World Health Organization (WHO) has given the guidelines that the current pandemic might go long, and the virus may become endemic in our society. As per the statistics of WHO, seven million people (as of 8th June 2020) have already been infected with coronavirus disease-2019 (COVID-19) and expected to increase exponentially in some of the regions (WHO, 2020). The coronavirus is a single-stranded RNA virus that typically affects the vertebrates and causes lethal effects (Fung & Liu, 2019). The transmission of the SARS-CoV-2 virus between different host species is generally through respiratory droplets, in aerosol form, and fomites (Zhou et al., 2020). Due to the genetic proximity of this novel Betacoronavirus, it is similar to the previous outbreaks of severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) (Zhu et al., 2020). The diverse spike protein of SARS-CoV-2 makes it more lethal than previous SARS and MERS family viruses. Among the various routes for its treatment/prevention by the genetic code information of Coronavirus, reported by the Chinese research group in March 2020 (Ren et al., 2020) provided an early opportunity for scientists across the globe to develop the vaccine for the COVID-19 disease at earliest. However, it is too early to assume success for the COVID-19 vaccine because the development of vaccination is tricky; it may take time to manufacture an effective and safe vaccine on a large scale. Alternatively, to the current situation, drug repurposing seems to be an effective drug discovery strategy by utilizing the existing drugs and natural plant products for the treatment of the SARS-CoV-2 outbreak. Drug repurposing strategy can potentially shorten the time and as well as reduce the cost compared to develop novel drug discovery and their clinical trials (Cheng, 2019; Cheng et al., 2016, 2017),. Overall, the world research community is repurposing known FDA approved drugs and improving them by conjugation or synthesizing potential analogs. Remdesivir (Beigel et al., 2020), Hydroxychloroquine (Colson et al., 2020), Chloroquine (Gao et al., 2020), Favipiravir (T-705) (Coomes & Haghbayan, 2020), and Galidesivir (BCX4430) (Li & De Clercq, 2020) are some of the most successful repurposed small molecule drugs which has shown broad-spectrum activity against Coronavirus.
These Repurposed drugs had initially shown good results; however, there were some major limitations as well, likewise potency, effectiveness, specificity to viral targets, and in some cases, side effects due to non-specificity, etc. Interestingly, combination therapy of approved drugs suggested for being an effective strategy to treat corona infection. Recently, Hydroxychloroquine (HCQ), a well-known drug for malaria, has been repurposed, which brought attention to potentially helping patients to recover from COVID-19. Also, it has been used in the combination therapy and reaches to the trials such as; Hydroxychloroquine with Mivermectin (Patrì & Fabbrocini, 2020), Hydroxychloroquine with Azithromycin (Magagnoli et al., 2020), Hydroxychloroquine with Nitazoxanide (Amawi et al., 2020), etc. However, these have not shown expected outcomes and associated with issues like high mortality rates and increased frequency of irregular heartbeat. As per WHO reports, these HCQ based combination therapies took major setbacks after not performing significantly in clinical trials. Hence in present work, we propose a drug conjugate of Noscapine along with major antiviral drugs in repurposing trials (Chloroquine, Hydroxychloroquine, Umifenovir, Favlplravir, and Galidesivir). Noscapine is an approved antitussive drug for the treatment of cough through simulating the lung linings. It is known to inhibit bradykinin enhanced cough response (Ebrahimi et al., 2003). We are aware of the fact that the primary target for SARS-CoV is the lungs, and noscapine has been widely used to inhibit bradykinin, eventually inhibiting inflammation and lung damage (Ebrahimi, 2020). Hence it makes it sense to conjugate Noscapine with antiviral drugs, which can decrease the severe symptoms due to the lung damage; this can overcome the setbacks of previous combinations therapies, including HCQ conjugations, by attenuating the high mortality rates due to COVID-19. Noscapine is known to obtain from the naturally occurring Opium Poppy (Papaver Sominiferum) medicinal plant, and it is characteristically a non-toxic phthalide-isoquinoline alkaloid (Aneja et al., 2006).
We aimed to design the combinatorial therapy against the main protease (Mpro or 3CLpro) enzyme, a key regulator of the life cycle of the coronavirus. Mpro is essential for the replication of the virus as it processes the polyproteins that are translated from the viral RNA to yield functional viral proteins (Chen et al., 2020). This makes the Mpro enzyme a potential drug target to combat coronaviruses. This fact has led to some of the research groups to develop a potential inhibitor to the protease (Mengist et al., 2020; Zhang et al., 2020),. These inhibitors prevent the replication of the virus after entering into the host cells. Mpro protease inhibitors are reported to modify the polypeptides essential for SARS-COV-2 infection and progression in the host. Also, our group has recently reported the potent interaction of Noscapine analogs and subsequent inhibition of the Mpro protease of the SARS-CoV-2 (Kumar, Kumari, et al., 2020).
With detailed literature, we designed and analyzed the noscapine conjugates with antiviral drugs (chloroquine, hydroxychloroquine, favipiravir, and galidesivir that are currently being clinically tested (Table 1). The molecular binding affinity of conjugates to inhibit the Mpro has been evaluated using high throughput computational assays. The molecular dynamics studies have been widely used in modern drug discovery and development. Computer-aided drug design has been employed to annotate the molecular binding mechanism of drugs to their specific targets (Singh et al., 2019; Sood et al., 2018),. In many studies, advanced molecular docking approach has been harnessed to screen an extensive library, define the binding affinity of drugs and to elucidate the involved molecular interactions in the complexation of the ligand with receptor protein (Kumar et al., 2018; Chaudhary et al., 2020),. Molecular dynamics (MD) analysis helps to insight into the binding mechanism of the drugs through the Root-mean-square deviation (RMSD), radius of gyration binding energy calculations, and relevant calculations (Kumar, Sood, Sharma, et al., 2020; Kumar, Sood, Tomar et al., 2020; Singh et al., 2016; Sood et al., 2018).,. With employing these advanced computational approaches, deigned noscapine conjugations with antiviral drugs were examined to derive the highly efficient combination against the Mpro of SARS-CoV-2 for further validation with experimental lab assays.
Table 1.
