

Abstract
ADAM proteases are multi domain transmembrane metalloproteases that cleave a range of cell surface proteins and activate signaling pathways implicated in tumor progression, including those mediated by Notch, EFGR, and the Eph receptors. Consequently, they have emerged as key therapeutic targets in the efforts to inhibit tumor initiation and progression. To that end, two main approaches have been taken to develop ADAM antagonists: (i) small molecule inhibitors, and (ii) monoclonal antibodies. In this mini-review we describe the distinct features of ADAM proteases, particularly of ADAM10 and ADAM17, their domain organization, conformational rearrangements, regulation, as well as their emerging importance as therapeutic targets in cancer. Further, we highlight an anti-ADAM10 monoclonal antibody that we have recently developed, which has shown significant promise in inhibiting Notch signaling and deterring growth of solid tumors in pre-clinical settings.
Keywords: ADAM10, ADAM17, Integrins, Therapeutic antibody, Notch signaling
1. Background
Proteolytic processing and subsequent release of membrane-bound proteins is a post-translational mechanism that governs the biological activity of the cleaved molecules [1–3]. This process, referred to as ‘protein ectodomain shedding’, is mediated by membrane-anchored ADAM metalloproteases [4–9], which shed a wide range of membrane-attached proteins implicated in growth factor signaling, cell migration and cell-adhesion [10]. In this mini-review, we will discuss the salient features of this unique class of proteases, the regulation of their activity via conformational rearrangements in their ectodomains and their potential as therapeutic targets during cancer progression. We will also discuss a novel anti-ADAM10 monoclonal antibody that has made significant headway towards treatment of solid tumors in pre-clinical setting.
ADAMs contain several conserved domains: an N-terminal signal sequence followed by a pro-domain, a metalloprotease domain (MP), a disintegrin domain (D), a cysteine-rich domain (C), an EGF-like domain (except ADAM10 and ADAM17), a transmembrane domain and a cytoplasmic domain [11]. The signal sequence guides the inactive ADAM proenzymes into the secretory pathway [12,13]. The prodomain acts as a chaperone during trans-Golgi transport and also inhibits premature MP domain activation [14,15]. The MP domain confers zinc-dependent proteolytic activity and contains a consensus HEXGHXXGXXHD motif that coordinates the zinc atom [16]. Despite being well studied, the substrate-specificities of the ADAMs are poorly understood [17]. They seem to be dictated, at least in part, by the ADAM D and C domains, which mediate protein-protein, and specifically substrate-enzyme, interactions. The D domains of ADAMs are also implicated in integrin-mediated cell-adhesion. In the case of ADAM15, a consensus RGD sequence in the D domain [17] is needed for interaction with the α5β1 and α5β3 integrins, while the importance of the C domain for proper substrate cleavage has been documented for ADAM13 and ADAM17 [8,18]. Swapping of the C domain of ADAM13 with that of ADAM10 leads to loss of function and failure to induce hyperplasia in Xenopus laevis embryos [19], deterring major developmental events. Likewise, a chimeric construct of ADAM17, with its D + C domains replaced by those of ADAM10, fails to shed interleukin (IL)-1R–II from the cell surface of the fibroblast cell line EC-2 [18].
2. Conformational regulation of ADAMs and intracellular signaling
Cleavage by ADAM proteases is regulated at many levels [8,11]. It is known that ADAM-mediated cleavage is essential for G-protein-coupled [20,21] or stress-induced [22] EGF receptor transactivation, and it is modulated by intracellular signaling in response to growth factors, phorbol esters, and changes in tyrosine kinase activity [8]. Interestingly, while ADAM family members harbor cytoplasmic protein-docking motifs [8], cytoplasmically-truncated ADAM10 and ADAM17 are fully functional [23], suggesting only a minor role for the cytoplasmic domains in controlling downstream signaling.
ADAM17 is suggested to be regulated by cysteine oxidation that modulates the conformation of its extracellular domain [24,25]. Furthermore, the notion that the ADAM activity is regulated by the presence of two distinct conformations “active” or “open” and “latent” or “closed” (Fig. 1) is supported by the crystal structures of related snake venom metalloproteinases. Indeed, these structures reveal two radically different conformations resulting from alternative disulfide-bonding arrangements of the D + C region [26,27].
Fig. 1.
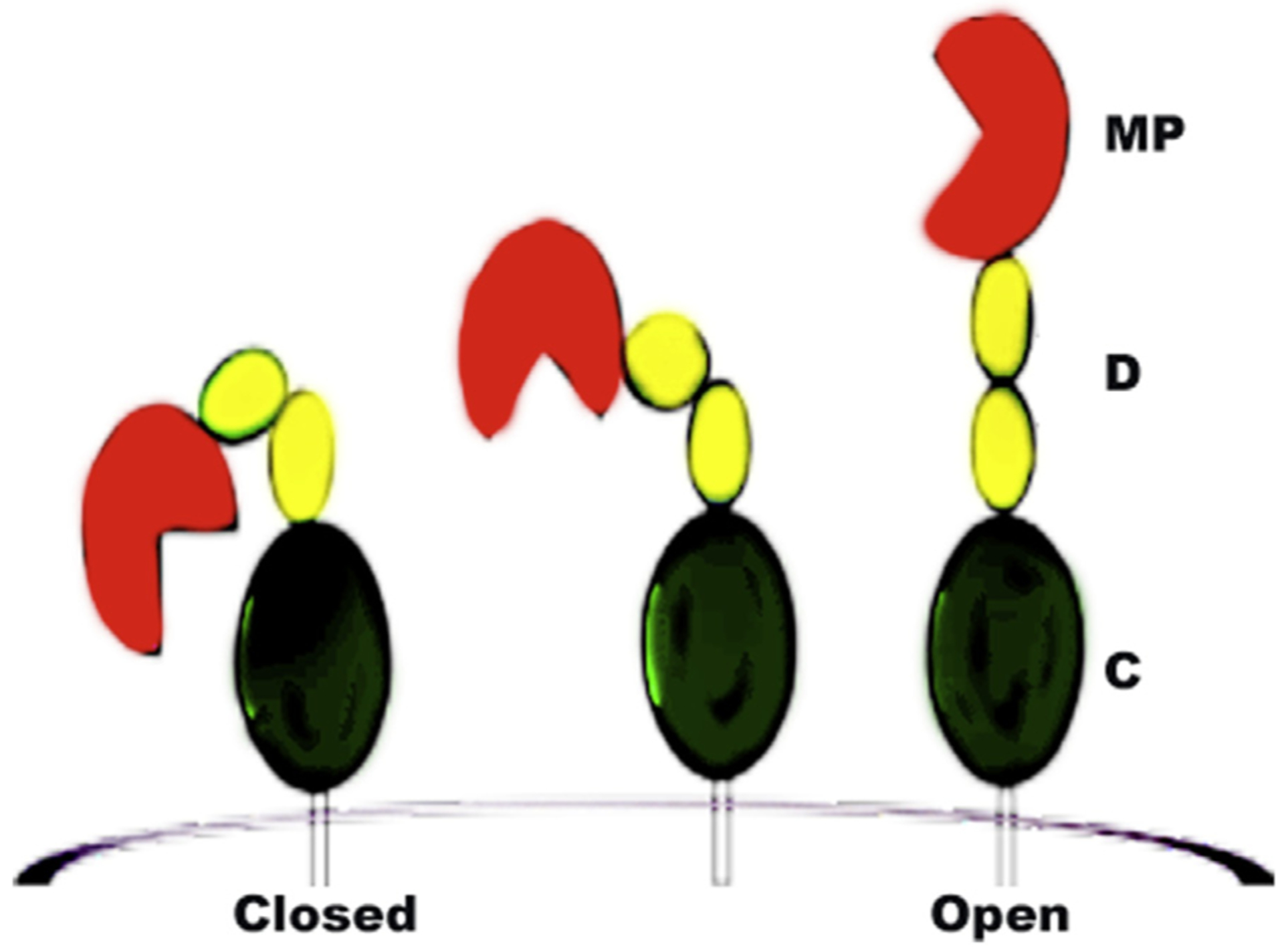
ADAM domains and proposed conformations. The ‘closed’ and ‘open’ ectodomain conformations of ADAM10 and ADAM17 are proposed to reflect alternate disulfide bonding arrangements of the D + C region. Both ADAM10 and ADAM17 contain a CxxCC motif, a consensus site for PDI-mediated disulphide exchange. Thus, their ectodomain conformations are influenced by redox effects.
