

A reduction in the synthesis of the neuromodulator histamine has been associated with Tourette's syndrome and obsessive-compulsive disorder. Symptoms of these disorders are thought to arise from a dysfunction or aberrant development ofcorticostriatal circuits. Here, we investigated how histamine affects developing corticostriatal circuits, both acutely and longer-term, during the first postnatal weeks, using patch-clamp and field recordings in mouse brain slices (C57Bl/6, male and female).
Keywords: basal ganglia, cortex, development, histamine, striatum, synaptic plasticity
Abstract
A reduction in the synthesis of the neuromodulator histamine has been associated with Tourette's syndrome and obsessive-compulsive disorder. Symptoms of these disorders are thought to arise from a dysfunction or aberrant development ofcorticostriatal circuits. Here, we investigated how histamine affects developing corticostriatal circuits, both acutely and longer-term, during the first postnatal weeks, using patch-clamp and field recordings in mouse brain slices (C57Bl/6, male and female). Immunohistochemistry for histamine-containing axons reveals striatal histaminergic innervation by the second postnatal week, and qRT-PCR shows transcripts for H1, H2, and H3 histamine receptors in striatum from the first postnatal week onwards, with pronounced developmental increases in H3 receptor expression. Whole-cell patch-clamp recordings of striatal spiny projection neurons and histamine superfusion demonstrates expression of functional histamine receptors from the first postnatal week onwards, with histamine having diverse effects on their electrical properties, including depolarization of the membrane potential while simultaneously decreasing action potential output. Striatal field recordings and electrical stimulation of corticostriatal afferents revealed that histamine, acting at H3 receptors, negatively modulates corticostriatal synaptic transmission from the first postnatal week onwards. Last, we investigated effects of histamine on longer-term changes at developing corticostriatal synapses and show that histamine facilitates NMDA receptor-dependent LTP via H3 receptors during the second postnatal week, but inhibits synaptic plasticity at later developmental stages. Together, these results show that histamine acutely modulates developing striatal neurons and synapses and controls longer-term changes in developing corticostriatal circuits, thus providing insight into the possible etiology underlying neurodevelopmental disorders resulting from histamine dysregulation.
SIGNIFICANCE STATEMENT Monogenic causes of neurologic disorders, although rare, can provide opportunities to both study and understand the brain. For example, a nonsense mutation in the coding gene for the histamine-synthesizing enzyme has been associated with Tourette's syndrome and obsessive-compulsive disorder, and dysfunction of corticostriatal circuits. Nevertheless, the etiology of these neurodevelopmental disorders and histamine's role in the development of corticostriatal circuits have remained understudied. Here we show that histamine is an active neuromodulator during the earliest periods of postnatal life and acts at developing striatal neurons and synapses. Crucially, we show that histamine permits NMDA receptor-dependent corticostriatal synaptic plasticity during an early critical period of postnatal development, which suggests that genetic or environmental perturbations of histamine levels can impact striatal development.
Introduction
In the adult brain, the neuromodulator histamine controls many processes, including sleep-wakefulness and levels of attention and vigilance, among others (Schwartz et al., 1991; Haas and Panula, 2003; Panula and Nuutinen, 2013). The main neuronal sources of histamine are the histaminergic neurons located in the tuberomamillary nucleus (TMN) of the hypothalamus, which possess widespread axonal arborizations and release histamine throughout the brain (Inagaki et al., 1988). Released histamine acts mainly at three distinct metabotropic histamine receptors expressed in the CNS: the H1, H2, and H3 receptors, which are each coupled to distinct intracellular pathways and regulate many cellular processes (Haas and Panula, 2003). Not only do the histaminergic neurons in the TMN start synthesizing histamine prenatally, the young developing brain also contains additional transient sources of histamine, such as raphe serotonergic neurons (Auvinen and Panula, 1988; Vanhala et al., 1994), ependymal cells lining the ventricle, and mast cells (Nissinen and Panula, 1995; Panula et al., 2014) among others (Zecharia et al., 2012), but the functional role for histamine during early brain development remains largely unknown.
A rare mutation in the gene coding for the histamine-synthesizing enzyme, histidine decarboxylase, which results in overall lower levels of histamine synthesis, was described in a group of patients diagnosed with Tourette's syndrome and obsessive-compulsive disorder (Ercan-Sencicek et al., 2010). The symptoms seen in these disorders are thought to arise from dysfunction of the basal ganglia (Mink, 2001; Albin, 2006; Yael et al., 2015); an interconnected network of subcortical nuclei important in motor behavior and cognitive function (Graybiel et al., 1994; Grillner et al., 2005). The striatum is the main input nucleus of the basal ganglia, which exhibits extensive modulation by histamine in adulthood (Doreulee et al., 2001; Ellender et al., 2011; Castellan Baldan et al., 2014; Bolam and Ellender, 2016; Rapanelli et al., 2017a), and much evidence points to alterations in cortico-striatal-thalamo-cortical loops in Tourette's syndrome (Felling and Singer, 2011; McNaught and Mink, 2011). A series of in vivo studies using mice with reduced brain histamine levels, either through gene KO (Castellan Baldan et al., 2014; Abdurakhmanova et al., 2017) or acutely through chemogenetic manipulation of histaminergic neurons (Rapanelli et al., 2017a,b), have shown that this can recapitulate some key symptoms (e.g., increases in repetitive behaviors) as a result of changes in striatum. However, these studies were focused on adult animals, which limits our understanding of histamine's role during early striatal development as well as in relation to neurodevelopmental disorders.
Here we explored whether histamine modulates the properties of the main neurons of the striatum: the GABAergic spiny projection neurons (SPNs) and their synaptic inputs during the earliest periods of postnatal life when SPNs are maturing and synapses are forming (Tepper et al., 1998; Kozorovitskiy et al., 2012; Peixoto et al., 2016, 2019; Krajeski et al., 2019). Experiments were performed during the first postnatal week (i.e., postnatal day 3–6) when most striatal SPNs have been born but excitatory synaptic inputs are thought to be minimal, the second postnatal week (i.e., postnatal 9–12) when excitatory inputs are undergoing a period of rapid maturation, and young adulthood (i.e., postnatal day 21–35) when the striatal SPNs and circuits are thought to be approaching maturity (Tepper et al., 1998; Khazipov et al., 2004; Dehorter et al., 2011; Kozorovitskiy et al., 2012; Peixoto et al., 2016, 2019) with mice reaching sexually maturity at 35 d (Foster et al., 1983). We find that histamine widely modulates both the electrical properties of striatal neurons as well as the developing corticostriatal synapses, during the earliest periods of postnatal life. Importantly, we find that histamine has a key role in permitting longer-term synaptic changes at corticostriatal synapses during an early critical period of development. Overall, these results suggest that altered histamine levels, as a result of genetic mutations or environmental factors, could acutely, can affect developing corticostriatal circuits.
Materials and Methods
Animals
All experiments were conducted on C57Bl/6 WT mice of both sexes with ad libitum access to food and water. Experiments were designed to use littermates for the various developmental age ranges within single experiments to control for effects of litter sizes and maternal care factors that could affect degree of neuronal and circuit maturity. All mice were bred and individually ventilated cages housed in a temperature-controlled animal facility (normal 12:12 h light/dark cycles) and used in accordance with the UK Animals (Scientific Procedures) Act (1986).
Slice preparation and recording conditions
Acute striatal slices were made from postnatal animals between postnatal day (P) P3–P6, P9–P12, or P21–P35. A small number of mice older than P35 were used for a subset of additional experiments, and results obtained have been reported where appropriate. The mice were anesthetized with isoflurane and then decapitated. Coronal 350–400 µm slices were cut using a vibrating microtome (Microm HM650V). Slices were prepared in aCSF containing the following (in mm): 65 sucrose, 85 NaCl, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 25 NaHCO3, and 10 glucose, pH 7.2–7.4, bubbled with carbogen gas (95% O2/5% CO2). Slices were immediately transferred to a storage chamber containing aCSF (in mm) as follows: 130 NaCl, 3.5 KCl, 1.2 NaH2PO4, 2 MgCl2, 2 CaCl2, 24 NaHCO3, and 10 glucose, pH 7.2–7.4, at 32°C and bubbled with carbogen gas until used for recording. Field recordings were performed in striatal slices transferred to an interface chamber (Scientific Systems Design) and continuously superfused with aCSF bubbled with carbogen gas with the same composition as the storage solution (32°C and perfusion speed of 2 ml/min). Recordings were made using glass pipettes, pulled from standard wall borosilicate glass capillaries and containing aCSF using a Multiclamp 700A amplifier (Molecular Devices), filtered at 4 kHz and acquired at 10 kHz using a X-series USB-6341 A/D board (National Instruments) and WinWCP software (University of Strathclyde, RRID:SCR_014713). Recordings from single neurons were made in striatal slices in a submerged recording chamber (Slicescope Pro 1000, Scientifica) continuously superfused with aCSF bubbled with carbogen gas with the same composition as the storage solution (32°C and perfusion speed of 2 ml/min). Whole-cell current-clamp recordings were performed using glass pipettes, pulled from standard wall borosilicate glass capillaries (to minimize dialysis of cytosolic components, we used 6-8 mΩ resistance pipettes), and containing for whole-cell current-clamp (in mm) as follows: 110 potassium gluconate, 40 HEPES, 2 ATP-Mg, 0.3 Na-GTP, 4 NaCl, and 4 mg/ml biocytin (pH 7.2–7.3; osmolarity, 290-300 mosmol/L). Recordings were made using a Multiclamp 700B amplifier and filtered at 4 kHz and acquired at 10 kHz using an InstruTECH ITC-18 analog/digital board and WinWCP software (University of Strathclyde, RRID:SCR_014713) at 10 kHz.