Antiviral drugs for repurposing analysis trial for SARS-CoV-2.
| Repurposed Drug | Chemical Structure | Biological activity | Mode of action |
|---|---|---|---|
| Chloroquine | 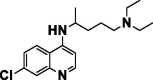 |
Anti-malarial | Inhibits action of heme polymerase, inhibits terminal glycosylation of ACE2 (SARS-COV-2) |
| Hydroxychloroquine | 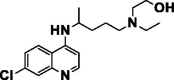 |
Anti-malarial | Inhibits terminal glycosylation of ACE2 (SARS-COV-2) |
| Favipiravir (T-705) | 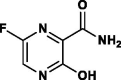 |
RNA-polymerase inhibitor | Prodrug, Selective viral RNA-polymerase inhibitor |
| Galidesivir (BCX-4430) | 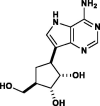 |
RNA-polymerase inhibitor | Viral RNA polymerase binder |
| Umifenovir (Arbidol) | 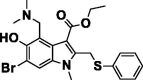 |
JAK kinase inhibitor | Membrane haemagglutinin fusion inhibitor in influenza viruses |
2. Methodology
2.1. Drug target main protease (mpro) of SARS-CoV-2 retrieval and structural assessments
The main protease regulatory enzyme of SARS-CoV-2 has been reported to play an essential role in coronavirus infection and progression in the host (Xue et al., 2008). To design the drug therapy against Mpro, a three-dimensional crystal structure of Mpro was retrieved from the protein data bank (PDB ID 6LU7). To design the highly specific targeting drugs, we have employed an existing binding site of Mpro to N3 inhibitor. After that, domain traits of Mpro crystal structure was assessed by CATH database (https://www.cathdb.info/), and domain annotation was determined using the Pfam database. The protein stability and secondary structural properties of Mpro were examined using the DSSP server (https://swift.cmbi.umcn.nl/gv/dssp/DSSP_3.html). Further to proceed to the molecular dynamics studies, the 3 D structure of Mpro was analyzed for its stereochemical and physicochemical properties through Ramachandran plot and 3 D structure local quality estimations (Laskowski et al., 1993; Morris et al., 1992),.
2.2. Noscapine based antiviral conjugates designing and optimization
We have designed the Noscapine based antiviral cognates with antiviral drugs in trials (Chloroquine, Umifenovir, Hydroxychloroquine, Favlplravir, and Galidesivir) (Liu et al., 2020). Noscapine is reported possess the appeased effects on lung and respiratory infection with its antitussive properties (Kim et al., 2019). Noscapine conjugates were drawn using the Chemdraw software and assessed for their compatibility and deformation.
2.3. Molecular docking analyses of noscapine based antiviral conjugates with SARS-CoV-2 mpro
The molecular interaction analyses of all conjugates were performed with the Mpro to illustrate their binding affinity. To perform the docking calculations, the target Mpro was prepared and optimized by the Whatif server (Vriend, 1990). Mpro was confined to add missing hydrogen atom, amino acid chain, any other unwanted chain and removing the water molecules. Similarly, ligand files of noscapine-antiviral conjugates were prepared and optimized for any missing atom and chain. The molecular docking was performed using the Hex 8.0 fast Fourier algorithm-based module (Macindoe et al., 2010). By incorporating the binding sites, grid box in all axes to Mpro was formed and ligand files were inserted to the server. The receptor grid was set to 0.6 Å for X, Y, and Z coordinates of Mpro structure. The translational step, twist range of 360° and protein flip ranges were set as per the molecular docking manual.
Moreover, to strengthen our results, we redocked the high-rank noscapine conjugate with another molecular docking tool with the Swissdock server. Swissdock works on the basis of the EADock DSS algorithm with curated scripts of receptors and ligands (Grosdidier et al., 2011). From the obtained various docked conformation of Mpro-conjugates, the model with the lowest energy was analyzed for the binding mechanism by protein-ligand profiles (Laskowski & Swindells, 2011; Salentin et al., 2015),. The model with the lowest docked energy score in all drug conjugates was selected for molecular dynamics simulation analysis.
2.4. Explicit solvent molecular dynamics simulation analyses of lead conjugation with mpro
To mechanistic insights into the binding of the conjugate (Nos-Hcq), the molecular dynamics simulation analysis was performed. MD simulations were implemented through GROMACS v5.0 under the force field GROMOS96 54a7 having water model SPC216 along with the time step of 1 fs for 100 ns (Yang et al., 2019). We investigated the atomic motions, conformational changes, structural stability, and binding energy contributing to the binding of the ligand to the target molecule. In an explicit water solvent, internal atomic motion simulation was initiated. The binding complex of Nos-Hcq conjugate was solvated in the octahedron system and stretched to 10 Å in all directions. The complex operation was minimized through four steps, and all atoms were fixed. In the minimization process 2000 steepest descent steps were executed, and all atoms were relaxed at 300 K with one atmospheric pressure in boundary states. NPT ensembles, along with periodic boundary conditions, were utilized to carry out MD simulations. A cut-off of about 12 Å was used in order to manage the Vander Waals forces. The Particle Mesh Ewald model having a cut-off of 14 Å was further utilized to calculate the electrostatic interactions. The obtained trajectory for 100 ns MD simulation run was analyzed for root mean square deviation (RMSD), root mean square fluctuations (RMSF), hydrogen bond analyses, solvent accessible surface area analysis (SASA) and radius of gyration (Rg) plot analysis (Grant et al., 2006). Also, the native contacts, along with the principal component analysis (PCA), cross-correlation map analyses were performed throughout the simulation run (McGibbon et al., 2015). Moreover, the binding energy calculation lead conjugate with Mpro was investigated through the reaction coordinates analysis. It determined the ligand compatibility with threshold energy and the progression of ligand for stabilized binding with the target receptor. A total of 1000 frames of interactions complex trajectory was analyzed for binding energy computation. The equation employed for the calculation is
ΔGcomplex, ΔGreceptor, ΔGligand represents the binding energy of complex, binding energy receptor, binding energy of ligand.
3. Results and discussion
3.1. Sars CoV-2 mpro enzyme as a potential target
SARS coronavirus is an enveloped positive-stranded RNA virus, which majorly affects the respiratory system and enteric system (Graham et al., 2013). SARS-CoV-2 infection and progression in the host are regulated by multiple structural proteins. Among various structural proteins, the main protease enzyme of coronavirus is reported to play significant roles in viral replication through proteolytic machinery and involved in transcription, translation, and amplification of viral proteins (Paules et al., 2020). Mpro enzyme is of size 306 amino acids and possesses high similarity with protease enzyme from different human and animal by sequence analysis. The protein annotation results depicted Mpro has various roles in coronavirus viz; a nucleic acid-binding domain of SARS coronavirus, polyprotein cleavage domain, coronavirus endopeptidase, corona NSP16, NSP7 and NSP9 replication implication regulatory roles, reported through Pfam annotation database.
Moreover, 3 D structural analysis of Mpro by the CATH server showed it consisted of beta-barrel and alpha domain (Figure 1). The secondary structural analysis by the DSSP server showed it comprised of 24% alpha-helices (10 helices, 75 residues) and 27% beta-barrel sheets (19 strands). These significant analyses along with the literature survey, suggested the Mpro enzyme as a potential target to design the COVID-19 therapeutic drugs.
Figure 1.

(A) Three-dimensional crystal structure of Mpro enzyme of coronavirus-19 and depiction of three major domains in the circles (B) Ramachandran plot assessment of Mpro enzyme, (C) Secondary structure analyses of Mpro protein (D) Local quality estimation of the 3 D structure of Mpro enzyme with native structures.