Like other proteins undergoing protein disulfide isomerase (PDI)-mediated thiol disulfide exchange, ADAM17 contains a conserved CxxCC motif, and mild reducing or oxidizing conditions alter its enzymatic activity [24]. Interestingly, facilitating disulfide exchange via a thiol isomerase was reported to favor the inactive ADAM17 conformation, while inhibiting the isomerase enhances the ADAM17 activity [25]. Other studies suggest that reactive oxygen species (ROS) also regulate the activity of ADAM proteases through similar conformational changes in their ectodomains [28–33]. An important source of ROS at cell membranes is a family of NADPH oxidases (NOXs) that get stimulated by activated receptor tyrosine kinases (RTKs), including EGF, PDGF and insulin receptors [34]. It has been elegantly demonstrated that an elevated level of ROS due to oxidative stress, common in tumors [35], stimulates ADAM-mediated shedding [30], cross-activation of RTKs [29], and contributes to tumor resistance to therapy [36,37]. Furthermore, an elevated level of ROS inhibits protein tyrosine phosphatases (PTPs) by reversible oxidation of their conserved active-site cysteine residue [38], which in turn increases RTK activity. The oxidation-dependent changes in the conformations of ADAM10 and ADAM17 suggest that RTK-stimulated ROS production could directly activate ADAMs. This implies a mechanism where conformational changes in the ADAM ectodomains mediate ADAM-RTK cross activation.
3. Oncogenic substrates of ADAM10 and ADAM17
ADAM10 and ADAM17 shed various cell-surface proteins and activate signaling pathways important in oncogenic development. Prominent oncogenic substrates include receptors/ligands of the erbB, Eph and Notch families, cytokines, cadherins, FAS ligand, Slit and L-selectin [39]. ErbB ligands are initially cell-surface tethered and their proteolytic release by ADAM10 and ADAM17 [40] is a key regulatory switch to trigger downstream autocrine signaling that drives tumor progression [39,41,42]. The erbBs display de-regulated signaling in many human cancers due to overexpression and/or mutations, including in some of the most intractable and common tumors, with EGFR and HER2 prominent in breast, ovarian and colon cancer [43]. In addition, elevated ADAM17-mediated cleavage of erbB2/erbB4 in tumors is associated with constitutive receptor activity and poor prognosis and undermines receptor-targeted therapies [44,45].
Eph receptors and their membrane-bound ephrin ligands control cell migration and positioning during normal and oncogenic development [46,47]. We documented that ADAM10 binds to Eph/ephrin receptor/ligand complexes and cleaves the ephrin ligands in trans. This disrupts the Eph/ephrin molecular tethers between cells, causing de-adhesion and retraction [48], as well as termination of Eph signaling. This function of ADAM10 was found to be dependent on the Eph kinase activity [48]. ADAM10, thereby, acts as a switch between cell-cell adhesion and segregation in response to the Eph phosphorylation status. This is important in the context of Eph-dependent oncogenesis, where aberrant Eph expression can contribute to tumorigenesis [46,47].
Notch receptors (Notch1–4) are single pass transmembrane proteins that undergo regulated intramembrane proteolysis by ADAM10 and γ-secretase. The proteolytic steps are triggered by Notch binding to DLL/Jagged cell-surface-attached ligands. This initiates ADAM10-mediated shedding of the ligand [49] and receptor ectodomains [50]. Subsequently, release of the Notch intracellular domain (NICD) by γ-secretase activates transcription of target genes, a pathway that is conserved in all phyla [51]. Notch signaling controls cell fate and differentiation, proliferation, epithelial-mesenchymal transition (EMT) and cell survival, while de-regulated Notch signaling promotes progression of both hematological and solid cancers [52]. Notch activity is particularly associated with a ‘stem-like’ phenotype in a subpopulation of tumor cells termed as cancer stem cells (CSCs) or tumor initiating cells (TICs). The CSCs not only initiate and sustain tumor growth, but also promote metastasis, chemoresistance, and angiogenesis [53,54]. This is especially evident in colorectal cancer (CRC) [55], where endothelial-expressed Notch ligands promote paracrine signaling and stem cell phenotype in tumor cells [56]. Accordingly, inhibition of Notch depletes CSCs and inhibits metastasis [57–60]. Notch signaling and CSCs are also associated with drug resistance, and inhibition of Notch signaling is widely reported to increase sensitivity to both chemo- and targeted therapies [61–65]. Importantly, it has been demonstrated that ADAM10 gene deletion is embryonic lethal in mice and the embryonic defects match the defects observed in Notch deficient animals. This suggests that the abnormalities associated with the absence of ADAM10 arise due to a failure to activate Notch signaling [57].
4. ADAM proteases as therapeutic targets
Since the ADAM proteases are involved in ectodomain shedding in a wide range of disease-related pathways, they are considered to be attractive targets for drug development. Studies with hepatocellular carcinoma cell lines highlight the importance of ADAM10 as anti-tumor therapeutic target, showing that downregulation of ADAM10 using siRNA markedly suppresses cell proliferation, cell migration and cell invasion in vitro, as well as tumor growth in vivo [66]. In addition, ADAM10 is over-expressed in oral squamous cell carcinoma (OSCC), where it contributes to OSCC invasion by interacting with integrin αvβ6 [67]. Likewise, ADAM17 is over-expressed in esophageal squamous cell carcinoma [68] and plays a significant role in the progression of glioma by upregulating the EGFR-P13K-AKT pathway [69]. Furthermore, microarray analysis revealed that ADAM17 is over-expressed on the surface of several types of cancer cells, including ovarian, breast and prostate [70]. In ovarian cancer, ADAM10 and ADAM17 cleave the calcium independent cell adhesion molecule Nectin-4 [71]. The cleaved ectodomain of Nectin-4 facilitates formation of spheroids that eventually invade the mesothelial layer to establish tumors at secondary sites [72].
ADAM9, another member of the family, is overexpressed in several cancers, including ovarian, clear cell carcinoma [73], lung adenocarcinoma [74], glioma, gastric, breast, and liver cancers [75]. In liver cancer, ADAM9 was shown to evade tumor immunity by cleaving the MHC class 1 polypeptide–related sequence A. Interestingly, it was established that ADAM9 interacts with β1 integrins (but does not cleave them) promoting melanoma, thus highlighting the importance of nonproteolytic ADAM functions in cancer progression [76]. Likewise, the cysteine-rich domain of ADAM12 is necessary for tumor cell adhesion [77]. The expression levels of ADAM12 were found to be significantly elevated in small cell lung cancer patients [78]. Amongst other cancer types, ADAM12 is responsible for cavernous sinus invasion in pituitary adenomas [79]. Owing to the low expression levels of ADAM12 in normal tissues, as compared to its high expression levels in several cancers (including both mRNA and protein), ADAM12 can be a useful biomarker [80]. ADAM15 mRNA and protein levels are increased in prostate cancer, where it triggers angiogenesis and angioinvasion of tumor cells causing unrestrained tumor growth [81]. In ovarian cancer, ADAM15 disrupts the binding of αvβ3 integrin to its extracellular matrix ligand vitronectin, thereby regulating tumor migration and invasion. Mechanistically, the RGD-binding motif of ADAM15 interacts with αvβ3 integrin and loosens the adhesion of tumor cells to the underlying matrix [82]. ADAM8 promotes growth and progression of triple negative breast cancer (TNBC) that lacks estrogen, progesterone and HER-2 receptor gene expression. In pancreatic cancer cells, ADAM8 enhances the binding of β1 integrin to endothelial cells. Thus, it promotes tumor cell migration and invasion by augmenting the ERK1/2 signaling pathway [83,84]. Recently, ADAM23, was found to be involved in breast cancer progression and dissemination of circulating tumor cells [85]. ADAM28 promotes lung metastasis by cleaving and inactivating the von Willebrand factor, VWF. VWF is a proapoptotic protein and its inactivation prolongs cancer cell survival and metastasis [86,87]. In TNBC, ADAM33 has been reported to be downregulated by DNA hypermethylation, suggesting that this protease could be potentially used as a biomarker for TNBC and other basal-like breast cancers [88].
Interestingly, some of the catalytically ‘dead’ human ADAM proteases, such as ADAM11 and ADAM29, were shown to promote integrin α4-dependent adhesion of immune cells. The studies revealed that the disintegrin domains of these ADAM proteases mediate cell-cell adhesions via interactions with the integrins α4β1 and α4β7 [89].
The ADAMTS (A Disintegrin and Metalloprotease domains with thrombospondin motifs) are secreted, multi-domain endopeptidases, and close relatives of the ADAMs [90]. They have been implicated in both oncogenic and tumor-suppressive signaling events. Specifically, they regulate the inflammatory microenvironment of tumors. For instance, ADAMTS4 causes macrophage infiltration in the environs of colorectal cancer [91]. Elevated expression of ADAMTS 1, 8, 9 and 18 were also observed in pancreatic cancer patient samples [92]. Table 1 documents the various ADAM proteases implicated in major cancer types.
Table 1.