Stimulation and recording protocols of field recordings
Activation of excitatory cortical afferents was performed using a bipolar stimulating electrode (FHC) or a glass electrode placed in the deep layers of the cortex at a distance to the striatum to avoid direct activation of striatal neurons. Electrical stimulating pulses were given using a DS2 Isolated Stimulator (Digitimer) ranging from 10 to 100 V and 200 μs in duration and stimulation strength was chosen to give half-maximum fEPSPs as recorded from a glass electrode placed in dorsal striatum. All recordings were made in the presence of the GABAA-receptor antagonist SR95531 (gabazine; 200 nm). The stability of fEPSPs during recordings was assessed for 20–30 min before start of experiments, and only recording conditions where the fEPSP amplitude changed <10% were accepted. Cortical afferents were activated every 5–10 s in both drug superfusion and synaptic plasticity experiments. Trains of stimulations consisted of 6 pulses given at 20 Hz repeated at 30 s intervals up to 5 times. Long-term synaptic plasticity protocols consisted of stimulation of cortical afferents at 5–10 s intervals for a minimum of 5 min to obtain a stable baseline measurement of fEPSP amplitude, after which afferents were transiently stimulated using a theta-burst stimulation (TBS) paradigm consisting of brief bursts of stimulation at either 10 or 50 Hz. These bursts consisted of 13 pulses given at 10 or 50 Hz and were repeated 20 times with a 200 ms interval. After this protocol, afferents were activated every 5–10 s for at least 40 min to assess potential longer-term changes in fEPSP amplitude.
Stimulation and recording protocols of single neuron recordings
Hyperpolarizing and depolarizing current steps were used to assess the intrinsic properties of recorded SPNs in dorsal striatum, including input resistance and spike threshold (using small incremental current steps) as well as the properties of action potentials (amplitude, frequency, and duration). Currents step ranges for P3–P6 slices were as follows: –50 to 50 pA; for P9–P12: –100 to 100 pA; and for P21–35: –500 to 500 pA. These ranges of currents were chosen to allow sufficient depolarization of SPNs, taking in consideration changes in input resistance and observations of depolarization block and action potential failure in SPNs. A distinction between striatal SPNs and interneurons was made based on our previous experience (Ellender et al., 2011; Krajeski et al., 2019) and involved a combined assessment of resting membrane potential, input resistance, delay to firing, and overall action potential frequency; and if in doubt, neurons were excluded from experiments and/or data analysis. The stability of whole-cell recordings was assessed for 1–3 min before the start of experiments, including measurements of input resistance and holding current and if these changed during this period by >10% the neuron was discarded.
Analysis of recordings
Data were analyzed offline using custom written programs in Igor Pro (Wavemetrics, RRID:SCR_000325). The input resistance was calculated from the observed membrane potential change after hyperpolarizing the membrane potential with a set current injection. The membrane time constant was calculated as the time it took to reach 63% of the final membrane potential change after hyperpolarizing the membrane potential. The resting membrane potential was obtained continuously during recordings, and the first 5 min of recordings in aCSF was compared with the last 5 min of recordings in the presence of histamine. Individual recording sweeps exhibiting large noise fluctuations were manually removed from analysis. The rheobase was the minimal current injection needed for a striatal SPN to generate an action potential. Time to first spike was the time between the start of the depolarizing step and the emergence of the first observed action potential. The spike rate was calculated from the total number of action potentials observed at the indicated depolarizing step. Other intrinsic properties were calculated from cellular responses after depolarizing steps forP3–P6 at 40 pA, for P9–P12 at 90 pA, and for P21–P35 at 400 pA. The interspike interval (ISI) was the time between two subsequent action potentials, with the first ISI referring to the time between the first and second action potential. The action potential amplitude was taken from the peak amplitude of the individual action potential relative to the average steady-state membrane depolarization during positive current injection. Action potential duration was taken as the duration between the upward and downward stroke of the action potential at 25% of the peak amplitude. fEPSP properties were analyzed from average fEPSPs derived from at least 20 sweeps. fEPSP recordings often included a clear stimulation artifact followed by a first negativity (so-called N1), which reflects direct activation of neurons and axons presynaptic fiber volley, and a second negativity (so-called N2), which reflects synaptic transmission (Malenka and Kocsis, 1988; Flagmeyer et al., 1997); the latter was used for fEPSP measurements. Measurements include peak amplitude (measured as the difference between the prestimulus voltage and the peak of the fEPSP), duration (measured from the start of the upward/downward stroke of the event until its return to the pre-event baseline), rise time (time between 20% and 80% of the peak amplitude), and decay time (measured as the time from peak amplitude until the event returned to baseline). The short-term plasticity at cortical afferents was derived from the amplitude of each fEPSP during train stimulation divided by the amplitude of the first response.
Histologic analyses
Histaminergic afferents were revealed in fixed brain sections using a modification of a previously described protocol (Ellender et al., 2011). In brief, P3–P6, P9–P12, or P21–P35 C57Bl/6 mice were anesthetized with isoflurane, and brains removed and washed in ice-cold cutting aCSF and immediately transferred to ice-cold 4% 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide HCl (Thermo Fisher Scientific, catalog #22981) in 0.1 m PB, pH 6.0, and kept at 4°C for 3 d followed by further fixation in 4% PFA in 0.1 m PBS for 2 d. Sagittal and coronal vibratome sections (40 µm) were collected and subjected to antigen retrieval by heating at 80°C in 10 mm sodium citrate, pH 6.0, for 20-30 min. Sections were then blocked with 10% normal goat serum (Jackson ImmunoResearch Laboratories, AB_2336990 catalog #005-000-121, lot #142539) in PBS containing 1% Triton X-100 for 1 h, at room temperature, followed by incubation for 4 d with 1:500 anti-rabbit histamine antibody (Immunostar, AB_572245, catalog #22939, lot #1532001) in PBS at 4°C under gentle agitation. After this, sections were washed and incubated in 1:500 biotinylated goat-anti-rabbit IgG (Invitrogen, catalog #B2770, lot #1870403) overnight at 4°C. The sections were then washed and incubated in Vectastain ABC Elite (Vector Laboratories, AB_2336827, catalog #PK-6100, lot #ZF0425) for 4 h at room temperature under gentle agitation, followed by incubation with 1:1000 DAPI for 10 min. Sections were washed thoroughly and then incubated overnight at 4°C in 1:1500 ZyMax anti-streptavidin-Cy3 fluorophore (Invitrogen, catalog #438315, lot #1001066A). Finally, all sections were mounted in Vectashield (Vector Laboratories, catalog #H-1000, lot #ZF0409), and images were captured with a DM5000B epifluorescence microscope (Leica Microsystems) using Openlab software (PerkinElmer Life and Analytical Sciences). Images were processed in Adobe Photoshop CS3 and Adobe Illustrator CS3. Measurements of histamine axonal length and intensity were performed using single representative images consisting of 1 mm2 regions of cortex and dorsal striatum containing histamine axons using ImageJ software. Axonal fluorescence signal was measured as the difference in signal between background (average of 30+ points outside of axonal fibers) and the signal within axonal fibers (average of 30+ points inside of axonal fibers) and is expressed as percentage above background. Axons were detected manually in images, but the fluorescent signal within axons had to pass a threshold of 2 SDs above the background signal. Total axonal length was derived from manually traced axons within the 1 mm2 regions. Axonal arborizations were manually counted during tracing of axons.