Moreover, prior to molecular binding analyses the 3 D structure of the Mpro enzyme was assessed for structural conformations and physicochemical by Ramachandran plot and Saves server. The Ramachandran plot analyses depicted 90.6% residues are in the favorable region, and 9.1% residues and 0.4% residues are in the outlier region of the plot. The local quality assessment showed good structural quality factors with high homology (0.1 to 0.7 Å) with the native structures in database of 160,954 known protein structures.
3.2. Molecular binding analyses of noscapine conjugate with antiviral drugs
With Noscapine significant roles in lung linings and dilation and Coronavirus movements into the lung cells evokes the cytokine storm and leads to neutrophilic infiltration and other implication (Martin & Ernst, 2003). Hence, it was essential to investigate the effectiveness of antitussive noscapine in conjugation with antiviral drugs against Mpro. We chose and designed the noscapine conjugation with potential antiviral drugs (Chloroquine, Umifenovir, Hydroxychloroquine, Favlplravir and Galidesivir) using the Chemdraw and simultaneously assessed their applicability and conformations. For molecular docking, both receptor Mpro and ligand files of noscapine conjugates were prepared for molecular docking using the Whatif prepdock module and ligprep server. For performing the molecular docking, binding site of Mpro enzyme-N3 inhibitor crystal structure, recently submitted to the protein data bank, was analyzed for binding sites and insights into the molecular mechanism of inhibition by drugs (Jin et al., 2020). Similar binding sites were employed for performing the molecular docking of the noscapine conjugations with the target Mpro of coronavirus using the Hex 8.0 and SwissDock servers.
The molecular docking was performed using Hex 8.0, which works based on a fast Fourier transform by SPF shape-density correlations, and results were recalculated with electrostatic association additionally. First, we have performed the Molecular docking analysis of drug alones which depicted the high binding score for Hydroxychloroquine with −301.08 kJ/mol, among all drugs. Further we assessed their drug likeliness properties of all drugs, none of them violated Lipinski rule of five except Galidesivir which has six hydrogen donor groups (Table 2). Taking these results altogether, we performed the docking for all conjugates. Interestingly, obtained results showed the strong binding of noscapine conjugates with Mpro of coronavirus. The Nos-Hcq conjugate depicted the most substantial binding with an energy score of −410.09 kJ/mol, among all conjugates. Besides, to assess the effectivity of noscapine (singlet state) against the Mpro, it was also docked using the same grid, which showed the binding score of −292.42 kJ/mol, larger than the known molecule N3 molecule bound to Mpro with binding score of −260.78 kJ/mol. It can be seen from the molecular binding analyses that hydroxychloroquine depicted the high binding score in drug alone analyses, whose binding efficiency was enhanced by conjugation of noscapine by about double with a docking score of −410.09 kJ/mol to Mpro (Table 3). Moreover, lead conjugation of Nos-Hcq does not found to follow the drug likeliness Lipinski rule of five for high molecular weight and hydrogen bond acceptor groups with combinations of two drugs. In addition, to strengthen our results, we redocked the all conjugates with Mpro using the SwissDock module. The outcomes affirmed the highest efficacy of Nos-Hcq conjugate with binding ΔG energy score of −10.01 kcal/mol and combined full fitness score of −1025.72 kcal/mol, among all noscapine conjugates (Figure 2). With these significant results, it can be attributed that Nos-Hcq conjugate has a high potential to bind the target Mpro enzyme and further can be used as effective therapeutics for SARS-CoV-2.
Table 2.
Molecular docking analysis of drug alone and further drug likeliness analyses.
| S. No. | Compound Name | HEX (fft shape + dars + 3) | Estimated ΔG (kcal/mol) | Drug likeliness (Lipinski rule of Five) |
|||
|---|---|---|---|---|---|---|---|
| MW (<500) | Log P (<5) | Hb Donor (<5) | Hb Acceptor (<10) | ||||
| 1. | Hydroxychloroquine | −301.08 kJ/mol | −8.56 kcal/mol | 335 Dalton | 1.62 | 2 | 4 |
| 2. | Chloroquine | −299.76 kJ/mol | −8.28 kcal/mol | 319 Dalton | 4.03 | 1 | 3 |
| 3. | Favipiravir | −210.74 kJ/mol | −6.16 kcal/mol | 157 Dalton | −0.57 | 5 | 5 |
| 4. | Galidesivir | −260.50 kJ/mol | −7.79 kcal/mol | 264 Dalton | −0.64 | 6 | 6 |
| 5. | Umifenovir | −200.1 kJ/mol | −6.08 kcal/mol | 476 Dalton | 3.69 | 1 | 5 |
| 6. | Noscapine | −292.42 kJ/mol | −8.42 kcal/mol | 413.42 Dalton | −0.053 | 5 | 6 |
Table 3.
Molecular docking analysis of noscapine and noscapine based antiviral conjugates with Mpro of SARS-CoV-2.
| S. No. | Compound Name | Structure of Conjugates | HEX (fft shape + dars + 3) | Estimated ΔG (kcal/mol) | Swiss Dock Full Fitness (kcal/mol) | Mol. Wt. (Daltons) |
|---|---|---|---|---|---|---|
| 1. | Noscapine | 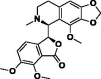 |
−292.42 kJ/mol | −8.42 kcal/mol | −1060.28 kcal/mol | 413.42 |
| 2. | Nos-Hydroxy Chloroquine (Nos-Hcq) |
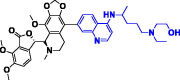 |
−410.09 kJ/mol | −10.01 kcal/mol | −1025.72 kcal/mol | 712.83 |
| 3.. | Nos- Chloroquine (Nos-Cq) |
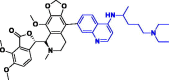 |
−398.46 kJ/mol | −9.58 kcal/mol | −1033.10 kcal/mol | 696.93 |
| 4. | Nos-Favlplravir (T-705) (Nos-Fav) | 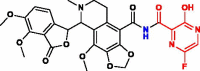 |
−293.70 kJ/mol | −9.17 kcal/mol | −1035.09 kcal/mol | 596.51 |
| 5. | Nos-Umifenovir (Nos-Ufr) | 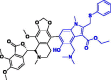 |
−379.74 kJ/mol | −8.33 kcal/mol | −1011.51 kcal/mol | 809.92 |
| 6. | Nos-Galidesivir (BCX-4430) (Nos-Gdr) |
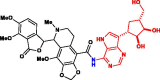 |
−378.08 kJ/mol | −9.05 kcal/mol | −1034.89 kcal/mol | 703.69 |
Figure 2.

Binding complexation of Mpro (ribbon view in sky blue color) with different conjugate ligands (A) Mpro-Noscpapine complex, noscapine in yellow color (B) Mpro-Nos-Hydroxychloroquine complex, Nos-Hcq conjugate in red color (C) Mpro-Nos-Chloroquine complex, Nos-Cq conjugate in coco color (D) Mpro-Nos-Favlplravir complex, Nos-Fav conjugate in blue color (E) Mpro-Nos-Umifenovir color, Nos-Umi conjugate in orange color (F) Mpro-Nos-Galidesivir complex, Nos-Gdr conjugate in black color.