ADAM proteases and associated major cancer types.
| ADAM/ADAMTS | Cancer types | References |
|---|---|---|
| ADAM8 | Triple negative breast cancer, Pancreatic | [83,84] |
| ADAM9 | Ovarian, lung, glioma, breast, gastric, liver | [73–76] |
| ADAM12 | Small cell lung, pituitary adenomas | [77–80] |
| ADAM15 | Ovarian | [81,82] |
| ADAM10 | Colon, hepatocellular, oral squamous cell carcinoma | [66,67,114] |
| ADAM17 | Esophageal, glioma, ovarian, breast, prostate | [68–71] |
| ADAM23 | Breast | [85] |
| ADAM28 | Lung | [86,87] |
| ADAM33 | Breast | [88] |
| ADAMTS4 | Colorectal | [91] |
| ADAMTS 1, 8, 9, 18 | Pancreatic | [92] |
Owing to the importance and therapeutic potential of ADAM proteases in cancer progression, attempts have been made to develop ADAM inhibitors to impede cancer growth. The two major approaches taken so far are discussed in the following paragraphs.
5. Small molecule ADAM inhibitors
Most of the small molecule ADAM inhibitors described to date are hydroxamate-based and are either specific for ADAM17 or selective for both ADAM10 and ADAM17. These inhibitors chelate the zinc from the active site of the proteases, thereby rendering them inactive. Amongst the dual inhibitors for ADAM10 and ADAM17, INCB3619 was one of the first to be investigated for anti-cancer activity [42,93,94]. In in vitro assays, the compound reduces the shedding of an array of EGFR ligands, which increases the susceptibility of non-small cell lung cancer cells to the EGFR tyrosine kinase inhibitor gefitinib [42]. Unlike the first-generation inhibitors, INCB3619 is remarkably free of musculoskeletal toxicity in animal models. INCB3619 also inhibits non-small cell lung, breast, head and neck carcinomas in animal models [42,93,94]. Of late, another structurally related inhibitor, INCB7839, underwent early clinical trials in HER-2 positive breast cancer patients with no evidence of musculoskeletal side effects or bone marrow toxicity. This particular inhibitor decreases the shedding of EGFR and HER-2 ligands [95]. Besides the INC class of dual inhibitors, the PF-series, including PF-5480090, was found to be specific for ADAM17 and does not inhibit ADAM10 [96]. PF-5480090 inhibits proliferation of a wide panel of breast cancer cell lines independent of their molecular subtype [97]. Specifically, in the case of TNBC where ADAM17 is expressed at higher level than in non-TNBC, PF-5480900 significantly decreases (i) the release of the EGFR ligand TGF alpha and (ii) the levels of phosphorylated EGFR, leading to proliferation inhibition of TNBC [97]. In addition to small molecule compounds, it has been shown that small hairpin RNAs, specific to ADAM17, significantly inhibit cell proliferation, invasion and tumor growth of non-small cell lung cancer in vitro [98].
Little progress has been made with regard to ADAM10-specific small molecule inhibitors. One such molecule, GI254023X, blocks migration and invasion of breast cancer cell lines with hardly any effect on proliferation. In cell-based assays, GI254023X impedes shedding of interleukin receptors and chemokine ligands [99–101]. Amongst the other ADAM proteases, a six-amino acid cyclic peptide, designated as BK-1361, inhibits the activation of ADAM8 and decreases tumor burden and metastasis of implanted pancreatic tumor cells. Specifically, it downregulates the ERK1/2 signaling pathway [83,84]. No small molecule inhibitors have been reported for ADAM28, though in animal models of lung cancer, ADAM28-targeting siRNA inhibits metastasis [86]. ADAM15 is known to be functionally associated with the progression of bladder cancer. Indeed, knock-down of ADAM15 in a human xenograft model of bladder cancer causes 45% tumor regression compared to controls [102]. A novel ADAM15-specific sulfonamide inhibitor, designed by structural modeling of the catalytic domain, also decreases tumor growth in animal model of bladder cancer [102].
6. Monoclonal antibodies targeting the ADAMs
The second approach for therapeutic intervention entails the generation of monoclonal antibodies (mAbs) targeting the ADAMs. Compared to small molecule inhibitors, mAbs can be highly target-specific, thereby reducing the risk of toxic side effects. Of late, a mAb that binds to both the catalytic (MP) and disintegrin-cysteine rich domains of ADAM17 has been shown to have significant therapeutic potential [103,104]. The mAb D1 (A12) inhibits tumor growth in an animal model of ovarian cancer and blocks the shedding of ADAM17 substrates, including TNF-alpha, TGF-alpha, HG-EGF and amphiregulin [103–105]. In a mouse model, the mAb is highly stable in circulation and retains its ability to bind ADAM17 over a prolonged period of time [103]. Another anti-ADAM17 mAb, MEDI3622, inhibits cancer progression in several different pre-clinical models [106]. Mutagenesis and molecular docking studies revealed that this mAb recognizes a unique surface loop present in the MP domain of ADAM17 [107]. In cell–based studies, MEDI3622 blocks the release of known ADAM17 substrates, including amphiregulin, epiregulin and heregulin [106].
In another study, a mAb (A300E) recognizing the membrane-proximal cysteine-rich region of human ADAM17 was reported. It was further modified to generate a recombinant scFv (single-chain variable fragment) containing the variable regions of the heavy and light chains. Using this scFv, a bispecific antibody was constructed that recognizes ADAM17 on tumor cells and CD3 on T-cells. In the presence of primary peripheral blood mononuclear cells, this bispecific antibody specifically kills prostate tumor cells expressing ADAM17 on the cell surface [108]. A300E, when coupled to doxorubicin or Pseudomonas exotoxin A, is able to kill the MDA-MB-231 TNBC cell line in an ADAM17-dependent manner [109]. Amongst the other ADAM proteases, a monoclonal antibody raised against the catalytic site of ADAM9 suppresses cancer cell proliferation and exhibits anti-tumor activity in a gastric cancer xenograft model [110]. ADAM8 was found to promote breast cancer development and brain metastasis in mouse models, and a commercial antibody to ADAM8 blocks transendothelial migration of breast cancer cells in in vitro assays [111].
7. Mechanism of action and therapeutic potential of the 8C7 anti-ADAM10 mAb
Though ADAM10 is implicated in several different cancers, including breast, pancreatic, melanoma and bladder carcinoma, no ADAM10 specific mAb has been developed so far for therapeutic intervention. Furthermore, Notch is the most prominent tumor-related ADAM10 target, and previous attempts to inhibit tumorigenic Notch signaling by blocking the γ-secretase activity or disrupting the formation of Notch-ligand complexes caused intestinal toxicity [112]. Nevertheless, Notch-inhibitory mAbs that do not exhibit toxicity have proved to be promising cancer therapeutic leads [113]. We, therefore, generated a highly specific anti-ADAM10 mAb (8C7) for the potential treatment of solid tumors. This antibody binds to the cysteine-rich domain of ADAM10 and targets not only the rapidly dividing cells at the invasive font of the tumors, but also the undifferentiated tumor stem cells. We documented that 8C7 mAb primarily acts via inhibiting Notch signaling in tumors. It also effectively prevents tumor relapse after chemotherapy in pre-clinical models without exhibiting any dose-limiting toxicity in animals [114].
Our in vitro and in vivo data indicate that 8C7 recognizes an active conformation of ADAM10 prevalent in tumors, as opposed to the inactive/latent ADAM10 ectodomain conformation present in normal tissues. It should be noted that currently there is no structural information regarding the open, activated ADAM10 conformation, and only the structure of the closed, autoinhibited conformation has been reported, where access to the proteinase active site is blocked [115]. The transition to the active ADAM conformation, as discussed in the previous sections of this review, is dependent on disulfide isomerization and oxidative conditions of the tumor microenvironment [29]. Consequently, in tumor cells, oxidizing conditions promote binding of the 8C7 mAb to ADAM10, while reducing conditions inhibit binding.
The crystal structure of 8C7 bound to the ADAM10 D + C domain [114] revealed the ADAM10 epitope recognized by 8C7. This ADAM10 region is stabilized by two intra-molecular disulfide bonds, C594–C639 and C632–C645, which are part of a CxxCC motif, a consensus PDI target sequence (Fig. 2). Mutating the CxxCC motif to AxxAC abrogates binding of 8C7 to ADAM10.
Fig. 2.