qRT-PCR
Acute brain slices (400 µm) from P3-P6, P9-P12, or P21-P35 C57Bl/6 mice were made as previously described with the addition of thorough cleaning of all tools and the vibratome with RNaseZAP (Sigma Millipore, catalog #R2020) and wiping the surfaces using RNase Decontamination Wipes (Thermo Fisher Scientific, catalog #AM9786). Brain slices containing the striatum were collected with a brush and transferred into a 1.5 ml RNase-free tube containing 700-1000 µl of RNAlater (Invitrogen, catalog #AM7020) and incubated at 4°C for at least 24 h. After incubation, the brain slices were removed from RNAlater solution and transferred to a Petri dish pretreated with RNase ZAP and containing sterile cutting aCSF. The striatum was dissected under a light microscope, and the sections of striatum were collected in a QIAshredder column (QIAGEN, catalog #7905A) containing 300 µl of PureLink lysis buffer solution (Invitrogen, catalog #12183018A). The striatal tissue was centrifuged at 20,000 × g (Eppendorf Centrifuge, model 5424) to disrupt the tissue. After this, 1 volume of 70% ethanol was added to the homogenate and vortexed to disperse any precipitate. The full volume was transferred to a PureLink spin cartridge and centrifuged (15 s, 13,000 × g). Further washing steps were performed according to the PureLink manufacturer's instructions. Elution of mRNA was performed with 50 µl of the manufacture-provided elution buffer. mRNA quantity and quality were assessed using a Nanodrop spectrophotometer (ND-1000), and a 1% agarose gel was used to confirm mRNA quality. A cDNA library was generated from 1 µg of striatal mRNA using the Super Script IV VILO kit (Thermo Fisher Scientific, catalog #11766050). The genomic DNA digestion, RT, and no-RT reactions were prepared according to the manufacturer's instructions. cDNA samples were kept at −80°C until needed. For the qRT-PCR, three pairs of primers targeting the exon-intron region of the histamine H1, H2, H3, and H4 receptor transcripts as well as the β-actin transcript were designed using the NCBI primer blast web server (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and verified using the oligo analyzer Beacon Designer (PRIMER Biosoft International); they were initially tested for amplification cycle and specificity, and a single pair was used for all future experiments. Primers used for qRT-PCR experiments include the following: for H1 (forward: TGACCAGACCTTGAGCCAGCCCAAAATG and reverse: AGACCTGCTTCTCGCTTTGACTTTGCCC), H2 (forward: CCACAACTCTCACAAAACTTCC and reverse: CTTCTCCTCCTGCCATCTAC), H3 (forward: TGCACAGGTATGGGGTGGGTGAG and reverse: CCAAGGACGCTGAAGACGCTGATG), H4 (forward: TGGAAGAACAGCACGAACACAAAGGAC and reverse: GAAATAAGCCACAGAGATGACAGGAAGCAG) and β-actin (forward: CCAGCCTTCCTTCTTGGGTATC and reverse: CTTTACGGATGTCAACGTCACAC). The primers were resuspended according to the manufacturer's instructions to a concentration of 100 μm. A primer aliquot was used to obtain a 2 μm stock solution. A mix was prepared for each reaction, containing 5 µl of PowerUp SYBER Green Master Mix (Thermo Fisher Scientific, catalog #100029284), 1 µl of the forward primer, 1 µl of the reverse primer (final concentration of 200 nm each primer, 1:10 dilution from the 2 μm stock solution), and 1 µl of nuclease free water; 2 µl of the cDNA (∼1 ng) of each striatal age group (P3–P6, P9–P12, or P21–P35) was added to the corresponding wells in a 96-well plate, for a final volume of 10 µl per reaction. β-actin was used as housekeeping gene, and samples were prepared following the same procedure. After this, the 96-well plate was covered with a Microseal B seal (Bio-Rad, catalog #MSB1001) and centrifuged (1000 × g, 10 s). The qRT-PCR was performed following the PowerUp SYBER Green manufacturer's instructions, and results were detected with a CFX96 real-time PCR detection system (Bio-Rad, catalog #184-5384). Data were collected from the striatal mRNA of 3 mice per age group (three biological replicates). Each experiment was designed to test for H1, H2, H3, and H4 receptor mRNA expression in the same age group, with each receptor being measured by triplicate (technical replicates). Data analysis was performed using the ΔCt method, and normalized against the respective β-actin control samples. A 0.05 ΔCt−1 cutoff was set, which corresponds to the minimum ΔCt value within a window comprising the β-actin Ct values and the maximum 40× Ct amplification cycles. In addition, a threshold of 100 RFU was set for accepting an amplification curve.
Experimental design and statistical analysis
All data are presented as mean ± SEM. n indicates the number of neurons, brain sections, RT-PCR samples, and the number of C57Bl/6 WT mice of both sexes tested. Statistical tests were all two-tailed and performed using SPSS 17.0 (IBM, RRID:SCR_002865) or GraphPad Prism version 5.0 (GraphPad Software, RRID:SCR_002798). Continuous data were assessed for normality, and appropriate parametric (ANOVA, paired t test, and independent t test) or nonparametric (Mann–Whitney U, Wilcoxon Signed Rank, and Kruskal-Wallis) statistical tests were applied.
Drugs and chemicals
Drugs used in electrophysiological experiments included histamine (10 μm), the H3 receptor antagonist thioperamide (10 μm), the NMDA receptor antagonist D-AP5 (50 μm), and the GABAA receptor antagonist SR95531 (200 nm). A concentration-response pilot experiment for histamine (range of 1-100 μm), reducing the amplitude of electrically evoked corticostriatal EPSPs, gave an IC50 of 1.2 μm with a maximum effect at 5 μm (Ellender et al., 2011). All drugs were obtained from Tocris Biosciences.
Results
The striatum contains histaminergic afferents from the second postnatal week onwards
The early postnatal mouse brain contains several sources of histamine, with the main neuronal source being the histaminergic neurons of the TMN, which start producing histamine prenatally and continue to do so throughout postnatal life (Auvinen and Panula, 1988; Vanhala et al., 1994; Nissinen and Panula, 1995). In adulthood, it has been shown that histaminergic neurons of the TMN innervate extensive regions of the brain, including the striatum (Haas and Panula, 2003). Here we explored whether histaminergic afferents are present in the cortex as well as the striatum of younger C57Bl/6 mice at P3–P6, P9–P12, and P21–P35 (n = 3 mice for each age range; Fig. 1A). A polyclonal antibody against histamine was used to label histamine-containing structures in 40-μm-thick sections of 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide HCl-fixed brains and produced clear labeling of histaminergic neurons in the TMN (Fig. 1B). In P3–P6 mice, histaminergic afferents were detected in the cortex (fluorescence signal expressed as percentage above background: 56.18 ± 14.32%) but not in the striatum (Fig. 1C,F). In contrast, histamine immunoreactivity could be readily seen in both cortex and striatum at P9–P12 (cortex: 69.76 ± 30.15%; striatum: 89.56 ± 30.95%) and consisted of thin axonal structures with minimal arborization (cortex, axonal length: 102.01 ± 9.16 µm; no arborization and striatum, axonal length: 67.68 ± 15.95 µm; arborization: 0.33 ± 0.33; Fig. 1D,F). In P21–P35 mice, histamine-containing fibers were observed in both striatum and the cortex at higher densities and were significantly increased for striatum (cortex, axonal length: 126.02 ± 18.08 µm; arborization: 0.66 ± 0.33; and striatum, axonal length: 111.3 ± 6.88 µm; arborization: 3.33 ± 0.33; axonal length striatum: P9-P12 vs P21–P35, F(2,6) = 31.28, p = 0.049; arborization striatum: P9–P12 vs P21–P35, F(2,6) = 45.50, p = 0.0006, ANOVA; n = 3 mice; Fig. 1E,F). These results suggest that histaminergic fibers in the cortex are present just after birth and that, by P9-P12, both cortex and striatum contain histaminergic axonal structures.
Figure 1.

Histaminergic innervation of the developing striatum. A, Schematic representation indicating developmental periods used for histamine labeling experiments. B, Histamine immunoreactivity (red) reveals histaminergic neurons in the TMN of the hypothalamus (red square in diagram) of a P21 mouse. All sections were stained with DAPI to facilitate delineation of anatomic structures; and for display purposes levels, brightness and contrast were optimized. C, At P3–P6, sparse histamine immunoreactive fiber staining was found in the cortex (top, white arrowheads), but not in the striatum (bottom). The diffuse weak signal in the striatum results from autofluorescence of blood vessels in striatum. D, At P9–P12, histamine immunoreactive fibers were found in both the striatum and the cortex (white arrowheads) and were also found at a higher density at (E) P21–P35. F, Quantification of histaminergic axonal length, fluorescence, and arborizations in both cortex (white, left) and striatum (blue, right). Cortical histaminergic innervation was present at birth, and axons did not exhibit a significant increase in length, fluorescence, or arborizations during postnatal development (all p > 0.05). Striatal histaminergic innervation was only detected at P9–P12 (axonal length P3–P6 vs P9–P12, F(2,6) = 31.28, p = 0.0074, and axonal fluorescence P3–P6 vs P9–P12, F(2,6) = 5.90, p = 0.034). ec, External capsule. ns, non-significant, ***p < 0.001; **p < 0.01; *p < 0.05; ANOVA.
Changes in relative expression levels of histamine receptor transcripts in the striatum
We next investigated to what extent histamine receptors are expressed in the striatum and whether expression levels change dynamically across postnatal development. The striatum was dissected from acute brain slices at the three developmental periods, followed by extraction of striatal mRNA and qRT-PCR to assess transcript levels for the H1, H2, H3, and H4 histamine receptors (Fig. 2A,B). Products of the qRT-PCR produced clear single bands of predicted sizes (Fig. 2C). Transcript levels were compared with the housekeeping gene β-actin, which exhibited minimal change across postnatal development (Ct values at P3-P6: 17.59 ± 0.10; P9-P12: 17.90 ± 0.20; and P21–P35: 18.40 ± 0.34, p = 0.14, Kruskal-Wallis; n = 12/3 mice in each age group; Fig. 2C,D).
Figure 2.