3.3. Noscapine conjugates-Mpro binding domain analyses
The detailed characterization of contacts of noscapine conjugates is vital to recognize the interaction pattern of ligands for stringent binding to target Mpro. The protein-ligand profiler was employed to determine the non-covalent contacts of complex systems. The Mpro structural domain analysis by the CATH server showed it forms majorly three ligand-binding domains and one additional domain. The globular structure analysis by Chimera modeling application suggested the plausibly first domain from Ser1-Pro96, second binding domain Pro99-Asp197, and third binding domain Thr198-Gln306. The obtained results showed the all conjugates bind in close proximity of the catalytic domain of Mpro, as per the N3 drug interaction pattern. Among all combinations, Nos-Hcq depicted the strongest binding in all three domains of Mpro protein. It has significant molecular contacts with the domain-2 and domain-3 with slight interaction with domain-1 of Mpro. Molecular interaction detailed analysis showed that the Nos-Hcq conjugate formed the four hydrogen bonds (5 A-LYS-2.81 Å, 137 A-LYS-3.01 Å, 199 A-THR-1.74 Å, and 289 A-ASP-1.94 Å), two salt bridge contacts (288 A-GLU-3.81 Å, 290 A-GLU-3.25 Å) and multiple hydrophobic interactions (137 A-LYS, 199 A-THR, 239 A-TYR, 286 A-LEU, 287 A-LEU, and 290 A-GLU) (Table 4) (Figure 3). Interestingly, the lead conjugation was found to bind firmly to the catalytic domains (residues close to domain-2; 130-190) of the Mpro enzyme. Moreover, the binding network of lead conjugation showed the extended interactions towards the third domain of Mpro and strengthening the binding. More similar to Nos-Hcq conjugate, Nos-Cq conjugate also found to bind to three domains of Mpro, possibly due to structural similarity of both. Nos-Cq complex showed that it forms the two hydrogen bonds (131 A-ARG-3.42, 199 A-THR-2.39), wherever NoS-Hcq formed the four hydrogen bonds, possible answer to the high efficacy towards Mpro.
Table 4.
Molecular binding analysis of all noscapine based conjugates through protein interaction profiler.
| S. No. | Conjugates | Hydrogen Bond | Hydrophobic interactions | π-Cation Interactions | Salt Bridges | Water Bridges |
|---|---|---|---|---|---|---|
| 1. | Nos-Hcq | 5A-LYS-2.81 137A-LYS-3.01 199A-THR-1.74 289A-ASP-1.94 |
137A-LYS-3.95 199A-THR-3.28 239A-TYR-3.44 286A-LEU-3.87 287A-LEU-3.99 290A-GLU-3.64 |
- | - | 288A-GLU-3.81 290A-GLU-3.25 |
| 2. | Nos-Cq | 131A-ARG-3.42 199A-THR-2.39 |
5A-LYS-3.28 137A-LYS-3.30 199A-THR-3.87 286A-LEU-3.99 289A-ASP-3.87 |
5A-LYS-4.36 | 290A-GLU-5.30 | 288A-GLU-3.49-3.14 |
| 3. | Nos-Fav | 105A-ARG-3.69 107A-GLN-2.41 110A-GLN-3.06 111A-THR-2.53 151A-ASN-1.93 152A-ILE-2.41 158A-SER-2.74 158A-SER-3.18 |
104A-VAL-3.83 106A-ILE-3.60 106A-ILE-3.45 151A-ASN-3.25 |
– | – | 102A-LYS-4.09 111A-THR-2.89 151A-ASN-4.02 151A-ASN-3.64 |
| 4. | Nos-Ufr | 131A-ARG-3.36 137A-LYS-2.89 289A-ASP-2.82 289A-ASP-2.26 |
137A-LYS-3.87 137A-LYS-3.56 137A-LYS-3.84 141A-LEU-3.79 |
172A-HIS-5.37 131A-ARG-4.13 137A-LYS-3.77 |
288A-GLU-3.67 | 5A-LYS − 3.91 288A-GLU-2.74 290A-GLU-3.69 |
| 5. | Nos-Gdr | 131A-ARG-3.02 137A-LYS-2.38 199A-THR-2.30 238A-ASN-2.30 287A-LEU-2.06 289A-ASP-2.07 290A-GLU-3.52 |
199A-THR-3.77 238A-ASN-2.86 286A-LEU-3.66 290A-GLU-3.96 |
– | 236A-LYS-5.47 | 290A-GLU-3.02 |
Figure 3.

Noscapine-Antiviral conjugates to Mpro binding domain analysis, surface view of the binding groove of Mpro in sky blue color in Leftside, and conjugate binding pattern in domains in Rightside (A) Mpro-Nos-Hydroxychloroquine complex (B) Mpro-Nos-Chloroquine complex C) Mpro-Nos-Favlplravir complex (D) Mpro-Nos-Umifenovir complex (E) Mpro-Nos-Galidesivir complex.
Moreover, other noscapine conjugates (Nos-Fav, Nos-Ufr, and Nos-Gdr) were found to interact with one or two domains, only with optimal contacts. Nos-Fav conjugate binds with Mpro through eight hydrogen bonds (105 A-ARG-3.69, 107 A-GLN-2.41, 110 A-GLN-3.06, 111 A-THR-2.53, 151 A-ASN-1.93, 152 A-ILE-2.41, 158 A-SER-2.74, 158 A-SER-3.18) but to single domain-2 only, causing the less affinity comparatively with Nos-Hcq. Similarly, Nos-Ufr formed the four hydrogen bonds (131 A-ARG-3.36, 137 A-LYS-2.89, 289 A-ASP-2.82, 289 A-ASP-2.26) but confined to domain-2 and domain-3 only. Nos-Gdr binds with although seven hydrogen bonds (131 A-ARG-3.02, 137 A-LYS-2.38, 199 A-THR-2.30, 238 A-ASN-2.30, 287 A-LEU, 2.06, 289 A-ASP-2.07, 290 A-GLU-3.52) however, it also remained confined to domain-2 and domain-3 (Figure 4). From these contact analyses of all conjugates, it can be attributed that Nos-Hcq conjugate has high affinity and wide molecular contacts for major domains of Mpro, among all conjugates. Moreover, it was further studied through molecular dynamics to get insight into inhibitory patterns and efficacy to combat the Mpro enzyme of coronavirus.
Figure 4.

Noscapine conjugates Mpro binding molecular contacts analysis (A) Mpro-Nos-Chloroquine complex (B) Mpro-Nos-Favlplravir complex (C) Mpro-Nos-Hydroxychloroquine complex (D) Mpro-Nos-Umifenovir complex (E) Mpro-Nos-Galidesivir complex.