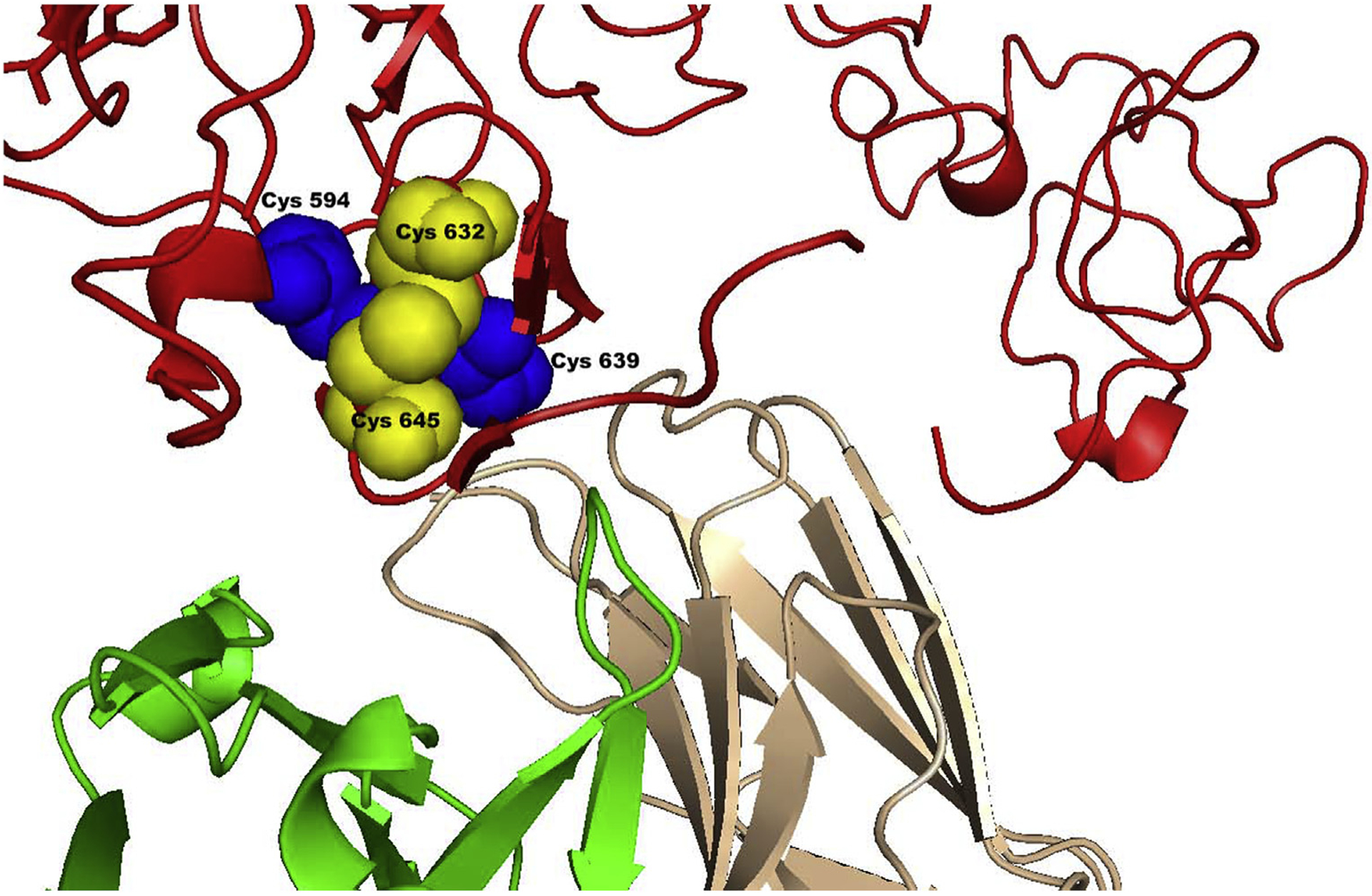
A close up-view of the disulfide-bonding pattern in the mAb (8C7)-bound ADAM10. The disulfide forming cysteines are shown as blue and yellow spheres. The ADAM10 D and C domains are represented in red. The heavy chain of the 8C7 mAb is in green and the light chain in wheat [114].
To understand how the 8C7 mAb inhibits ADAM10 function, the ADAM10–8C7 complex structure was compared to the structures of full-length snake venom metalloproteases VAP1 and Atragin, with similar MP + D + C domain architectures [26,27]. This comparison (Fig. 3A) shows that the MP domain, residing within the concave side of the D + C region, occupies the same space where the 8C7 mAb binds. It is, therefore, likely that 8C7 displaces the ADAM10 MP domain upon binding, thus inhibiting substrate cleavage on the cell surface. The recent crystal structure of the complete ADAM10 ectodomain corroborates this hypothesis (Fig. 3B). In that structure, ADAM10 adopts a closed conformation, with the MP and the D + C domains positioned similarly to what was observed in the Atragin structure [115].
Fig. 3.
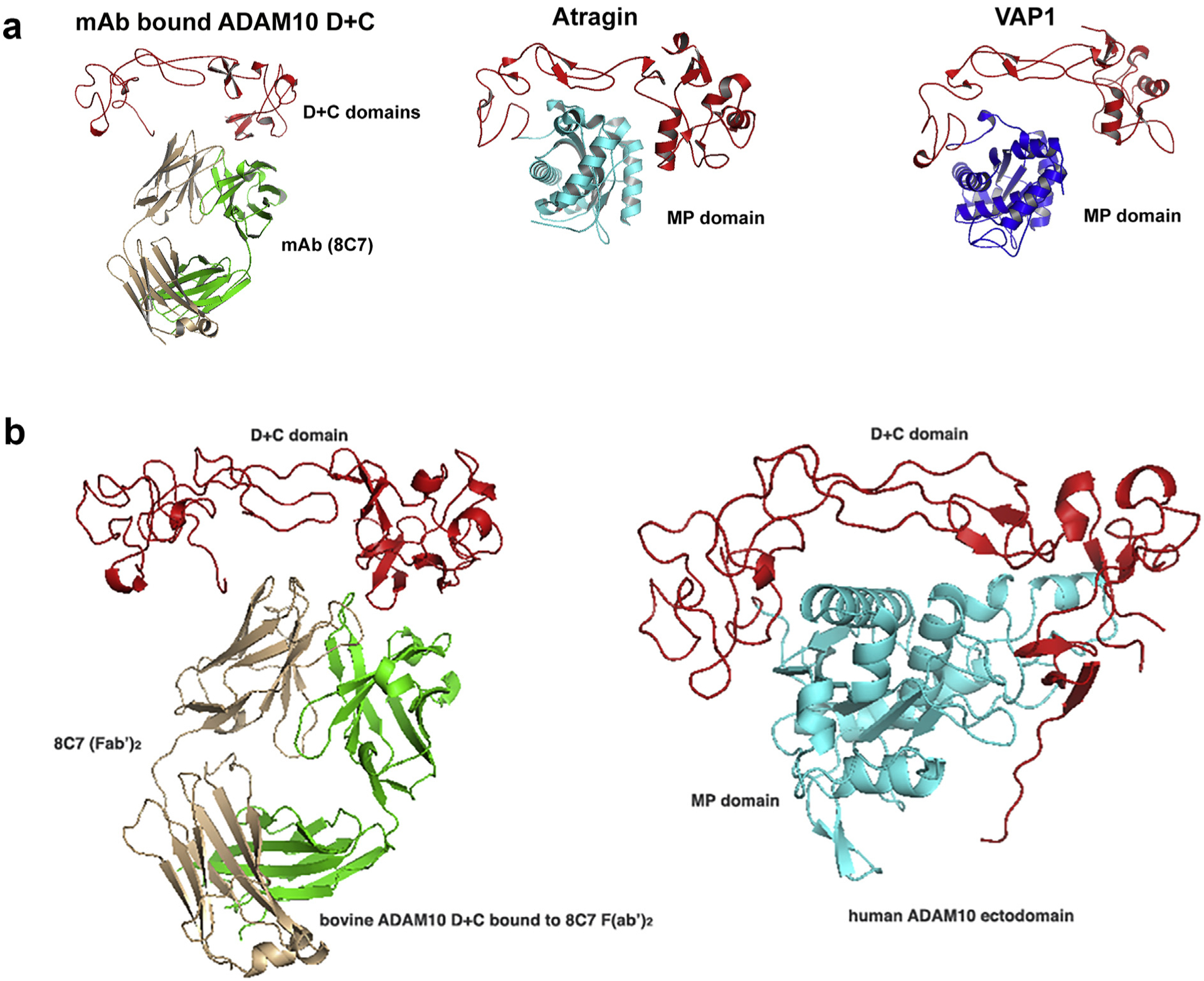
Comparison of the mAb 8C7-bound ADAM10 D + C structure with the structures of the related snake venom metalloproteinases Atragin and VAP1 (3A), as well as the structure of the complete ADAM10 ectodomain (3B) [115].