Dynamic changes in transcript levels of histamine receptors in the developing striatum. A, The striatum of both hemispheres was dissected from acute brain slices of P3–P6, P9–P12, and P21–P35 C57Bl/6 mice, and mRNA was extracted and processed for qRT-PCR. B, Representative raw amplification curves from single qRT-PCR experiments for each developmental period. The housekeeping gene β-actin (gray gradient) appeared at a similar cycle (Ct) throughout the different ranges, while the histamine receptor amplification cycles changed across development. Note the leftward shift of the histamine H3 receptor curve (blue) at P9–P12 and P21–P35 while the H1 (red) and H2 (green) receptor amplification curves remain relatively constant. Note also the lack of a significant amplification curve for the H4 receptor (orange). qRT-PCR amplification curves obtained from the no-RT samples (bottom panels) served as a control. C, Representative gel of RT-PCR products showing single bands at expected sizes for each of the transcripts in the different developmental periods. Predicted band sizes are for the H1 receptor: 175 bp; H2 receptor: 88 bp; H3 receptor: 187 bp; H4 receptor: 117 bp; and β-actin: 66 bp. D, Consistency of the β-actin cycle (Ct) amplification values at the three developmental periods. E, Transcript levels of the histamine receptors in the striatum. While the levels of transcripts for the H1 and H2 histamine receptors remained relatively constant during postnatal development, the levels for H3 receptor transcripts increased more than twofold by P9–P12 and increased further by P21–P35. No significant transcript levels for the H4 histamine receptor were detected. Values are mean ± SEM of three independent biological replicates, and each biological replicate consisted of three technical replicates (see Materials and Methods). RFU, Relative fluorescence units. ns, non-significant, **p < 0.01; *p < 0.05; ANOVA.
We found that transcripts for the H1 receptor (ΔCt−1 values, P3–P6: 0.10 ± 0.02; P9–P12: 0.12 ± 0.01; and P21–P35: 0.11 ± 0.01; n = 3 mice in each age group, F(2,6) = 1.19, p = 0.36, all vs P3–P6 group, one-way ANOVA), and transcripts for the H2 receptor remained predominantly constant throughout the developmental periods studied, with only a slight but significant increase for the H2 receptor from P3–P6 to P21–P35 (ΔCt −1 values, P3–P6: 0.09 ± 0.003; P9–P12: 0.10 ± 0.002; and P21–P35: 0.11 ± 0.005, F(2,6) = 9.33, p = 0.009, P3–P6 vs P21–P35; n = 3 mice in each age group, one-way ANOVA). In contrast, transcripts for the H3 histamine receptor showed a rapid and significant increase from the first to the second postnatal week, and a further increase from the second postnatal week onwards (ΔCt−1 values, P3–P6: 0.10 ± 0.02; P9–P12: 0.22 ± 0.02; P21–P35: 0.25 ± 0.02; F(2,6) = 21.89, P3–P6 vs P9–P12 p = 0.004 and P3–P6 vs P21–P35 p = 0.0015, one-way ANOVA; n = 3 mice in each age group; Fig. 2D). We did not detect significant expression levels for the H4 receptor at any of the developmental periods (ΔCt−1 values, P3–P6: 0.03 ± 0.02; P9–P12: 0.01 ± 0.03; P21–P35: 0.02 ± 0.03, all vs P3-P6 group, F(2,6) = 0.99, p = 0.42, one-way ANOVA; n = 3 mice in each age group), suggesting that these receptors are not expressed in the striatum (Schneider and Seifert, 2016). In addition, we did not observe amplification in the no-RT reactions for either histamine receptors or β-actin (Fig. 2B). Together, these results suggest that the striatum already contains transcripts coding for the various histamine receptors from the first postnatal days onwards, and, second, that expression levels are dynamic throughout development as exemplified by significant increases in transcript levels for the H2 and H3 histamine receptors.
Histamine modulates the electrical properties of developing striatal SPNs
The observation that the striatum during the first postnatal weeks contains both histaminergic afferents and transcripts coding for the main histamine receptors suggests that released histamine has the potential to act on developing striatal neurons and synapses. Therefore, we next investigated whether the intrinsic electrical properties of striatal SPNs were modulated by histamine by performing whole-cell patch-clamp recordings of SPNs at P3–P6, P9–P12, and P21–P35 in combination with superfusion of histamine (5 μm). The concentration of histamine was chosen based on previous experimental data (Ellender et al., 2011) (see Materials and Methods) and in accordance with previous studies (Jafri et al., 1997; Brown and Haas, 1999; Atzori et al., 2000; Doreulee et al., 2001; Yu et al., 2009; Ellender et al., 2011; Zhuang et al., 2018), reporting a concentration-response and IC50/EC50 values of ∼2–3 μm with maximum effects seen at ∼5 μm. A combination of criteria was used to verify that recordings were made from SPNs (see Materials and Methods) and likely consisted of both D1 and D2 SPNs. Potential changes in the electrical properties of SPNs were measured by comparing the first 5 min in aCSF with the last 5 minafter superfusion of histamine (5 μm). Histamine modulated the intrinsic electrical properties of SPNs in complex ways from the earliest postnatal days onwards; at P3–P6, superfusion of histamine mainly led to a significant decrease in the input resistance (aCSF: 992.41 ± 65.94 mΩ and histamine: 647.68 ± 72.79 mΩ, p = 0.0002, paired t test; n = 13/5 mice; Table 1; Fig. 3A), and a hyperpolarization of the resting membrane potential (aCSF: −70.44 ± 0.08 mV and histamine: −74.47 ± 0.08 mV, p = 0.0001, paired t test; Fig. 3A). Despite these changes, histamine did not significantly modulate the frequency of action potentials elicited by injecting increasing amounts of positive current (at 40 pA aCSF: 14.58 ± 1.78 Hz and histamine: 11.09 ± 2.21 Hz, F(1,56) = 1.28, p = 0.26, two-way ANOVA; Fig. 3A). In contrast, in the second postnatal week at P9-P12 as well as during later developmental periods, histamine superfusion did not alter the input resistance of the SPNs (P9–P12, aCSF: 397.1 ± 23.34 mΩ and histamine: 345.7 ± 25.35 mΩ, p = 0.06, paired t test; n = 22/7 mice and P21-P35, aCSF: 113.4 ± 10.62 mΩ and histamine: 109.4 ± 10.16 mΩ, p = 0.64, paired t test; n = 14/8 mice; Fig. 3B,C); and during these periods, histamine superfusion now led to a significant depolarization of SPNs (P9–P12, aCSF: −71.61 ± 0.14 mV and histamine: −67.50 ± 0.06 mV, p = 0.0001, paired t test; n = 22/7 mice and P21-P35, aCSF: −70.99 ± 0.21 mV and histamine: −65.96 ± 0.15 mV, p = 0.0001, paired t test; n = 14/8 mice; Fig. 3C,D). Notwithstanding the observed depolarization of SPNs, the frequency of action potentials elicited by injecting increasing amounts of positive current was reduced at both P9-P12 (aCSF: 13.14 ± 4.05 Hz and histamine: 9.07 ± 2.99 Hz, F(1,158) = 20.09, p = 0.0001, two-way ANOVA; Fig. 3C) and P21–P35 (aCSF: 32.2 ± 5.6 Hz and histamine: 26.5 ± 3.82 Hz, F(1,110) = 9.99, p = 0.002, two-way ANOVA; Fig. 3D). The reduction in action potential frequency was concurrent with an overall increase in theduration of the ISIs, which was particularly prominent at P9–P12 (% increase vs aCSF, P3–P6: 10.09 ± 0.15%, p = 0.15; P9–P12: 41.97 ± 18.46%, p = 0.02; and P21–P35: 8.71 ± 2.01%, p = 0.59, paired t test; Table 1). Last, histamine modulated the membrane time constant of developing striatal SPNs and led to a shortening in the duration of the membrane time constant at P3–P6 (aCSF: 36.32 ± 1.56 ms and histamine: 23.90 ± 2.79 ms, p = 0.0029, paired t test; n = 12/5 mice) and a minor prolongation at P21–P35 (aCSF: 2.67 ± 0.16 ms and histamine: 3.49 ± 0.37 ms, p = 0.008, pairedt test; n = 16/8 mice; Table 1). We did not find other histamine-mediated changes in the intrinsic electrical properties of striatal SPNs (Table 1).
Table 1.