3.4. Molecular dynamic simulation analyses of Nos-Hcq conjugate with mpro
The binding affinity of the Nos-Hcq conjugate was investigated by the molecular dynamics simulation analyses for 100 ns. The stable binding of Nos-Hcq along with protein dynamics in the target Mpro was assessed through the GROMOS96 54a7 force field with water model SPC216 consisting time step of 1 fs. The obtained trajectory was solvated in the octahedron box in an explicit water state to provide sustainable conformations. The system minimization was done for 2000 steps by the steepest descent program and further 300 steps by conjugated gradient method through inserted force fields. The root means square deviation (RMSD), root means square fluctuation (RMSF), and Radius of gyration (Rg) were computed for the resulting trajectory to assess the stable binding of lead combination (Nos-Hcq) with Mpro of coronavirus. Moreover, to compare and determine the boosting power and synergetic contribution, MD simulation was also performed for the singlet state of Mpro. The steep RMSD variation can be considered an implication of a malleable and instinctive protein. The RMSD (average backbone range of complex system) for the frames of native contacts with target Mpro depicted the stable binding of lead Nos-Hcq conjugate to Mpro with fluctuation in the range of 0.2-0.55 nm and for Mpro singlet state fluctuation in the range of 0.15- 0.81 nm (Figure 5). Interestingly, RMSD of the complex system was more stabilized than the native form of target receptor Mpro.
Figure 5.

Protein complex MD simulation analyses of Nos-Hcq conjugation (A) Root mean square deviation plot of Mpro receptor with Nos-Hcq conjugation, green color native Mpro protein, purple Mpro-NosHcq complex (B) Radius of gyration analysis of interacting complex of Mpro with Nos-Hcq conjugation, green color native Mpro protein, purple Mpro-NosHcq complex (C) Root mean square fluctuation plot of Mpro receptor with Nos-Hcq conjugation, green color native Mpro protein, purple Mpro-NosHcq complex (D) SASA analyses of Mpro receptor with Nos-Hcq conjugation, green color native Mpro protein, purple Mpro-NosHcq complex, (E) Secondary structure analysis of Mpro in the native state (F) Secondary structure analysis of Mpro with Nos-Hcq conjugation for throughout the 100 ns MD simulation.
Moreover, the secondary structure analysis showed the compelling flexibility in terminal residues and other loops of beta linker segments of the Mpro (residues 189-2998) to about 0.21 nm and Mpro-conjugate (residues 130-140 and 189-299) to about 0.1-0.2 nm. Also, we found the residues 130 to 299 have comparatively lesser discrepancies in RMSF statistics, which are corresponding to the interacting segment of the Mpro-conjugate complex after MD simulation. Interestingly, the binding region for lead conjugation before MD simulation was also within the residues 137 to 298, which remains the same after simulation, suggesting a strong binding of Nos-Hcq towards the Mpro and provides stability to the Mpro-conjugate. After that, the obtained trajectory was computed to evaluate the Radius of gyration of the Mpro and compared with the noscapine singly. The lead combination with the Mpro protein showed the Rg fluctuation in the range of 2.12-2.26 nm and for Mpro in range of 2.14– 2.33 nm and demonstrated the stable and robust binding of Nos-Hcq with Mpro (Figure 5).
Furthermore, similar outcomes were obtained by SASA computation, which represented the solvent-accessible protein surface, and various orientations through the folding and proceeding to alterations in buried and exposed regions of the surface area of the protein complex. Similar observations were determined through SASA analysis representing the solvent defined protein surface and its orientation through folding, making the alterations in the exposed and buried regions of the surface area of proteins. SASA values for both Mpro and binding system were about 155 nm/S2/N and 153 nm/S2/N after 7 ns and 15 ns, respectively. Also, it remained the same till 100 ns in case of Mpro singlet native state, whereas in the complexed state with the conjugate, it reduces slightly about to 145 nm/S2/N after 75 ns and further remains same as before till 100 ns (Figure 5). Here, Mpro-NosHcq solvation profile shows a convincing SASA value suggesting a stable structure and sturdy binding. Also, the secondary structure analysis of complex trajectory and native Mpro trajectory was performed by employing the DSSP tool of Gromacs. The cluster analyses of both depicted the conformational changes before and after the simulation run. Mpro-NosHcq and Mpro were consisted of mainly conserved β-sheets along with loops of β linker segments and other random coil regions infused with the various bend, α-helices, turn and β bridges. The observations of both plot analyses showed the Mpro rationale unfolded and implemented surface with β-sheets to bind stably with lead conjugation Nos-Hcq.
Binding energy landscape
MM/PB (GB) surface area (SA) calculations were performed to estimate the lead conjugation binding affinities to Mpro protein. The system was found to be the most favorable in terms of the sum of various binding energy computations. MM/GBSA free binding energy calculations were executed with two fifty frames dynamically equilibrated for the stabilized trajectory of the Mpro-NosHcq complex system between 75 ns to 80 ns. The electrostatic energy distribution in molecular mechanics was obtained through the high Vander Waal energy (VDW) −203.87 ± 15.81 kJ/mol, electrostatic energy −1077.91 ± 171.34 kJ/mol and total binding energy of −1281.78 ± 179.25 kJ/mol, which are considered as most significant contributor in the binding of ligand (Nos-Hcq conjugation) to the target protein. The polar solvation energy (PBSOL) was calculated to be −8915.14 ± 210.92 kJ/mol, which added a vital contribution to the stable binding of the ligand to target protein.
Further, the stability and flexibility of the binding of Nos-Hcq with Mpro were strengthening with the stabilized run of trajectory with set NVT parameters. The average temperature for both native Mpro and in conjugation was close to 300 Kelvin; average pressure was found to be close to 1.4 bars and an average density of 993.5 kg/m3 (Figure 6). These outcomes through 100 ns MD simulation suggested the stable binding of lead combination with significant contribution of binding energy to the target Mpro of the coronavirus. Also, the energy work profile of ligand binding and Mpro protein dynamics was investigated by SMD trajectory based on Jarzynski relationships in molecular dynamics simulation. The reaction coordinate work profile demonstrated the contribution of binding energies in potential interaction. The calculated work profile for ligand binding through the reaction coordinates stated the stabilized coupling with energy contribution reached a free energy profile of 100 kcal/mol at an RC value of 33 Å and sustained throughout binding with the less flexible atomic motions. In the early stages, it reached to the maximum value and stabilized at 100 kcal/mol with hydrogen bonds and reached to work profile of 130 kcal/mol at RC 70 Å in later stages. The atomic energy profiles for Mpro-Nos-Hcq reaction coordinates showed an average radius of 1.88 Å, with overall Priority of conjugated reaction progression about 91.84%. Along with the binding energy calculations, hydrogen bond consistency and dynamic contribution of bound Nos-Hcq revealed the involvement of a high number of hydrogen bonds throughout the simulation run for both native Mpro and complex system (Figure 6). Mpro formed an average of 550 hydrogen bonds with the lead combination during the simulation and played an essential role in the selectivity of intermolecular interactions. These outcomes through the MD simulation signified for the stable binding of Nos-Hcq conjugation in correlation with molecular docking results.