7.1. Future prospects
Targeting the MP domains of ADAM proteases, particularly of ADAM10 and ADAM17, for the treatment of various diseases, has been unsuccessful so far mostly because the proteinase active site is highly conserved, resulting in toxicity due to the interactions of drugs with other matrix metalloproteinases. Recently, it has been shown that the non-catalytic ADAM10 and ADAM17 domains, specifically the D and C domains, confer substrate-specificities, and it is expected that future efforts will concentrate on targeting these domains for successful therapeutic intervention. This endeavor could also be extrapolated to other ADAM family members, which differ from ADAM10 and ADAM17 in that they contain an additional EGF domain. For instance, ADAM9, ADAM12, ADAM13 and ADAM15 [19,75,77,82] bind to a variety of integrins via their non-catalytic domains promoting adhesion of tumor cells. Likewise, the ancillary disintegrin, cysteine-rich and thrombospondin domains of ADAMTS dictate substrate-specificity, and thus regulate the diverse functions of these secreted proteases [90]. Structural studies have already shown specific molecular regions and surfaces outside of the ADAM MP domain that mediate ADAM-substrate interactions, providing attractive targets for novel small-molecule and antibody therapeutics. Furthermore, a large and ever-growing amount of DNA sequencing data from healthy individuals and patients provides a great opportunity to not only understand better the molecular mechanisms of ADAM-mediated signaling, but to also identify and/or validate therapeutic target regions on the molecular surface of the ADAM family members. One such source is the sequencing data from the Center for Molecular Oncology, Memorial Sloan Kettering Cancer Center [disseminated through cBioportal, http://www.cbioportal.org/ [116,117]]. The use of genomic and proteomic data should also provide in the future the basis for “personalized/precision medicine” as well as novel “targeted therapeutics” developments [118,119].
Acknowledgements and Funding
Our studies are supported by National Institutes of Health [R21CA185930 and R01NS038486 to D.B.N and 5RO1NS096956-03/ RES510708 to JPH]; The Experimental Therapeutics Center of Memorial Sloan-Kettering, support from Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research [2013-2015 and 2018-2020 to D.B.N]; Functional Genomics Initiative [2015-2017 and 2019-2020 to D.B.N.]
Abbreviations:
- MP
Metalloprotease domain
- D
Disintegrin domain
- C
Cysteine rich domain
- VEGF
Vascular endothelial growth factor
- OSCC
Oral squamous cell carcinoma
- ScFv
Single chain variable fragment
- PDI
Protein disulfide isomerase
- TNBC
Triple negative breast cancer
- MMP
Matrix metalloproteinase
Footnotes
Conflicts of interest
The authors declare that they have no competing interest.
References
- [1].Hooper NM, Karran EH, Turner AJ, Membrane protein secretases, Biochem. J 321 (Pt 2) (1997) 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hooper NM, Turner AJ, Protein processing mechanisms: from angiotensin-converting enzyme to Alzheimer’s disease, Biochem. Soc. Trans 28 (2000) 441–446. [PubMed] [Google Scholar]
- [3].Massague J, Pandiella A, Membrane-anchored growth factors, Annu. Rev. Biochem 62 (1993) 515–541. [DOI] [PubMed] [Google Scholar]
- [4].Becherer JD, Blobel CP, Biochemical properties and functions of membrane-anchored metalloprotease-disintegrin proteins (ADAMs), Curr. Top. Dev. Biol 54 (2003) 101–123. [DOI] [PubMed] [Google Scholar]
- [5].Black RA, White ADAMs JM, Focus on the protease domain, Curr. Opin. Cell Biol 10 (1998) 654–659. [DOI] [PubMed] [Google Scholar]
- [6].Kheradmand F, Werb Z, Shedding light on sheddases: role in growth and development, Bioessays: News Rev. Mol. Cell. Develop. Biol 24 (2002) 8–12. [DOI] [PubMed] [Google Scholar]
- [7].Moss ML, Bartsch JW, Therapeutic benefits from targeting of ADAM family members, Biochemistry 43 (2004) 7227–7235. [DOI] [PubMed] [Google Scholar]
- [8].Primakoff P, Myles DG, The ADAM gene family: surface proteins with adhesion and protease activity, Trends Genet. 16 (2000) 83–87. [DOI] [PubMed] [Google Scholar]
- [9].Schlondorff J, Blobel CP, Metalloprotease-disintegrins: modular proteins capable of promoting cell-cell interactions and triggering signals by protein-ectodomain shedding, J. Cell Sci 2 (Pt 21) (1999) 3603–3617. [DOI] [PubMed] [Google Scholar]
- [10].Klein T, Bischoff R, Active metalloproteases of the A Disintegrin and Metalloprotease (ADAM) family: biological function and structure, J. Proteome Res 10 (2011) 17–33. [DOI] [PubMed] [Google Scholar]
- [11].Blobel ADAMs CP, Key components in EGFR signalling and development, Nat. Rev. Mol. Cell Biol 6 (2005) 32–43. [DOI] [PubMed] [Google Scholar]
- [12].Anders A, Gilbert S, Garten W, Postina R, Fahrenholz F, Regulation of the alpha-secretase ADAM10 by its prodomain and proprotein convertases, FASEB (Fed. Am. Soc. Exp. Biol.) J.: Off. Publ. Fed. Am. Soc. Exper. Biol 15 (2001) 1837–1839. [DOI] [PubMed] [Google Scholar]
- [13].Loechel F, Overgaard MT, Oxvig C, Albrechtsen R, Wewer UM, Regulation of human ADAM 12 protease by the prodomain. Evidence for a functional cysteine switch, J. Biol. Chem 274 (1999) 13427–13433. [DOI] [PubMed] [Google Scholar]
- [14].Fahrenholz F, Gilbert S, Kojro E, Lammich S, Postina R, Alpha-secretase activity of the disintegrin metalloprotease ADAM 10. Influences of domain structure, Ann. N. Y. Acad. Sci 920 (2000) 215–222. [DOI] [PubMed] [Google Scholar]
- [15].Seals DF, Courtneidge SA, The ADAMs family of metalloproteases: multidomain proteins with multiple functions, Genes Dev. 17 (2003) 7–30. [DOI] [PubMed] [Google Scholar]
- [16].Wolfsberg TG, White JM, ADAM Metalloproteinases, Handbook of Proteolytic Enzymes, Academic Press, London, 1998, pp. 709–714. [Google Scholar]
- [17].White ADAMs JM, Modulators of cell-cell and cell-matrix interactions, Curr. Opin. Cell Biol 15 (2003) 598–606. [DOI] [PubMed] [Google Scholar]
- [18].Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA, Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme, J. Biol. Chem 275 (2000) 14608–14614. [DOI] [PubMed] [Google Scholar]
- [19].Smith KM, Gaultier A, Cousin H, Alfandari D, White JM, DeSimone DW, The cysteine-rich domain regulates ADAM protease function in vivo, J. Cell Biol 159 (5) (2002) 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A, EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF, Nature 402 (1999) 884–888. [DOI] [PubMed] [Google Scholar]
- [21].Yan Y, Shirakabe K, Werb Z, The metalloprotease Kuzbanian (ADAM10) mediates the transactivation of EGF receptor by G protein-coupled receptors, J. Cell Biol 158 (2002) 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fischer OM, Hart S, Gschwind A, Ullrich A, EGFR signal transactivation in cancer cells, Biochem. Soc. Trans 31 (2003) 1203–1208. [DOI] [PubMed] [Google Scholar]
- [23].Janes PW, Wimmer-Kleikamp SH, Frangakis AS, Treble K, Griesshaber B, Sabet O, Grabenbauer M, Ting AY, Saftig P, Bastianens PI, Lackmann M, Cytoplasmic relaxation of active Eph controls ephrin shedding by ADAM10, PLoS Biol. 7 (2009) e1000215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang Y, Herrera AH, Li Y, Belani KK, Walcheck B, Regulation of mature ADAM17 by redox agents for L-selectin shedding, J. Immunol 182 (2009) 2449–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Willems SH, Tape CJ, Stanley PL, Taylor NA, Mills IG, Neal DE, McCafferty J, Murphy G, Thiol isomerases negatively regulate the cellular shedding activity of ADAM17, Biochem. J 428 (2010) 439–450. [DOI] [PubMed] [Google Scholar]