Histaminergic modulation of intrinsic electrophysiological properties of striatal SPNs across postnatal developmentaa
| aCSF | Histamine | p | |
|---|---|---|---|
| P3–P6 | |||
| Resting membrane potential (mV) | –70.73 ± 0.16 | –74.47 ± 0.08 | 1.00E-06 |
| Input resistance (mΩ) | 992.41 ± 65.94 | 647.68 ± 72.79 | 2.22E-04 |
| Membrane time constant (ms) | 36.32 ± 1.56 | 23.90 ± 2.79 | 0.0029 |
| Rheobase (pA) | 26.92 ± 2.56 | 33.76 ± 5.48 | 0.26 |
| Time to first spike (ms) | 168.75 ± 22.83 | 114.89 ± 35.57 | 0.36 |
| Spike rate 50 pA (Hz) | 16.92 ± 1.67 | 11.56 ± 2.06 | 0.16 |
| Spike rate 40 pA (Hz) | 14.58 ± 1.78 | 11.09 ± 2.21 | 0.82 |
| Spike rate 30 pA (Hz) | 11.8 ± 1.52 | 12.5 ± 1.76 | 0.99 |
| Spike rate 20 pA (Hz) | 10 ± 1.29 | 9.68 ± 1.33 | 0.99 |
| First ISI (ms) | 46.72 ± 4.72 | 51.65 ± 5.32 | 0.97 |
| Second ISI (ms) | 51.46 ± 4.84 | 59.05 ± 4.27 | 0.90 |
| Third ISI (ms) | 53.22 ± 2.95 | 55.03 ± 3.13 | 0.98 |
| Fourth ISI (ms) | 56.65 ± 2.57 | 61.54 ± 4.35 | 0.94 |
| First spike amplitude (mV) | 57.82 ± 3.74 | 65.41 ± 2.57 | 0.23 |
| Second spike amplitude (mV) | 50.09 ± 5.02 | 53.31 ± 5.75 | 0.54 |
| First spike duration (ms) | 3.37 ± 0.20 | 3.17 ± 0.19 | 0.88 |
| Second spike duration (ms) | 5.74 ± 0.74 | 4.37 ± 0.21 | 0.06 |
| P9–P12 | |||
| Resting membrane potential (mV) | –73.31 ± 0.07 | –67.35 ± 0.09 | 1.00E-04 |
| Input resistance (mΩ) | 459.84 ± 50.42 | 368.44 ± 28.69 | 0.06 |
| Membrane time constant (ms) | 18.13 ± 1.45 | 16.15 ± 1.17 | 0.22 |
| Rheobase (pA) | 52.27 ± 3.71 | 54.50 ± 4.89 | 0.77 |
| Time to first spike (ms) | 199.82 ± 12.75 | 170.86 ± 23.18 | 0.26 |
| Spike rate 100 pA (Hz) | 22.97 ± 1.66 | 16.97 ± 1.55 | 0.005 |
| Spike rate 80 pA (Hz) | 18.88 ± 1.90 | 13.68 ± 1.42 | 0.02 |
| Spike rate 60 pA (Hz) | 15.33 ± 1.67 | 9.37 ± 1.22 | 0.01 |
| Spike rate 40 pA (Hz) | 8.5 ± 0.91 | 5.35 ± 0.56 | 0.99 |
| First ISI (ms) | 56.8 ± 8.25 | 60.43 ± 9.82 | 0.77 |
| Second ISI (ms) | 52.62 ± 6.61 | 65.66 ± 8.41 | 0.72 |
| Third ISI (ms) | 48.38 ± 5.56 | 61.33 ± 4.08 | 0.42 |
| Fourth ISI (ms) | 41.96 ± 2.73 | 59.29 ± 4.73 | 0.03 |
| First spike amplitude (mV) | 68.53 ± 3.15 | 65.19 ± 2.52 | 0.64 |
| Second spike amplitude (mV) | 63.77 ± 3.21 | 55.75 ± 3.17 | 0.14 |
| First spike duration (ms) | 2.5 ± 0.19 | 2.3 ± 0.06 | 0.49 |
| Second spike duration (ms) | 3.35 ± 0.28 | 3.04 ± 0.11 | 0.33 |
| P21–P35 | |||
| Resting membrane potential (mV) | –70.99 ± 0.21 | –65.96 ± 0.15 | 1.00E-05 |
| Input resistance (mΩ) | 113.44 ± 10.62 | 109.38 ± 10.15 | 0.64 |
| Membrane time constant (ms) | 2.67 ± 0.16 | 3.49 ± 0.37 | 0.008 |
| Rheobase (pA) | 128.71 ± 17.21 | 112.30 ± 19.09 | 0.30 |
| Time to first spike (ms) | 109.70 ± 21.12 | 76.48 ± 15.24 | 0.19 |
| Spike rate 500 pA (Hz) | 46.07 ± 1.71 | 34.71 ± 2.14 | 0.0015 |
| Spike rate 400 pA (Hz) | 42.5 ± 2.15 | 33.36 ± 1.97 | 0.01 |
| Spike rate 300 pA (Hz) | 35.53 ± 2.80 | 28.75 ± 2.48 | 0.21 |
| Spike rate 200 pA (Hz) | 28.97 ± 2.46 | 22.39 ± 2.12 | 0.15 |
| Spike rate 100 pA (Hz) | 14.64 ± 2.09 | 13.92 ± 2.10 | 0.99 |
| First ISI (ms) | 17.67 ± 1.73 | 17.92 ± 2.14 | 0.99 |
| Second ISI (ms) | 19.25 ± 1.76 | 23.97 ± 2.59 | 0.75 |
| Third ISI (ms) | 21.31 ± 1.49 | 29.72 ± 2.48 | 0.96 |
| Fourth ISI (ms) | 22.83 ± 1.59 | 32.74 ± 3.55 | 0.18 |
| First spike amplitude (mV) | 75.80 ± 4.65 | 75.21 ± 4.52 | 0.99 |
| Second spike amplitude (mV) | 66.28 ± 4.37 | 59.86 ± 4.39 | 0.94 |
| First spike duration (ms) | 1.81 ± 0.12 | 1.69 ± 0.08 | 0.96 |
| Second spike duration (ms) | 2.51 ± 0.16 | 2.40 ± 0.13 | 0.98 |
aData are mean ± SEM, statistical comparisons by Student's t test and ANOVA.
Figure 3.
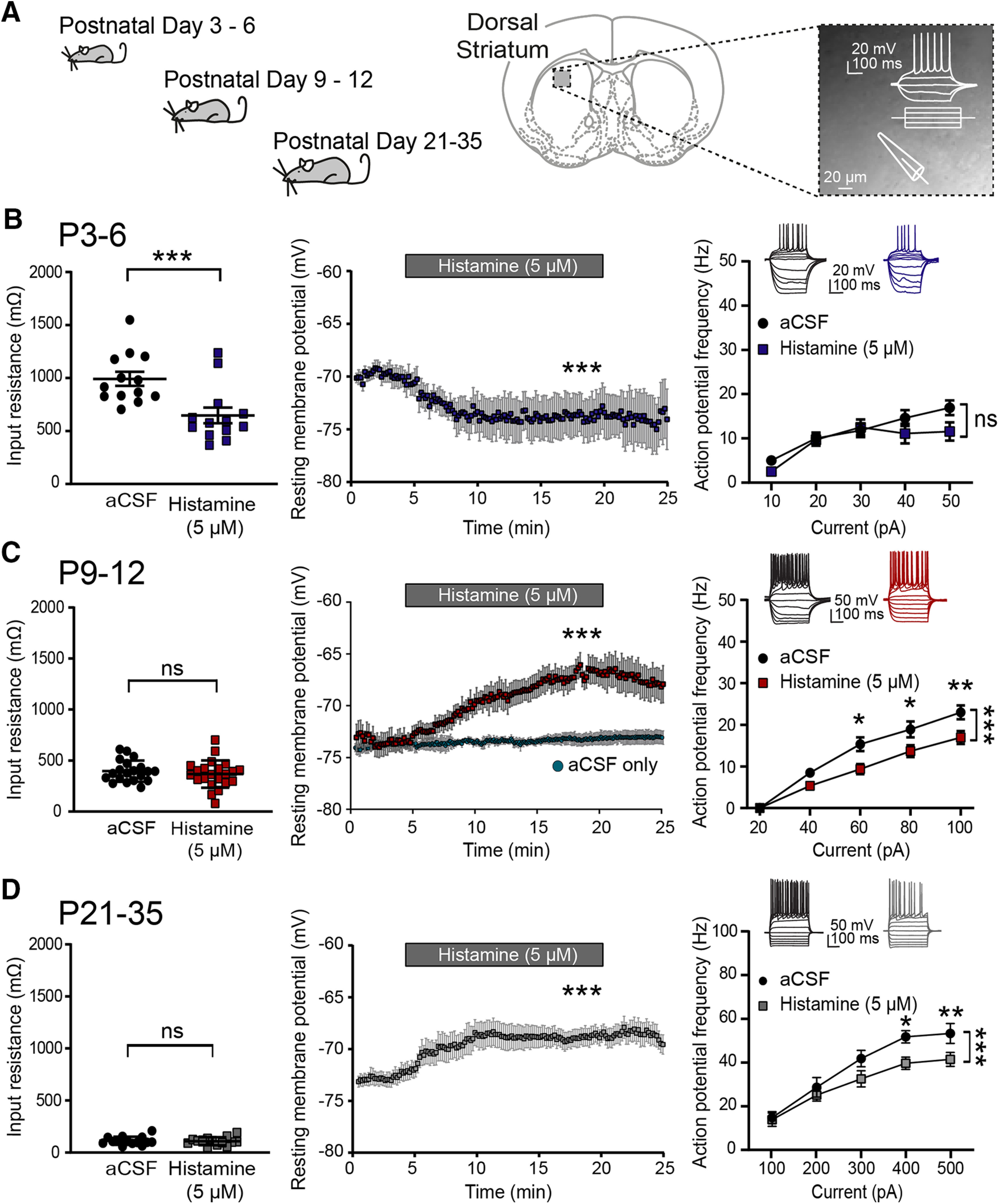
Histamine modulates the intrinsic electrical properties of developing striatal SPNs. A, The intrinsic electrical properties of developing SPNs were examined by performing whole-cell patch-clamp recordings in acute brain slices from P3–P6, P9–P12, and P21–P35 C57Bl/6 mice. B, At P3–P6, histamine superfusion lowered the input resistance (left) and led to a hyperpolarization of SPNs (middle). The action potential frequency of SPNs induced by positive current injection was unaffected (right). C, At P9–P12, histamine did not affect the input resistance but now led to a depolarization of SPNs and significantly decreased their action potential frequency. Little to no depolarization was observed in the absence of histamine superfusion (aCSF only; middle) during prolonged recordings from SPNs (1–5 min: −71.84 ± 0.50 mV; and 20–25 min: −70.80 ± 0.80 mV, p = 0.058, paired t test; n = 7/3). D, At P21–P35, histamine superfusion did not change the input resistance but led to a depolarization of SPNs and a significant decrease in action potential frequency. The indication of “histamine” refers to start of superfusion. Values are mean ± SEM for all datasets. Statistical p values are provided in Table 1. ns, non-significant, ***p < 0.001; **p < 0.01; *p < 0.05; ANOVA and paired t test.