Figure 6.

Mpro with Nos-Hcq conjugation binding energy analyses (A) GROMACS total energy analysis of complex system (B) Two-dimensional projection of simulated trajectory (C) Plot statistics showing the pressure variations of the system (D) Equilibrated temperature plot of stabilized system trajectory during the energy minimization (E) Graph depicting the density of complex system through the MD simulation (F) Hydrogen bond contributions to the complex system through the MD simulation, green color native Mpro protein, purple Mpro-NosHcq complex (G) Hydrogen bond statistics depicting the involvement of a large number of hydrogen bonds in the intermolecular binding of Mpro-NosHcq system.
3.5. Principal component and dynamical residues cross-correlation binding analysis
The principal component analysis (PCA) was performed to insight into the binding clusters variances analyses of the complex system. The binding cluster frames were categorized into two coordinate clusters in black and red color. PCA calculations of the atomic backbone of the complex system were computed by three conformations PC1, PC2, and PC3 by normal mode molecular dynamics. Obtained results showed that the PCA cluster possesses the highest variability of 30.2% by binding of lead combination to Mpro in terms of internal motions of trajectory. After that, PC2 statistics depicted the minimal variability with 14.43%, and consequently, PC3 calculations 9.64% and suggested the stabilized binding with Eigen scores and minimal atomic motions variability of Nos-Hcq in complexation with Mpro (Figure 7). Both the conformations in two groups in different PCA analyses were superimposed and aligned, as shown in Figure 7d. The PCA trajectory analyses showed a conformational change in clusters from the first blue cluster to white cluster progressively to red color cluster, similarly recovered from first cluster (black color) to second cluster (red color) through Simple Clustering in PC Subspace (Figure 8).
Figure 7.

Principal component analyses for Mpro binding to ligand Nos-Hcq (A) PC1, (B) PC2, (C) PC3 based on MD trajectories from the internal mode atomic motions in correlation with protein dynamics secondary structures. The secondary structures schematics are added to the top and bottom (D) Superimposed ligand conformations to the receptor main protease protein of coronavirus (E) The dynamical residue cross-correlation map.
Figure 8.

Principal component analysis of clusters by protein complex dynamics from internal modes (A and B). PCA resulting trajectory frames change from blue to white to red conformation during the MD simulation run and recovered by changing conformations from black to red color. The trajectory snapshots are divided into two different clusters of the color black and red through the top three PC1, PC2, and PC3 spaces, (C and D) superimposed conformations of two clusters.
Furthermore, the dynamical residue cross-correlation map (DCCM) analysis was performed, which depicted the pairwise correlation of atomic coordinates of Nos-Hcq to the Mpro through the magnitude of pairwise cross-correlation coefficient. The map showed the correlation residues (>0.8) of the complex system in blue color, and anti-correlated residues (<-0.4) depicted in red color. The highly pairwise correlated residues (light blue color) of complex trajectory in the map showed the stable binding of ligand file Nos-Hcq. The outcomes were also found in correlation with optimal secondary structures of complex protein dynamics during the MD simulation run. The binding ability of Nos-Hcq towards the Mpro and its essence for coronavirus for replication and regulation suggests the potency of Nos-Hcq and possible role in COVID-19 clinical drug development.
3.6. Proposed scheme for the chemical synthesis of Nos-Hcq conjugate
With the computational rationalization of lead conjugate Nos-Hcq against the Mpro, we propose the plausible scheme of chemical synthesis. The drug conjugates of Noscapine 1 and selected Re-purposed small molecules drugs depict the feasible tentative synthetic methodology (Scheme 1). Earlier in 2006, our research group developed a series of microtubule-interfering halogenated noscapine analogs that perturb mitosis in cancer cells followed by cell death. Iodo-Nos 2 was one of the crucial analogs of halogenated Noscapine. 9-Iodo-Nos was synthesized by the iodination of noscapine 1 using pyridine–iodine chloride in acetonitrile solvent with a 75% yield (Aneja et al., 2006). The target compound 5 can be achieved by the application of a well-known Stille metal cross-coupling reaction. Compound 3, organostannanes, which can act as a precursor for the Stille cross-coupling reaction, can be achieved via radical pathways in the presence of SnMe3, DIPEA (base) and MeCN (solvent) (Chen et al., 2016). The presence of the chloro group at Hydroxychloroquine 4 facile stille coupling reaction to take place at ease. The Pd(PPh3)4 catalytic system with copper(I) iodide and cesium fluoride in DMF is most effective for coupling iodides. Copper(I) iodide and cesium fluoride combination facile the synthesis of sterically hindered systems such as 5 (Figure 9).
Figure 9.

Proposed schematic diagram for the chemical synthesis of Nos-Hcq conjugate.
4. Conclusion
We report the efficient combinatorial therapy by conjugating the noscapine (antitussive drug) with potential hydroxychloroquine (Nos-Hcq) against the SARS-CoV-2, through the computational assays with insights into the experimental results. The conjugate Nos-Hcq showed the most definite binding affinity towards the Mpro, among the various noscapine based conjugates with (Chloroquine, Umifenovir, Favlplravir, and Galidesivir). Noscapine (natural source opium poppy) is deemed to form the conjugation with antiviral drugs in trials against SARS-CoV-2. The molecular docking assays depicted that by conjugation binding affinity score almost doubled with Nos-Hcq than with noscapine in the singlet state. The binding mechanism analyses of all conjugations illustrated that the lead conjugation (Nos-Hcq) has strong binding to three major domains of Mpro. Moreover, MD simulation analyses of the trajectory (Mpro-Nos-Hcq) for 100 ns in explicit solvent depicted the stable binding of Nos-Hcq conjugate to Mpro with minimal conformational variations in RMSD and Rg plots. We reveal the drug binding with three major domains of Mpro has high reaction coordinate completion efficacy (energy work profiles) with a significant contribution of immense binding energies and meticulous atomic level stabilization. The DCCP and PCA analyses confirmed the pivotal interactions and stabilize the binding of lead conjugation with Mpro. Besides rationalizing the combinatorial drug therapy, our study provides the fundamental plausible route of chemical synthesis of Nos-Hcq conjugate. This work paves the way for boosting the current medications into the effective combinational strategy to bind target Mpro strongly, which can enhance the inhibitory action of drugs and contributes to fighting against the pandemic COVID-19 with saving time, cost with minimal time screening in the current emergency. Further, the warranted lead drug conjugation needs to be validated experimentally in laboratories and clinical trials.
Notes
The authors declare no competing financial interest.
Acknowledgements
N. K. particularly thanks CSIR for Research Associate fellowship and University of Delhi for Guest Assistant Professor Opportunity to conduct research smoothly. A. A gratefully acknowledge DST-INSPIRE for the award of Senior Research Fellowship.