- [26].Guan HH, Goh KS, Davamani F, Wu PL, Huang YW, Jeyakanthan J, Wu WG, Chen CJ, Structures of two elapid snake venom metalloproteases with distinct activities highlight the disulfide patterns in the D domain of ADAMalysin family proteins, J. Struct. Biol 169 (3) (2010) 294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Takeda S, Igarashi T, Mori H, Araki S, Crystal structures of VAP1 reveal ADAMs’ MDC domain architecture and its unique C-shaped scaffold, EMBO J. 25 (2006) 2388–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Atapattu L, Lackmann M, Janes PW, The role of proteases in regulating Eph/ ephrin signaling, Cell Adhes. Migrat 8 (2014) 294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fischer OM, Giordano S, Comoglio PM, Ullrich A, Reactive oxygen species mediate Met receptor transactivation by G protein-coupled receptors and the epidermal growth factor receptor in human carcinoma cells, J. Biol. Chem 279 (2004) 28970–28978. [DOI] [PubMed] [Google Scholar]
- [30].Fischer OM, Hart S, Gschwind A, Prenzel N, Ullrich A, Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor, Mol. Cell. Biol 24 (2004) 5172–5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hattori M, Osterfield M, Flanagan JG, Regulated cleavage of a contact-mediated axon repellent, Science 289 (2000) 1360–1365. [DOI] [PubMed] [Google Scholar]
- [32].Rzymski T, Petry A, Kracun D, Reisz F, Pike L, Harris AL, Gorlach A, The unfolded protein response controls induction and activation of ADAM17/TACE by severe hypoxia and ER stress, Oncogene 31 (31) (2012) 3621–3634. [DOI] [PubMed] [Google Scholar]
- [33].Wang B, Mysliwiec T, Feller SM, Knudsen B, Hanafusa H, Kruh GD, Proline-rich sequences mediate the interaction of the Arg protein tyrosine kinase with Crk, Oncogene 13 (1996) 1379–1385. [PubMed] [Google Scholar]
- [34].Chiarugi P, Cirri P, Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction, Trends Biochem. Sci 28 (2003) 509–514. [DOI] [PubMed] [Google Scholar]
- [35].Benz CC, Yau C, Ageing, oxidative stress and cancer: paradigms in parallax, Nat. Rev. Cancer 8 (2008) 875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG, Targeting MET as a strategy to overcome crosstalk-related resistance to EGFR inhibitors, Lancet Oncol. 10 (2009) 709–717. [DOI] [PubMed] [Google Scholar]
- [37].Pillay V, Allaf L, Wilding AL, Donoghue JF, Court NW, Greenall SA, Scott AM, Johns TG, The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases, Neoplasia 11 (2009) 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Neel BG, Gu H, Pao L, The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling, Trends Biochem. Sci 28 (2003) 284–293. [DOI] [PubMed] [Google Scholar]
- [39].Murphy G, The ADAMS: signalling scissors in the tumour microenvironment, Nat. Rev. Cancer 8 (2008) 929–941. [DOI] [PubMed] [Google Scholar]
- [40].Harris RC, Chung E, Coffey RJ, EGF receptor ligands, Exp. Cell Res 284 (1) (2003) 2–13. [DOI] [PubMed] [Google Scholar]
- [41].Blobel CP, Carpenter G, Freeman M, The role of protease activity in ErbB biology, Exp. Cell Res 315 (4) (2009) 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhou B-BS, Petyon M, He B, Liu C, Girard L, Caudler E, Lo Y, Baribaud F, Mikami I, Reguart N, et al. , Targeting ADAM-mediated ligand cleavage to inhibit HER2 and EGFR pathways in non-small cell lung cancer, Cancer Cell 10 (1) (2006) 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yarden Y, Sliwkowski MX, Untangling the ErbB signaling network, Nat. Rev. Mol. Cell Biol 2 (2001) 127–137. [DOI] [PubMed] [Google Scholar]
- [44].Rio C, Buxbaum JD, Peschon JJ, Corfas G, Tumor necrosis factor alpha-converting enzyme is required for cleavage of erbB4/HER4, J. Biol. Chem 275 (14) (2000) 10379–10387. [DOI] [PubMed] [Google Scholar]
- [45].Wang SE, Xiang B, Guix M, Olivares MG, Parker J, Chung CH, Pandiella A, Artiega CL, Transforming growth factor beta engaged TACE and ErbB3 to activate phosphatidylinositol-3 kinase/Akt in ErbB2-overexpressing breast cancer and desensitizes cells to trastuzumab, Mol. Cell. Biol 28 (18) (2008) 5605–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nievergall E, Lackmann M, Janes PW, Eph-dependent cell-cell adhesion and segregation in development and cancer, Cell. Mol. Life Sci 69 (2012) 1813–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Pasquale EB, Eph receptors and ephrins in cancer: bidirectional signaling and beyond, Nat. Rev. Cancer 10 (2010) 165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Blobel CP, Himanen JP, Lackmann M, Nikolov DB, Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans, Cell 123 (2) (2005) 291–304. [DOI] [PubMed] [Google Scholar]
- [49].LaVoie MJ, Selkoe DJ, The notch ligands, jagged and delta, are sequentially processed by α-secretase and presenilin/γ-secretase and release signaling fragments, J. Biol. Chem 278 (2003) 34427–34437. [DOI] [PubMed] [Google Scholar]
- [50].Kopan R, Ilagan MXG, The canonical notch signaling pathway: unfolding the activation mechanism, Cell 137 (2009) 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bray SJ, Notch signalling: a simple pathway becomes complex, Nat. Rev. Mol. Cell Biol 7 (2006) 678–689. [DOI] [PubMed] [Google Scholar]
- [52].Ranganathan P, Weaver KL, Capobianco AJ, Notch signalling in solid tumours: a little bit of everything but not all the time, Nat. Rev. Cancer 11 (2011) 338–351. [DOI] [PubMed] [Google Scholar]
- [53].Espinoza I, Pochampally R, Xing F, Watabe K, Miele L, Notch signaling: targeting cancer stem cells and epithelial-to-mesenchymal transition, OncoTargets Ther. 6 (2013) 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fillipo G, Giancotti. Mechanisms governing metastatic dormancy and reactivation, Cell 155 (2013) 750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Miyamoto S, Rosenberg DW, Role of Notch signaling in colon homeostasis and carcinogenesis, Cancer Sci. 102 (2011) 1938–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, Tozzi F, Sceusi E, Zhou Y, Tachibana I, Maru DM, Hawke DH, Rak J, Mani SA, Zweidler-McKay P, Ellis LM, Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of jagged-1, Cancer Cell 23 (2013) 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lübke T, Lena Illert A, von Figura K, Saftig P, The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts, Hum. Mol. Genet 11 (21) (2002) 2615–2624. [DOI] [PubMed] [Google Scholar]
- [58].Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, Nikkhah G, Dimeco F, Piccirillo S, Vescovi AL, Eberhart CG, NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts, Stem Cells 28 (2010) 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].McGowan PM, Mullooly M, Caiazza F, Sukor S, Madden SF, Maguire AA, Pierce A, McDermott EW, Crown J, O’Donovan N, Duffy MJ, ADAM-17: a novel therapeutic target for triple negative breast cancer, Ann. Oncol 24 (2013) 362–369. [DOI] [PubMed] [Google Scholar]
- [60].Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, Bonal DM, Charytonowicz E, Gladoun N, de la Iglesia-Vicente J, Petrylak DP, Benson MC, Silva JM, Cordon-Cardo C, Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells, Cancer Cell 22 (2012) 373–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Fischer M, Yen WC, Kapoun AM, Wang M, O’Young G, lewicki J, Gurney A, Hoey T, Anti-DLL4 inhibits growth and reduces tumor-initiating cell frequency in colorectal tumors with oncogenic KRAS mutations, Cancer Res. 71 (2011) 1520–1525. [DOI] [PubMed] [Google Scholar]
- [62].Li J, Sainson RC, Oon CE, Turley H, Leek R, Sheldon H, Bridges E, Shi W, Snell C, Bowden ET, Wu H, Chowdhury PS, Russell AJ, Montgomery CP, Poulsom R, Harris HL, DLL4-Notch signaling mediates tumor resistance to anti-VEGF therapy in vivo, Cancer Res. 71 (2011) 6073–6083. [DOI] [PubMed] [Google Scholar]