Together, these data show that histamine can modulate the intrinsic electrical properties of developing striatal SPNs from the first postnatal days onwards. Overall, the main effect of histamine appears to be to change the membrane potential of SPNs in conjunction with a general reduction in their action potential output.
Histamine modulates excitatory glutamatergic transmission at developing corticostriatal synapses
We next considered whether histamine was able to modulate the excitatory synaptic inputs to the developing striatum. Previously, it was shown that histamine could negatively modulate synaptic transmission by acting at presynaptic H3 histamine receptors in adult mice (Doreulee et al., 2001; Ellender et al., 2011), but whether histamine modulates synaptic transmission at developing synapses also is unknown. We investigated the effect of histamine on transmission at developing corticostriatal synapses in acute brain slices kept in an interface chamber (Haas et al., 1979), at the interface between aCSF and humidified carbogen gas, and corticostriatal afferents were activated using a stimulating electrode placed in the cortex (bordering the external capsule) with fEPSPs recorded with a glass electrode placed in dorsal striatum (Fig. 4A). This recording configuration does not disturb the cytosolic components of SPNs (Lahiri and Bevan, 2020), as synaptic events are recorded extracellularly, and allows for stable and long duration recordings. Recorded fEPSPs are a reflection of the summed synaptic activity occurring at a large numbers of corticostriatal excitatory synapses, principally on striatal SPNs with a likely similar contribution of both D1 and D2 SPNs (Malenka and Kocsis, 1988; Flagmeyer et al., 1997). All recordings were performed in the presence of the GABAA receptor antagonist SR95531 (gabazine; 200 nm) to minimize recruitment of GABAergic afferents and facilitate isolation of glutamatergic afferents. Indeed, evoked fEPSPs could be blocked by the addition of the glutamatergic antagonists NBQX (40 μm) and D-AP5 (50 μm) to the aCSF (to 9.24 ± 8.76% of baseline amplitude; n = 5/3 mice; Fig. 4A). In these experiments, a slightly higher concentration of histamine was used (10 μm) to guarantee a sufficient concentration of histamine under these recording conditions, with aCSF predominantly flowing underneath slices, as has also been used extensively in previous studies (Jafri et al., 1997; Brown and Haas, 1999; Atzori et al., 2000; Doreulee et al., 2001; Yu et al., 2009; Ellender et al., 2011; Zhuang et al., 2018). Three main observations were made during these experiments. First, we found that the properties of corticostriatal fEPSPs were dynamic and changed during early postnatal development, similar to previous observations made from single SPNs (Krajeski et al., 2019), and include a progressive increase in amplitude and a reduction in the duration of evoked fEPSPs (Table 2). Second, we found that histamine (10 μm) superfusion rapidly decreased the amplitude of corticostriatal fEPSPs in all developmental periods (normalized change in fEPSP amplitude, P3-P6: to 87.95 ± 4.57%; P9-P12: to 83.87 ± 5.46%; and P21–P35: to 83.29 ± 4.04%: p = 0.046, p = 0.04,2 and p = 0.009, respectively, paired t test; n = 6/5 mice, 5/5 mice and 6/6 mice; Fig. 4B–D), and continued to do so in brain slices from older animals also (P35+: to 76.66 ± 7.76%, p = 0.008; n = 10/5 mice). Other properties of the fEPSPs were found to be unaffected by histamine superfusion (Table 3). Such reductions in fEPSP amplitude were not observed during control experiments consisting of continued superfusion with aCSF (normalized change in fEPSP amplitude, P3–P6: to 100.82 ± 2.22%; P9-P12: to 100.38 ± 1.48%; and P21–P35: to 103.93 ± 1.60%: p = 1.000, p = 0.715 and p = 0.109, Wilcoxon Signed Rank Test; n = 4/4 mice; n = 4/3 mice, and n = 3/3 mice; Fig. 4B–D). Last, to investigate at which receptors histamine might be acting, we superfused histamine together with the H3 receptor antagonist thioperamide, as histamine had previously been shown to regulate corticostriatal transmission through H3 histamine receptors in adult mice (Doreulee et al., 2001; Ellender et al., 2011). The potent and selective H3 receptor antagonist thioperamide acts in the nm range in cell-based assays (Arrang et al., 1987, 1995; Hew et al., 1990; Morisset et al., 2000; Molina-Hernández et al., 2001; Gbahou et al., 2006), in the μm range in brain slices studies (Arias-Montano et al., 2001) and does not produce expected behavioral effects in H3 receptor KO mice (Toyota et al., 2002); and for the reasons outlined above, a moderately high concentration of thioperamide (10 μm) was used to guarantee a sufficient concentration at slices under these recording conditions. These experiments revealed that, similar to observations in adult mice (Doreulee et al., 2001; Ellender et al., 2011), the reduction in fEPSP amplitude was not seen when histamine was superfused together with thioperamide (normalized change in fEPSP amplitude, P3–P6: to 95.19 ± 2.44%; P9–P12: to 100.39 ± 2.17%; and P21–P35: to 99.97 ± 3.70%: p = 0.096, p = 0.862, and p = 0.993, respectively, paired t test; n = 7/2 mice, 8/3 mice and 5/2 mice; Fig. 4B–D).
Figure 4.
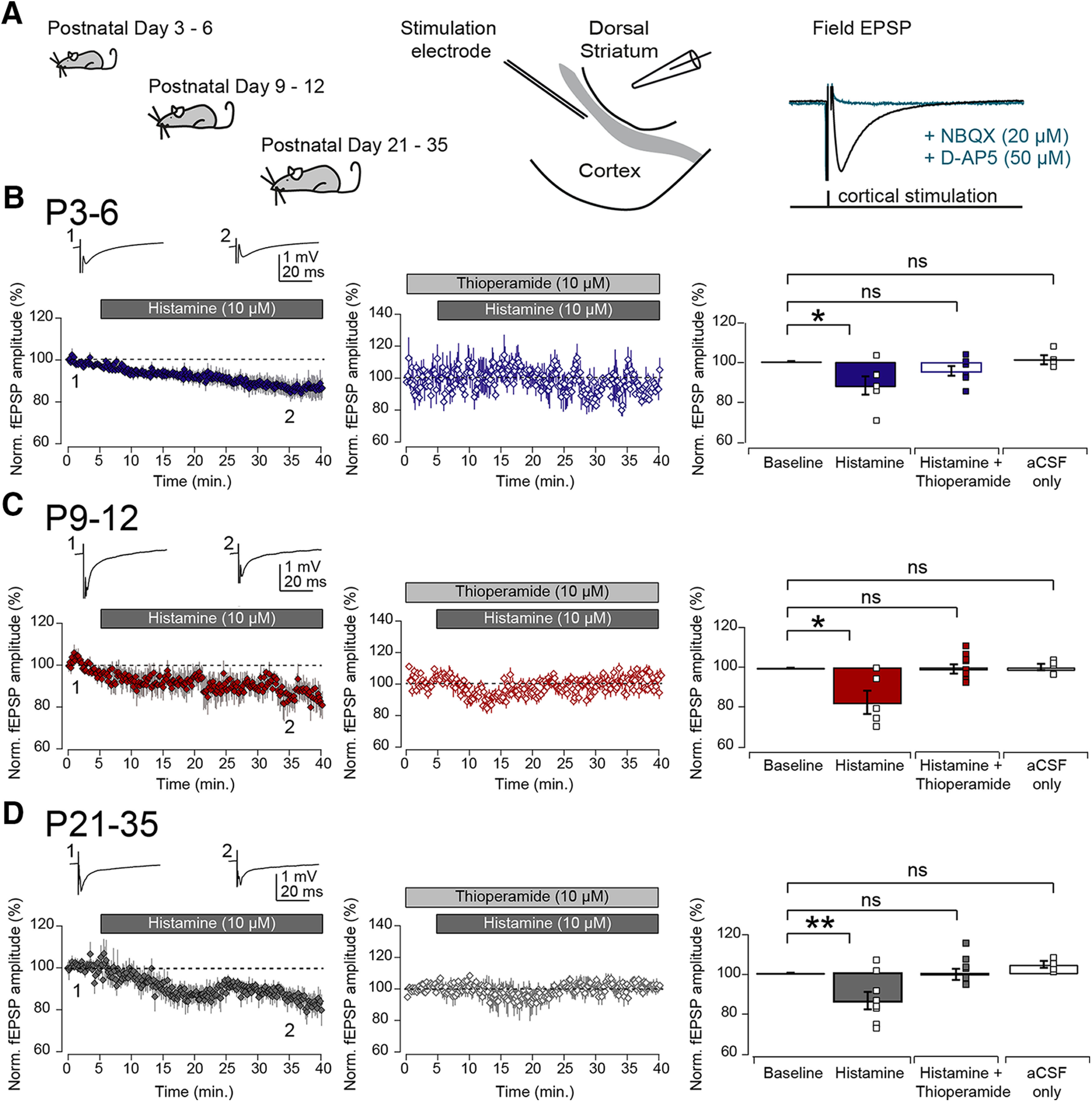
Histamine negatively modulates corticostriatal transmission during early postnatal periods. A, Acute coronal brain slices were made from C57Bl/6 mice at the indicated age ranges and slices kept in an interface recording chamber. Cortical afferents were stimulated electrically and corticostriatal fEPSPs recorded with a glass electrode placed in dorsal striatum. Evoked fEPSPs could be blocked by the addition of the glutamatergic antagonists NBQX (20 μm) and D-AP5 (50 μm, right). B, Left, At P3–P6, superfusion of histamine (10 μm) significantly reduced the peak amplitude of the evoked corticostriatal fEPSPs (top, two example fEPSP traces corresponding to time points indicated in the graph). Middle, The addition of the H3 receptor antagonist thioperamide (10 μm) to the aCSF blocked the histamine-mediated reduction in corticostriatal fEPSP amplitude. Right, Bar plots represent the effect of superfusion of histamine, histamine together with thioperamide, or aCSF-only on the corticostriatal fEPSP amplitude. Plotted values are normalized means of the first 5 min of recording for baseline versus the last 5 min of recordings. At both P9–P12 (C) and at P21–P35 (D), superfusion of histamine also led to a significant reduction in the amplitude of the corticostriatal fEPSP, which was also blocked by coapplication of the H3 receptor antagonist thioperamide and did not exhibit changes in aCSF-only controls. The indication of “histamine” refers to start of superfusion, which precedes histamine being present at the slice. ns, non-significant, **p < 0.01; *p < 0.05; paired t test and Wilcoxon signed rank test.