Authors contribution
£- NK and AA contributed equally. N.K, A.A., D.S., R.C., and N.S. designed the studies. N.K., D.S., and A.A, A.K., carried out the in silico experiments. N.K, A.A., A.G., and D.S. wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Amawi, H., Abu Deiab, G. A. I., A Aljabali, A. A., Dua, K., & Tambuwala, M. M. (2020). COVID-19 pandemic: An overview of epidemiology, parthenogenesis, diagnostics and potential vaccines and therapeutics. Therapeutic Delivery, 11(4), 245–268. 10.4155/tde-2020-0035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aneja, R., Vangapandu, S. N., Lopus, M., Viswesarappa, V. G., Dhiman, N., Verma, A., Chandra, R., Panda, D., & Joshi, H. C. (2006). Synthesis of microtubule-interfering halogenated noscapine analogs that perturb mitosis in cancer cells followed by cell death. Biochemical Pharmacology, 72(4), 415–426. 10.1016/j.bcp.2006.05.004 [DOI] [PubMed] [Google Scholar]
- Beigel, J. H., Tomashek, K. M., Dodd, L. E., Mehta, A. K., Zingman, B. S., Kalil, A. C., & Lopez de Castilla, D. (2020). Remdesivir for the treatment of Covid-19—preliminary report. New England Journal of Medicine. 10.1056/NEJMoa2007764 [DOI] [PubMed] [Google Scholar]
- Chaudhary, M., Kumar, N., Baldi, A., Chandra, R., Babu, M. A., & Madan, J. (2020). 4-Bromo-4’-chloro pyrazoline analog of curcumin augmented anticancer activity against human cervical cancer, HeLa cells: In silico-guided analysis, synthesis, and in vitro cytotoxicity. Journal of Biomolecular Structure and Dynamics, 38(5), 1335–1353. 10.1080/07391102.2019.1604266 [DOI] [PubMed] [Google Scholar]
- Chen, K., He, P., Zhang, S., & Li, P. (2016). Synthesis of aryl trimethylstannanes from aryl halides: An efficient photochemical method. Chemical Communications (Cambridge, England), 52(58), 9125–9128. 10.1039/C6CC01135G [DOI] [PubMed] [Google Scholar]
- Chen, Y., Liu, Q., & Guo, D. (2020). Emerging coronaviruses: Genome structure, replication, and pathogenesis. Journal of Medical Virology, 92(4), 418–423. 10.1002/jmv.25681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, F. (2019). In silico oncology drug repositioning and polypharmacology. In Krasnitz A. (Ed.), Cancer Bioinformatics. Methods in Molecular Biology, vol. 1878. New York, NY: Humana Press. 10.1007/978-1-4939-8868-6_15 [DOI] [PubMed] [Google Scholar]
- Cheng, F., Hong, H., Yang, S., & Wei, Y. (2017). Individualized network-based drug repositioning infrastructure for precision oncology in the panomics era. Briefings in Bioinformatics, 18(4), 682–697. 10.1093/bib/bbw051 [DOI] [PubMed] [Google Scholar]
- Cheng, F., Murray, J. L., & Rubin, D. H. (2016). Drug repurposing: New treatments for zika virus infection? Trends in Molecular Medicine, 22(11), 919–921. 10.1016/j.molmed.2016.09.006 [DOI] [PubMed] [Google Scholar]
- Colson, P., Rolain, J. M., Lagier, J. C., Brouqui, P., & Raoult, D. (2020). Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. International Journal of Antimicrobial Agents, 55(4), 105932. 10.1016/j.ijantimicag.2020.105932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coomes, E. A., & Haghbayan, H. (2020). Favipiravir, an antiviral for COVID-19. Journal of Antimicrobial Chemotherapy, 75(7), 2013–2014. ? 10.1093/jac/dkaa171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi, S. A. (2020). Noscapine, a possible drug candidate for attenuation of cytokine release associated with SARS‐CoV‐2. Drug Development Research. https://doi.org/DOI: 10.1002/ddr.21676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi, S. A., Zareie, M. R., Rostami, P., & Mahmoudian, M. (2003). Interaction of noscapine with the bradykinin mediation of the cough response. Acta Physiologica Hungarica, 90(2), 147–155. 10.1556/APhysiol.90.2003.2.7 [DOI] [PubMed] [Google Scholar]
- Fung, T. S., & Liu, D. X. (2019). Human coronavirus: Host-pathogen interaction. Annual Review of Microbiology, 73, 529–557. 10.1146/annurev-micro-020518-115759 [DOI] [PubMed] [Google Scholar]
- Gao, J., Tian, Z., & Yang, X. (2020). Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Bioscience Trends, 14(1), 72–73. 10.5582/bst.2020.01047 [DOI] [PubMed] [Google Scholar]
- Graham, R. L., Donaldson, E. F., & Baric, R. S. (2013). A decade after SARS: Strategies for controlling emerging coronaviruses. Nature Reviews. Microbiology, 11(12), 836–848. 10.1038/nrmicro3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant, B. J., Rodrigues, A. P., ElSawy, K. M., McCammon, J. A., & Caves, L. S. (2006). Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics, 22(21), 2695–2696. 10.1093/bioinformatics/btl461 [DOI] [PubMed] [Google Scholar]
- Grosdidier, A., Zoete, V., & Michielin, O. (2011). SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Research, 39(Web Server issue), W270–W277. 10.1093/nar/gkr366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., & Yang, H. (2020). Structure of Mpro from COVID19 virus and discovery of its inhibitors. Nature, 582(7811), 289–293. [DOI] [PubMed] [Google Scholar]
- Kim, D. E., Min, J. S., Jang, M. S., Lee, J. Y., Shin, Y. S., Park, C. M., Song, J. H., Kim, H. R., Kim, S., Jin, Y. H., & Kwon, S. (2019). Natural Bis-benzylisoquinoline alkaloids-tetrandrine, fangchinoline, and cepharanthine, inhibit human coronavirus OC43 infection of MRC-5 human lung cells. Biomolecules, 9(11), 696. 10.3390/biom9110696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, D., Kumari, K., Jayraj, A., Kumar, V., Kumar, V., Dass, S., Chandra, R., & Singh, P. (2020). Understanding the binding affinity of Noscapine with protease of COVID-19 using MD simulation at different temperature. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1752310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, N., Sood, D., Sharma, N., & Chandra, R. (2020). Multiepitope subunit vaccine to evoke immune response against acute encephalitis. Journal of Chemical Information and Modeling, 60(1), 421–433. 10.1021/acs.jcim.9b01051 [DOI] [PubMed] [Google Scholar]
- Kumar, N., Sood, D., Tomar, R., & Chandra, R. (2019). Antimicrobial peptide designing and optimization employing large-scale flexibility analysis of protein-peptide fragments. ACS Omega, 4(25), 21370–21380. 10.1021/acsomega.9b03035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, N., Tomar, R., Pandey, A., Tomar, V., Singh, V. K., & Chandra, R. (2018). Preclinical evaluation and molecular docking of 1, 3-benzodioxole propargyl ether derivatives as novel inhibitor for combating the histone deacetylase enzyme in cancer. Artificial Cells, Nanomedicine, and Biotechnology, 46(6), 1288–1299. 10.1080/21691401.2017.1369423 [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., & Swindells, M. B. (2011). LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. Journal of Chemical Information and Modeling, 51(10), 2778–2786. 10.1021/ci200227u [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S., & Thornton, J. M. (1993). PROCHECK: A program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, 26(2), 283–291. 10.1107/S0021889892009944 [DOI] [Google Scholar]