- [63].McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS, Chin KT, Partridge JC, Poole BB, Cheng KH, Daggett J, Cullen K, Kantoff E, Hasselbatt K, Berkowitz J, Muto MG, Berkowitz RS, Aster JC, Matulonis UA, Dinulescu DM, Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy, Proc. Natl. Acad. Sci 109 (2012) E2939–E2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Meng RD, Shelton CC, Li YM, Qin LX, Notterman D, Paty PB, Gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity, Cancer Res. 69 (2009) 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Timme CR, Gruidi M, Yeatman TJ, Gamma-secretase inhibition attenuates oxaliplatin-induced apoptosis through increased Mcl-1 and/or Bcl-xL in human colon cancer cells, Apoptosis: Int. J. Progr. Cell Death 18 (2013) 1163–1174. [DOI] [PubMed] [Google Scholar]
- [66].Liu S, Zhang W, Liu K, Ji B, Wang G, Silencing ADAM10 inhibits the in vitro and in vivo growth of hepatocellular carcinoma cancer cells, Mol. Med. Rep 11 (2015) 597–602. [DOI] [PubMed] [Google Scholar]
- [67].Jones AV, Lambert DW, Speight PM, Whawell SA, ADAM10 is over expressed in oral squamous cell carcinoma and contributes to invasive behaviour through a functional association with alphavbeta6 integrin, FEBS (Fed. Eur. Biochem. Soc.) Lett 587 (2013) 3529–3534. [DOI] [PubMed] [Google Scholar]
- [68].Liu HB, Zhu Y, Yang QC, Shen Y, Zhang XJ, Chen H, Expression and clinical significance of ADAM17 protein in esophageal squamous cell carcinoma, GMR 14 (2015) 4391–4398. [DOI] [PubMed] [Google Scholar]
- [69].Zheng X, Jiang F, Katakowski M, Lu Y, Chopp M, ADAM17 promotes glioma cell malignant phenotype, Mol. Carcinog 51 (2) (2012) 150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sinnathamby G, Zerfass J, Hafner J, Block P, Nickens Z, Hobeika A, Secord AA, Lyerly HK, Morse MA, Philip R, ADAM metalloprotease domain 17 (ADAM17) is naturally processed through major histocompatibility complex (MHC) class 1 molecules and is a potential immunotherapeutic target in breast, ovarian, and prostate cancers, Clin. Exp. Immunol 163 (3) (2011) 324–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Buchanan PC, Boylan KLM, Walcheck B, Heinze R, Geller MA, Argenta PA, Skubitz APN, Ectodomain shedding of the cell adhesion molecule Nectin-4 in ovarian cancer is mediated by ADAM10 and ADAM17, J. Biol. Chem 292 (15) (2017) 6339–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Boylan KL, Buchanan PC, Manion RD, Shukla DM, Braumberger K, Bruggemeyer C, Skubitz AP, The expression of Nectin-4 on the surface of ovarian cancer cells alters their ability to adhere, migrate, aggregate, and proliferate, Oncotarget 8 (6) (2017) 9717–9738 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ueno M, Shiomi T, Mochizuki S, Chijiiwa M, Shimoda M, Kanai Y, Kataoka F, Hirasawa A, Susumu N, Aoki D, Okada Y, ADAM9 is over-expressed in human ovarian clear cell carcinomas and suppresses cisplatin-induced cell death, Cancer Sci. 109 (2) (2018) 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kossmann CM, Annereau M, Thomas- Schoemann A, Nicco-Overney C, Chereau C, Batteux F, Alexandre J, Lemare F, ADAM9 expression promotes an aggressive lung adenocarcinoma phenotype, Tumour Biol. 39 (7) (2017) 1–11. [DOI] [PubMed] [Google Scholar]
- [75].Oria VO, Lopatta P, Schillig O, The pleiotropic roles of ADAM9 in the biology of solid tumors, Cell. Mol. Life Sci 75 (13) (2018) 2291–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Oria VO, Lopatta P, Schmitz T, Preca BT, Nyström A, Conrad C, Bartsch JW, Kulemann B, Hoeppner J, Maurer J, Bronsert P, Schilling O, ADAM 9 contributes to vascular invasion in pancreatic ductal adenocarcinoma, Mol. Oncol 13 (2019) 456–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Iba K, Albrechtsen R, Gilpin BJ, Loechel F, Wewer UM, Cysteine rich domain of human ADAM12 (meltrin alpha) supports tumor cell adhesion, Am. J. Pathol 154 (5) (1999) 1489–1501 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Shao S, Li Z, Gao W, Yu G, Liu D, Pan F, ADAM-12 as a diagnostic marker for the proliferation, migration and invasion in patients with small cell lung cancer PLoS One, 9 (Issue 1) (2014) e85936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wang J, Voellger B, Benzel J, Schlomann U, Nimsky C, Bartsch JW, Carl B, Metalloproteinases ADAM12 and MMP-14 are associated with cavernous sinus invasion in pituitary adenomas, Int. J. Cancer 139 (6) (2016) 1327–1339. [DOI] [PubMed] [Google Scholar]
- [80].Cheon DJ, Li AJ, Beach JA, Walts AE, Tran H, Lester J, Karlan BY, Orsulic S, ADAM12 is a prognostic factor associated with an aggressive molecular subtype of high-grade serous ovarian carcinoma, Carcinogenesis 36 (7) (2015) 739–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lucas N, Day ML, The role of the disintegrin metalloproteinase ADAM15 in prostate cancer progression, J. Cell. Biochem 15 (6) (2009) 967–974 106. [DOI] [PubMed] [Google Scholar]
- [82].Beck V, Herold H, Benge A, Luber B, Hutzler P, Tschesche H, ADAM15 decreases integrin alphavbeta3/vitronectin-mediated ovarian cancer cell adhesion and motility in an RGD-dependent fashion, Int. J. Biochem. Cell Biol 37 (3) (2005) 590–603. [DOI] [PubMed] [Google Scholar]
- [83].Romagnoli M, Mineva ND, Polmear M, Conrad C, Srinivasan S, Loussouarn D, Barillé-Nion S, Georgakoudi I, Dagg A, McDermott EW, Duffy MJ, McGowan PM, Schlomann U, Parsons M, Bartsch JW, Sonenshein GE, ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis, EMBO Mol. Med 6 (2) (2014) 278–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Schlomann U, Koller G, Conrad C, Ferdous T, Golfi P, Garcia AM, Barillé-Nion S, Georgakoudi I, Dagg A, McDermott EW, Duffy MJ, McGowan PM, Schlomann U, Parsons M, Bartsch JW, Sonenshein GE, ADAM8 as a drug target in pancreatic cancer, Nat. Commun 28 (2015) 6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zmetakova I, Kalinkova L, Smolkova B, Horvathova V, Cierna Z, Danihel L, Bohac M, Sedlackova T, Minarik G, Karaba M, Benca J, Cihova M, Buocikova V, Miklikova S, Mego M, Fridrichova I, A disintegrin and metalloprotease 23 hypermethylation predicts decreased disease-free survival in low-risk breast cancer patients, Cancer Sci. 110 (5) (2019) 1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Fourie AM, Coles F, Moreno V, Karlsson L, Catalytic activity of ADAM8, ADAM15, and MDC-L (ADAM28) on synthetic peptide substrates and in ectodomain cleavage of CD23, J. Biol. Chem 278 (33) (2003) 30469–30477. [DOI] [PubMed] [Google Scholar]
- [87].Mitsui Y, Mochizuki S, Kodama T, Shimoda M, Ohtsuka T, Shiomi T, Chijiiwa M, Ikeda T, Kitajima M, Okada Y, ADAM28 is overexpressed in human breast carcinomas: implications for carcinoma cell proliferation through cleavage of insulin-like growth factor binding protein-3, Cancer Res. 66 (20) (2006) 9913–9920. [DOI] [PubMed] [Google Scholar]
- [88].Manica GC, Ribeiro CF, Oliveira MA, Pereira IT, Chequin A, Ramos EA, Klassen LM, Zanata SM, Noronha L, Rabinovich I, Costa FF, Souza EM, Klassen G, Down regulation of ADAM33 as a predictive biomarker of aggressive breast cancer, Sci. Rep 7 (2017) 44414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wang L, Hoggard JA, Korleski ED, Long GV, Ree BC, Hensley K, Bond SR, Wolfsberg TG, Chen J, Zeczycki T, Bridges LC, Multiple non-catalytic ADAMs are novel integrin α4 ligands, Mol. Cell. Biochem 442 (2018) 29–38. [DOI] [PubMed] [Google Scholar]
- [90].Kelwick R, Desanlis I, Wheeler GN, Edwards DR, The ADAMTS (A disintegrin and metalloproteinase with thrombospondin motifs) family, Genome Biol. 16 (1) (2015) 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Chen J, Luo Y, Zhou Y, Qiu S, Cui R, Yu M, Qin J, Zhong M, Promotion of tumor growth by ADAMTS4 in colorectal cancer: focused on macrophages, Cell. Physiol. Biochem 46 (4) (2018) 1693–1703. [DOI] [PubMed] [Google Scholar]