Table 2.
Dynamic changes in the properties of corticostriatal fEPSP properties across postnatal developmentaa
| P3–P6 | P9–P12 | P21–P35 | |
|---|---|---|---|
| fEPSP amplitude (mV) | 0.65 ± 0.11 | 0.80 ± 0.10 | 1.63 ± 0.27 |
| fEPSP duration (ms) | 143.19 ± 19.26 | 148.35 ± 15.86 | 98.20 ± 24.93 |
| fEPSP rise time (ms) | 11.08 ± 4.05 | 9.82 ± 2.84 | 10.60 ± 3.45 |
| fEPSP decay time (ms) | 138.22 ± 16.72 | 138.53 ± 13.10 | 87.60 ± 21.62 |
| Short-term plasticity (2 vs 1) | 1.42 ± 0.13 | 1.01 ± 0.18 | 1.02 ± 0.07 |
| Short-term plasticity (3 vs 1) | 1.49 ± 0.15 | 0.97 ± 0.21 | 0.91 ± 0.08 |
| Short-term plasticity (4 vs 1) | 1.41 ± 0.24 | 0.93 ± 0.23 | 0.86 ± 0.10 |
| Short-term plasticity (5 vs 1) | 1.37 ± 0.28 | 0.95 ± 0.25 | 0.84 ± 0.11 |
| Short-term plasticity (6 vs 1) | 1.41 ± 0.28 | 0.96 ± 0.28 | 0.84 ± 0.12 |
aData are mean ± SEM.
Table 3.
Histaminergic modulation of corticostriatal fEPSP properties across postnatal developmenta
| aCSF | Histamine | p | |
|---|---|---|---|
| P3–P6 | |||
| fEPSP amplitude (mV) | 0.41 ± 0.10 | 0.34 ± 0.07 | 0.56 |
| fEPSP duration (ms) | 241.58 ± 39.55 | 247.41 ± 35.16 | 0.92 |
| fEPSP rise time (ms) | 30.02 ± 9.17 | 30.68 ± 8.59 | 0.94 |
| fEPSP decay time (ms) | 211.55 ± 30.99 | 216.74 ± 27.77 | 0.92 |
| Short-term plasticity (2 vs 1) | 1.48 ± 0.18 | 1.74 ± 0.30 | 0.48 |
| Short-term plasticity (3 vs 1) | 1.55 ± 0.20 | 1.89 ± 0.37 | 0.44 |
| Short-term plasticity (4 vs 1) | 1.47 ± 0.33 | 1.86 ± 0.48 | 0.52 |
| Short-term plasticity (5 vs 1) | 1.48 ± 0.38 | 1.84 ± 0.52 | 0.58 |
| Short-term plasticity (6 vs 1) | 1.52 ± 0.38 | 1.98 ± 0.59 | 0.53 |
| P9–P12 | |||
| fEPSP amplitude (mV) | 0.62 ± 0.15 | 0.50 ± 0.08 | 0.47 |
| fEPSP duration (ms) | 224.74 ± 48.24 | 225.52 ± 55.01 | 0.99 |
| fEPSP rise time (ms) | 24.22 ± 9.89 | 26.57 ± 10.94 | 0.88 |
| fEPSP decay time (ms) | 200.52 ± 38.41 | 198.96 ± 44.26 | 0.98 |
| Short-term plasticity (2 vs 1) | 1.01 ± 0.18 | 1.03 ± 0.12 | 0.93 |
| Short-term plasticity (3 vs 1) | 0.97 ± 0.21 | 0.91 ± 0.15 | 0.82 |
| Short-term plasticity (4 vs 1) | 0.93 ± 0.23 | 0.86 ± 0.20 | 0.83 |
| Short-term plasticity (5 vs 1) | 0.95 ± 0.25 | 0.87 ± 0.19 | 0.82 |
| Short-term plasticity (6 vs 1) | 0.96 ± 0.28 | 0.89 ± 0.21 | 0.83 |
| P21–P35 | |||
| fEPSP amplitude (mV) | 1.74 ± 0.75 | 1.50 ± 0.65 | 0.81 |
| fEPSP duration (ms) | 196.93 ± 58.46 | 188.62 ± 51.16 | 0.92 |
| fEPSP rise time (ms) | 18.56 ± 8.99 | 19.50 ± 9.22 | 0.94 |
| fEPSP decay time (ms) | 178.37 ± 49.69 | 169.12 ± 42.80 | 0.89 |
| Short-term plasticity (2 vs 1) | 0.92 ± 0.05 | 1.19 ± 0.21 | 0.26 |
| Short-term plasticity (3 vs 1) | 0.79 ± 0.07 | 1.15 ± 0.25 | 0.20 |
| Short-term plasticity (4 vs 1) | 0.71 ± 0.09 | 1.07 ± 0.21 | 0.17 |
| Short-term plasticity (5 vs 1) | 0.70 ± 0.08 | 0.97 ± 0.17 | 0.20 |
| Short-term plasticity (6 vs 1) | 0.67 ± 0.08 | 1.03 ± 0.20 | 0.14 |
aData are mean ± SEM; statistical comparisons by Mann–Whitney U test.
In conclusion, these results show that histamine is already able to modulate corticostriatal synaptic transmission from the first postnatal days onwards and does so by acting at H3 histamine receptors.
Histamine controls the induction of long-term synaptic plasticity at developing corticostriatal synapses
So far, our data show that the developing striatum receives histaminergic afferents and expresses functional histamine receptors, with superfusion of histamine acutely modulating the functional properties of developing striatal circuits. Alongside acute effects on neurons and circuits, we next asked whether histamine might also effect longer-term changes at striatal synapses, resulting in more persistent alterations in corticostriatal circuits. Two questions were next considered: (1) Do corticostriatal synapses exhibit long-term synaptic plasticity during the first weeks of postnatal development? (2) Is the induction of long-term synaptic plasticity during this period modulated by histamine? A number of studies have shown that mature corticostriatal synapses can undergo activity-dependent prolonged changes in synaptic strength using a variety of protocols (Fino et al., 2005; Kreitzer and Malenka, 2008; Kozorovitskiy et al., 2012; Fisher et al., 2017). We next investigated whether developing corticostriatal synapses exhibit long-term synaptic plasticity using two different stimulation patterns, consisting of a series of brief bursts at either 10 or 50 Hz (see Materials and Methods). As these brief bursts were repeated with 200 ms intervals (i.e., at theta frequency), such stimulation patterns are often referred to as TBS (Larson et al., 1986). TBS has been shown to be particularly effective at inducing long-term changes in synaptic strength at adult corticostriatal synapses (Hawes et al., 2013), reflecting activity patterns observed during naturalistic behavior (Tort et al., 2008), and 10 or 50 Hz frequencies of activity reflecting physiological activity patterns found in the young developing brain (Khazipov et al., 2004; Hanganu et al., 2006; Yang et al., 2009). Slices were kept at interface conditions as previously in the presence of the GABAA receptor antagonist SR95531 (200 nm), and cortical afferents were stimulated with a glass or bipolar stimulation electrode with fEPSP recordings made from a recording electrode placed in dorsal striatum (Fig. 5A). Although we observed brief post-tetanic potentiation, we find, at P3–P6, neither 10 Hz nor 50 Hz TBS stimulation resulted in long-term changes in synaptic efficacy (aCSF 10 Hz: 104.41 ± 6.22% and 50 Hz: 106.49 ± 7.46%, p = 0.51 and p = 0.42, paired t test, both n = 6/6 mice; Fig. 5B), and this was not altered by the presence of histamine in the recording aCSF (histamine 10 Hz: 96.25 ± 4.04% and 50 Hz: 100.20 ± 7.47%, p = 0.41 and p = 0.98, paired t test, both n = 5/5 mice; Fig. 5B). To confirm stability of fEPSP recordings, we also recorded the amplitude of fEPSPs for similar duration without TBS and found that fEPSP amplitudes stay constant over time (first 5 min: 100.1 ± 0.05% and last 5 min: 101.30 ± 1.10%, p = 0.27, paired t test; n = 11/10 mice of all age ranges; Fig. 5C).
