
Fig. 1.
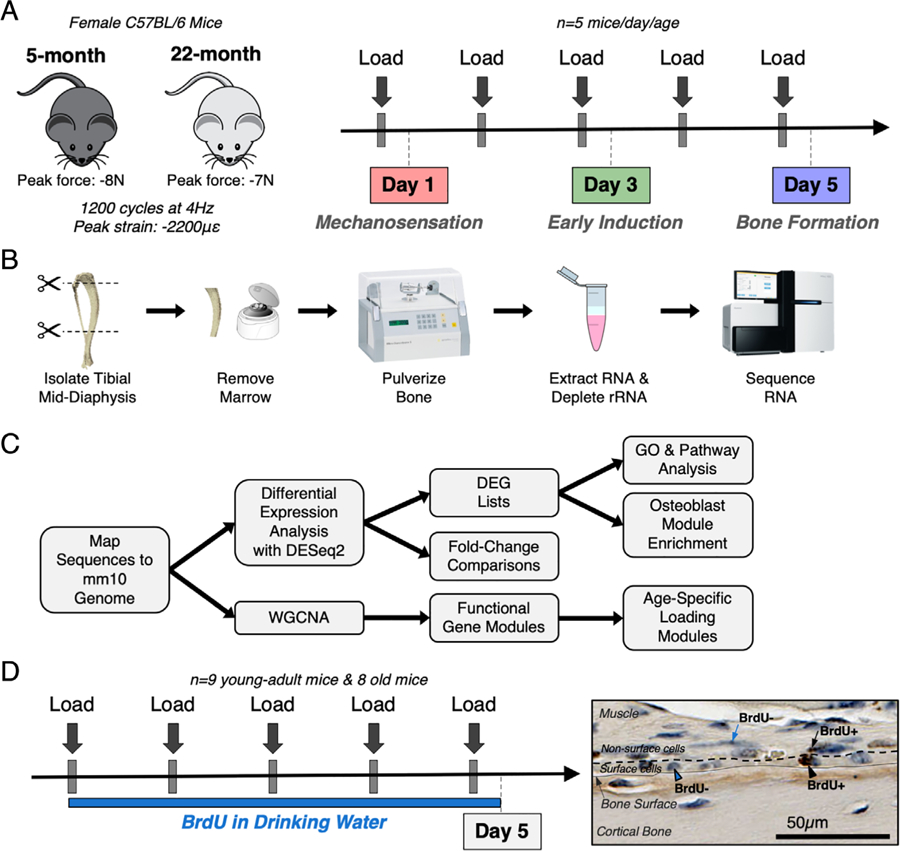
Experimental design for RNA-seq and cell proliferation studies. (A) Thirty female C57BL/6N mice (5 mice/day/age) were subjected to daily in vivo axial tibial compression for either 1 (day 1), 3 (day 3), or 5 (day 5) bouts and euthanized 4 hours after their final bout of loading. (B) The loaded (right) and non-loaded (left) tibial mid-diaphyses were isolated, removed of marrow, and pulverized. RNA was extracted and sequenced. (C) Sequencing reads were mapped to mm10 genome. First, differential expression analysis was performed using DESeq2 to obtain fold changes for each gene at every time point and lists of DEGs using a fold-change threshold of >1.5 or <0.67 and an FDR of 0.05. Pathway and GO analyses were performed using the DEG lists. Second, WGCNA was used to cluster genes into functional modules. (D) Seventeen female C57BL/6N mice (nine young-adult, eight old) were subjected to five bouts of daily in vivo axial tibial compression. Mice were given BrdU to label proliferating cells from the start of loading until euthanasia, 4 hours after their final bout. Surface and non-surface cells were counted as either BrdU+ or BrdU− using BIOQUANT. BrdU = bromodeoxyuridine; DEG = differentially expressed gene; FDR = false discovery rate; GO = gene ontology; WGCNA = weighted gene co-expression network analysis.