- Li, G., & De Clercq, E. (2020). Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nature Reviews. Drug Discovery, 19(3), 149–150. 10.1038/d41573-020-00016-0 [DOI] [PubMed] [Google Scholar]
- Liu, C., Zhou, Q., Li, Y., Garner, L. V., Watkins, S. P., Carter, L. J., Smoot, J., Gregg, A. C., Daniels, A. D., Jervey, S., & Albaiu, D. (2020). Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Central Science, 6(3), 315–331. 10.1021/acscentsci.0c00272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macindoe, G., Mavridis, L., Venkatraman, V., Devignes, M. D., & Ritchie, D. W. (2010). HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Research, 38(Web Server issue), W445–W449. 10.1093/nar/gkq311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magagnoli, J., Narendran, S., Pereira, F., Cummings, T., Hardin, J. W., Sutton, S. S., & Ambati, J. (2020). Outcomes of hydroxychloroquine usage in United States veterans hospitalized with Covid-19. Med. 10.1016/j.medj.2020.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, K. W., & Ernst, E. (2003). Antiviral agents from plants and herbs: A systematic review. Antiviral Therapy, 8(2), 77–90. [PubMed] [Google Scholar]
- McGibbon, R. T., Beauchamp, K. A., Harrigan, M. P., Klein, C., Swails, J. M., Hernandez, C. X., Schwantes, C. R., Wang, L. P., Lane, T. J., & Pande, V. S. (2015). MDTraj: A modern open library for the analysis of molecular dynamics trajectories. Biophysical Journal, 109(8), 1528–1532. 10.1016/j.bpj.2015.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengist, H. M., Fan, X., & Jin, T. (2020). Designing of improved drugs for COVID-19: Crystal structure of SARS-CoV-2 main protease M pro. Signal Transduction and Targeted Therapy, 5(1), 1–2. 10.1038/s41392-020-0178-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, A. L., MacArthur, M. W., Hutchinson, E. G., & Thornton, J. M. (1992). Stereochemical quality of protein structure coordinates. Proteins: Structure, Function, and Genetics, 12(4), 345–364. 10.1002/prot.340120407 [DOI] [PubMed] [Google Scholar]
- Patrì, A., & Fabbrocini, G. (2020). Hydroxychloroquine and ivermectin: A synergistic combination for COVID-19 chemoprophylaxis and treatment? Journal of the American Academy of Dermatology, 82(6), e221. 10.1016/j.jaad.2020.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paules, C. I., Marston, H. D., & Fauci, A. S. (2020). Coronavirus infections—More than just the common cold. JAMA, 323(8), 707–708. 10.1001/jama.2020.0757 [DOI] [PubMed] [Google Scholar]
- Ren, L.-L., Wang, Y.-M., Wu, Z.-Q., Xiang, Z.-C., Guo, L., Xu, T., Jiang, Y.-Z., Xiong, Y., Li, Y.-J., Li, X.-W., Li, H., Fan, G.-H., Gu, X.-Y., Xiao, Y., Gao, H., Xu, J.-Y., Yang, F., Wang, X.-M., Wu, C., … Wang, J.-W. (2020). Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study. Chinese Medical Journal, 133(9), 1015–1024. 10.1097/CM9.0000000000000722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F., & Schroeder, M. (2015). PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Research, 43(W1), W443–W447. 10.1093/nar/gkv315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, A., Kumar, N., Sood, D., Singh, S., Awasthi, A., Tomar, V., & Chandra, R. (2019). Designing of a novel indoline scaffold based antibacterial compound and pharmacological evaluation using chemoinformatics approach. Current Topics in Medicinal Chemistry, 18(23), 2056–2065. 10.2174/1568026619666181129125524 [DOI] [PubMed] [Google Scholar]
- Singh, V. K., Kumar, N., Kalsan, M., Saini, A., & Chandra, R. (2016). A novel peptide thrombopoietin mimetic designing and optimization using computational approach. Frontiers in Bioengineering and Biotechnology, 4, 69. 10.3389/fbioe.2016.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood, D., Kumar, N., Rathee, G., Singh, A., Tomar, V., & Chandra, R. (2018). Mechanistic interaction study of bromo-noscapine with bovine serum albumin employing spectroscopic and chemoinformatics approaches. Scientific Reports, 8(1), 1–11. 10.1038/s41598-018-35384-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood, D., Kumar, N., Singh, A., Sakharkar, M. K., Tomar, V., & Chandra, R. (2018). Antibacterial and pharmacological evaluation of fluoroquinolones: A chemoinformatics Approach. Genomics & Informatics, 16(3), 44–51. 10.5808/GI.2018.16.3.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriend, G. (1990). WHAT IF: A molecular modeling and drug design program. Journal of Molecular Graphics, 8(1), 52–56. 10.1016/0263-7855(90)80070-V [DOI] [PubMed] [Google Scholar]
- World Health Organization. ( 2020). Coronavirus disease (COVID-19) pandemic. https://www.who.int/emergencies/diseases/novel-coronavirus-2019
- Xue, X., Yu, H., Yang, H., Xue, F., Wu, Z., Shen, W., Li, J., Zhou, Z., Ding, Y., Zhao, Q., Zhang, X. C., Liao, M., Bartlam, M., & Rao, Z. (2008). Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. Journal of Virology, 82(5), 2515–2527. 10.1128/JVI.02114-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. F., Wang, F., Chen, Y. Z., Hao, G. F., & Yang, G. F. (2019). LARMD: Integration of bioinformatic resources to profile ligand-driven protein dynamics with a case on the activation of estrogen receptor. Briefings in Bioinformatics, bbz141. 10.1093/bib/bbz141 [DOI] [PubMed] [Google Scholar]
- Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., & Hilgenfeld, R. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science, 368(6489), 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., Hou, Y., Shen, J., Huang, Y., Martin, W., & Cheng, F. (2020). Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discovery, 6(1), 14–18. 10.1038/s41421-020-0153-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., Zhao, X., Huang, B., Shi, W., Lu, R., Niu, P., Zhan, F., Ma, X., Wang, D., Xu, W., Wu, G., Gao, G. F., Phil, D., & Tan, W. (2020). A novel coronavirus from patients with pneumonia in China, 2019. The New England Journal of Medicine, 382(8), 727–733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]