- [92].Kilic MO, Ayenikin B, Bozer M, Kara A, Haltas H, Icen D, Demicran K, Differentially regulated ADAMTS1, 8, 9, and 18 in pancreas adenocarcinoma, Przeglad Gastroenterol. 12 (4) (2017) 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Fridman JS, Caulder E, Hansbury M, Liu X, Yang G, Wang Q, Lo Y, Zhou BB, Pan AM, Thomas SM, Grandis JR, Zhuo J, Yao W, Newton RC, Freidman SM, Scherle PA, Vaddi K, Selective inhibition of ADAM metalloproteasesas a novel approach for modulating ErbB pathways in cancer, Clin. Cancer Res 13 (2007) 1892–1902. [DOI] [PubMed] [Google Scholar]
- [94].Witters L, Scherle P, Friedman S, Fridman J, Caulder E, Newton R, Lipton A, Synergistic inhibiton with a dual epidermal growth factor receptor/HER-2/neu tyrosine kinase inhibitor and a disintegrin and metalloprotease inhibitor, Can. Res 68 (17) (2008) 7083–7089. [DOI] [PubMed] [Google Scholar]
- [95].Newton RC, Bradley EC, Levy RS, Doval D, Bondarde S, Sahoo TP, Lokanatha D, Julka PK, Nagarkar R, Fridman SM, Targeting ADAM-mediated ligand cleavage to inhibit HER2 and EGFR pathways in non-small cell lung cancer. Cancer Cell, 10(1): 39–50 et al. Clinical benefit of INCB7839, a potent and selective ADAM inhibitor, in combination with trastuzumab in patients with metastatic HER2C breast cancer, J. Clin. Oncol. 28 (15_suppl) (2006) 3025 supplement, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zhang Y, Hegen M, Xu J, Keith JC, Jin G, Du X, Cummons T, Sheppard BJ, Sun YL, Rao VR, Wang Q, Xu W, Cowling R, Nickerson-Nutter CL, Gibbons J, Skotnicki J, Lin LL, Levin J. J Characterization of (2R, 3S)-2-([[4-(2-butynyloxy) phenyl] sulfonyl] amino)-N,3-dihydroxybutanamide, a potent and selective inhibitor of TNF-alpha converting enzyme, Int. Immunopharmacol 4 (2004) 1845–1857. [DOI] [PubMed] [Google Scholar]
- [97].McGowan PM, Simedrea C, Ribot EJ, Foster PJ, Palmieri D, Steeg PS, Allan AL, Chambers AF, Notch1 inhibition alters the CD44hi/CD24lo population and reduces the formation of brain metastases from breast cancer, Mol. Cancer Res 9 (2011) 834–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lv X, Li Y, Qian M, Ma C, Jing H, Wen Z, Qian D, ADAM17 silencing suppresses the migration and invasion of non-small cell lung cancer, Mol. Med. Rep 9 (5) (2014) 1935–1940. [DOI] [PubMed] [Google Scholar]
- [99].Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews VV, et al. , The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion, Blood 102 (2003) 1186–1195 (A). [DOI] [PubMed] [Google Scholar]
- [100].Ludwig A, Hundhausen C, Lambert MH, Broadway N, Andrews RC, Bickett DM, Leesnitzer MA, Becherer JD, Metalloproteinase inhibitors for the disintegrin-like ester inducible shedding of cell surface molecules, Comb. Chem. High Throughput Screen 8 (2005) 161–171. [DOI] [PubMed] [Google Scholar]
- [101].Moss ML, Stoeck A, Yan W, Dempsey PJ, ADAM10 as a target for anti-cancer therapy, Curr. Pharmaceut. Biotechnol 9 (2008) 2–8. [DOI] [PubMed] [Google Scholar]
- [102].Lorenzatti Hiles G, Bucheit A, Rubin JR, Hayward A, Cates AL, C Day K, EI-Sawy L, Kunju LP, Daignault S, Lee CT, Leibert M, Hussain M, Day ML, ADAM15 is functionally associated with the metastatic progression of human bladder cancer, PLoS One 11 (3) (2016) e0150138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Richards FM, Tape CJ, Jodrell DI, Murphy G, Anti-tumour effects of a specific anti-ADAM17 antibody in an ovarian cancer model in vivo, PLoS One 7 (7) (2012) e40597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Tape CJ, Willems SH, Dombernowsky SL, Stanley PL, Fogarasi M, Ouwehand W, McCafferty J, Murphy G, Cross-domain inhibition of TACE ectodomain, Proc. Natl. Acad. Sci. U. S. A 108 (14) (2011) 5578–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Huang Y, Benaich N, Tape C, Kwok HF, Murphy G, Targeting the sheddase activity of ADAM17 by an anti-ADAM17 antibody D1 (A12) inhibits head and neck squamous cell carcinoma cell proliferation and motility via blockage of bradykinin induced HERs transactivation, Int. J. Biol. Sci 10 (2014) 702–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Rios-Doria J, Sabol D, Chesebrough J, Stewart D, Xu L, Tammali R, Cheng L, Du Q, Schifferli K, Rothstein R, Leow CC, Heidbrink-Thompson J, Jin XX, Gao C, Friedman J, Wilkinson B, Damschroder M, Pierce AJ, Hollingsworth RE, Tice DA, Michelotti EF, A monoclonal antibody to ADAM17 inhibits tumor growth by inhibiting EGFR and non-EGFR-mediated pathways, Mol. Cancer Ther 14 (2015) 1637–1649. [DOI] [PubMed] [Google Scholar]
- [107].Peng L, Cook K, Xu L, Cheng L, Damschroder M, Gao C, Wu H, Dall’Acqua WF, Molecular basis for the mechanism of action of an anti-TACE antibody, mAbs 8 (8) (2016) 1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Yamamoto K, Trad A, Baumgart A, Huske L, Lorenzen I, Chalaris A, Grotzinger J, Dechow T, Scheller J, Rose-John S, A novel bispecific single-chain antibody for ADAM17 and CD3 induces T-cell-mediated lysis of prostate cancer cells, Biochem. J 445 (2012) 135–144. [DOI] [PubMed] [Google Scholar]
- [109].Trad AA, Hansen HP, Shomali M, Peipp M, Klausz K, Hedemann N, Yamamoto K, Mauermann A, Desel C, Lorenzen I, Lemke H, Rose-John S, Grötzinger J, ADAM17-overexpressing breast cancer cells selectively targeted by antibody-toxin conjugates, Cancer Immunol. Immunother 62 (3) (2013) 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Kim JM, Jeung HC, Rha SY, Yu EJ, Kim TS, Shin YK, Yamamoto K, Mauermann A, Desel C, Lorenzen I, Lemke H, Rose-John S, Grötzinger J, The effect of disintegrin-metalloproteinase ADAM9 in gastric cancer progression, Mol. Cancer Ther 13 (2014) 3074–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Conrad C, Götte M, Schlomann U, Roessler M, Pagenstecher A, Anderson P, Preston J, Pruessmeyer J, Ludwig AA, Li R, Kamm RD, Ritz R, Carl B, Nimsky C, Bartsch JW, ADAM8 expression in breast cancer derived brain metastases: functional implications on MMP-9 expression and transendothelial migration in breast cancer cells, Int. J. Cancer 142 (4) (2018) 779–791. [DOI] [PubMed] [Google Scholar]
- [112].Dikic I, Schmidt MH, Notch: implications of endogenous inhibitors for therapy, Bioessays 32 (2010) 481–487. [DOI] [PubMed] [Google Scholar]
- [113].Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, De leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, Schahin-Reed D, Dow GJ, Shelton A, Stawicki S, Watts RJ, Zhang J, Choy R, Howard PP, Kadyk L, Yan M, Zha J, Callahan CA CA, Hymowitz SG, Siebel CW, Therapeutic antibody targeting of individual Notch receptors, Nature 464 (2010) 1052–1057 2010. [DOI] [PubMed] [Google Scholar]
- [114].Atapattu L, Saha N, Chheang C, Eissman MF, Xu K, Vail ME, Hii L, Llerena C, Liu Z, Horvay K, Abud HE, Kusebauch U, Moritz RL, Ding BS, Cao Z, Rafii S, Ernst M, Scott AM, Nikolov DB, Lackman M, Janes PW, An activated form of ADAM10 is tumor selective and regulates cancer stem-like cells and tumor growth, J. Exp. Med 213 (9) (2016) 1741–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].M Seegar TC, Killingsworth LB, Saha N, Meyer PA, Patra D, Zimmerman B, Janes PW, Rubinstein E, Nikolov DB, Skiniotis G, Kruse AC, Blacklow SC, Structural basis for regulated proteolysis by the α-secretase ADAM10, Cell 171 (7) (2017) 1638–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N, The cBio Cancer Genomics Portal: an Open Platform for Exploring Multidimensional Cancer Genomics Data Cancer Discov vol 2, (2012), pp. 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N, Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal, Sci. Signal 6 (2013) pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Cheng PF, Dummer R, Levesque MP, Data mining the Cancer Genome Atlas in the era of precision cancer medicine, Swiss Med. Wkly 145 (2015) w14183. [DOI] [PubMed] [Google Scholar]
- [119].Sirintrapun SJ, Zehir A, Syed A, Gao J, Schultz N, Cheng DT, Translational bioinformatics and clinical research (biomedical) informatics, Surg. Pathol. Clin 8 (2015) 269–288. [DOI] [PubMed] [Google Scholar]