Figure 5.
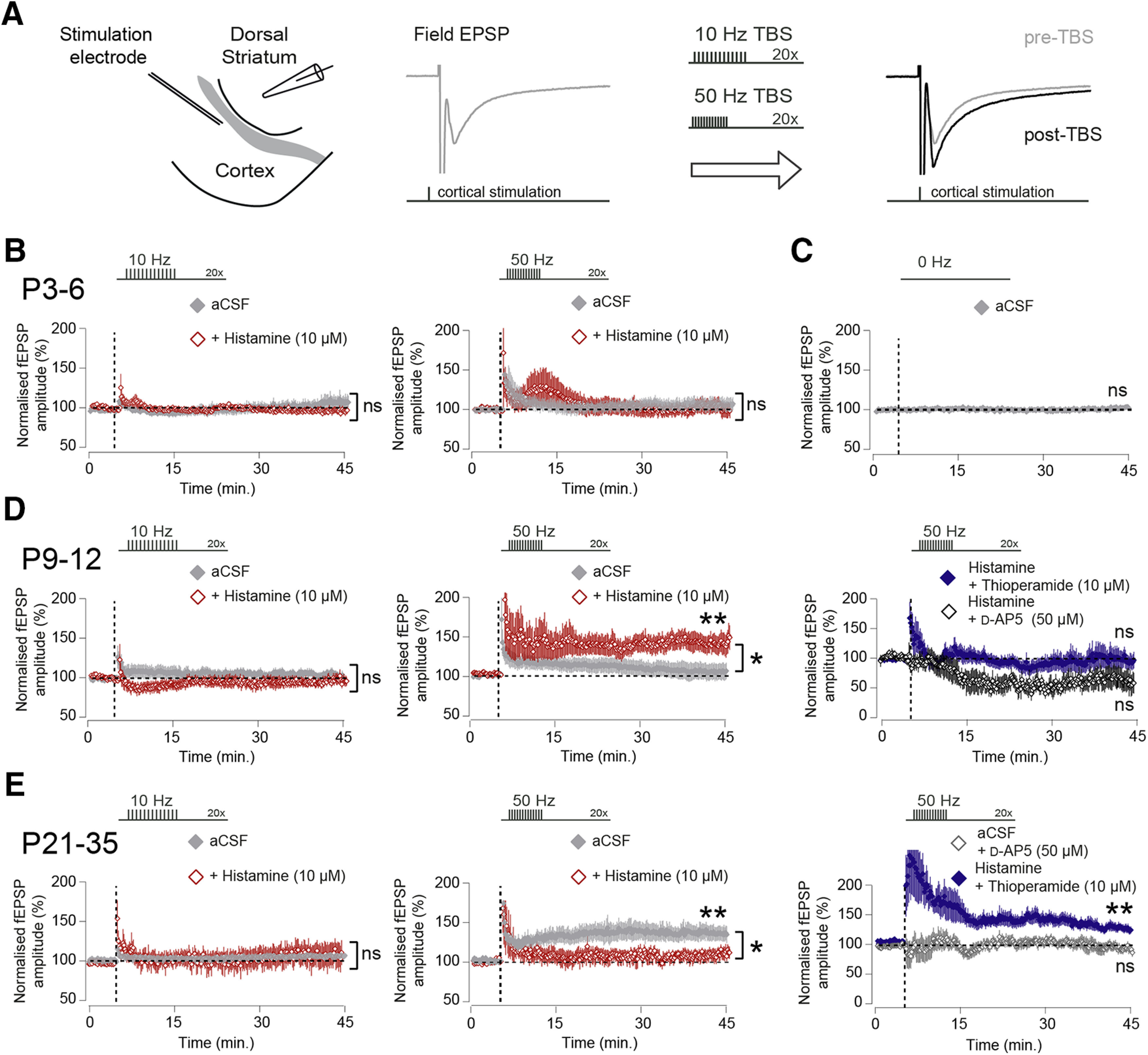
Histamine acting at H3 receptors has opposing effects on the induction of corticostriatal synaptic plasticity depending on developmental age. A, Diagram of the recording configuration consisting of coronal brain sections kept at interface conditions. Electrical stimulation in the cortex generated fEPSPs as recorded with a glass electrode placed in dorsal striatum. Stable measurements of corticostriatal fEPSP amplitude were followed by brief bursts of TBS stimulation, at either 10 or 50 Hz frequency, followed by continued measurements of corticostriatal fEPSP amplitude. B, At P3–P6, TBS at 10 Hz (left) or 50 Hz (right) did not result in significant changes in the amplitude of corticostriatal fEPSPs, nor was this modified in the presence of histamine (10 μm). C, Corticostriatal fEPSP amplitude was stable and constant during recording periods in the absence of TBS. D, At P9–P12, 10 Hz (left) or 50 Hz (middle) TBS did not lead to significant changes in the amplitude of corticostriatal fEPSPs in normal aCSF, but the presence of histamine facilitated the induction of LTP after 50 Hz TBS (red trace, middle, aCSF vs aCSF+histamine, p = 0.029, independent t test; n = 11/10 and 9/5 mice). The LTP induced by 50 Hz TBS was not observed with histamine in combination with the H3 receptor antagonist thioperamide (blue trace, right, 98.99 ± 15.12%, vs pre-TBS, p = 0.95, paired t test, 6/2 mice) or the NMDA receptor antagonist D-AP5 (black trace, right, 62.51 ± 17.45%, vs pre-TBS, p = 0.064, paired t test,5/2 mice). E, At P21–P35, TBS at 10 Hz did not lead to long-term changes in the fEPSP amplitude in aCSF or aCSF-containing histamine (left). In contrast, 50 Hz TBS produces robust potentiation of the fEPSP amplitude in normal aCSF (gray trace, middle), which was reduced in the presence of histamine (red trace, middle, aCSF vs aCSF+histamine, p = 0.047, independent t test; n = 5/5 and 4/4 mice). The synaptic plasticity observed after 50 Hz TBS in aCSF was blocked by the addition of the NMDA receptor antagonist D-AP5 (dark gray trace, right, vs pre-TBS, p = 0.75, paired t test; n = 10/5 mice), and the induction of LTP could be partly rescue by the addition of the H3 receptor antagonist thioperamide (blue trace, right, aCSF+histamine vs histamine+thioperamide, p = 0.17, independent t test; n = 4/4 and 9/3 mice). ns, non-significant, **p < 0.01; *p < 0.05; paired and independent t test.
Similarly, in the second postnatal week, both 10 and 50 Hz TBS stimulation did not result in significant changes in synaptic strength in aCSF (aCSF 10 Hz: 106.01 ± 4.19% and 50 Hz: 105.74 ± 10.29%, p = 0.19 and p = 0.59, paired t test; n = 9/8 mice and 11/10 mice; Fig. 5D). However, in this developmental period, the presence of histamine in the recording aCSF now led to the induction of a persistent LTP of synaptic efficacy using 50 Hz, but not 10 Hz, TBS stimulation (histamine 10 Hz: 94.31 ± 5.48%, vs pre-TBS p = 0.36, paired t test; n = 5/5 mice, and 50 Hz: 137.37 ± 11.65%, vs pre-TBS p = 0.0034, paired t test; n = 9/5 mice; Fig. 5D). We next investigated which receptors were responsible for the observed changes in synaptic strength and hypothesized that the H3 receptor, which exhibited a rapid increase in expression at P9–P12, might play a role in the observed synaptic plasticity. Indeed, the LTP revealed in the presence of histamine at P9–P12 was blocked by the addition of the H3 receptor antagonist thioperamide (10 μm) (50 Hz: 98.99 ± 15.12%, histamine vs histamine+thioperamide p = 0.048,independent t test; n = 9/5 mice and n = 6/2 mice; Fig. 5D). Furthermore, the observed synaptic potentiation at P9-P12 in the presence of histamine was also blocked by the NMDA receptor antagonist D-AP5 (50 μm) (50 Hz: 62.51 ± 17.45%, histamine vs histamine+D-AP5 p = 0.0031, independent t test; n = 9/5 mice and n = 5/2 mice; Fig. 5D).
Finally, in slices taken at P21–P35, 10 Hz TBS stimulation did not lead to changes in synaptic strength similar to all other age ranges (aCSF 10 Hz: 106.91 ± 2.30% and histamine 10 Hz: 109.67 ± 15.38%, vs pre-TBS p = 0.071 and p = 0.54, paired t test; n = 4/4 mice and n = 6/6 mice; Fig. 5E). However, at this developmental period, 50 Hz TBS stimulation reliably led to the induction of LTP of the fEPSP amplitude in standard recording aCSF (aCSF 50 Hz: 146.57 ± 12.97%, vs pre-TBS p = 0.0071, paired t test; n = 5/5 mice; Fig. 5E); and instead of facilitating potentiation, now the presence of histamine in the aCSF blocked the induction of LTP (histamine 50 Hz: 109.01 ± 10.28%, vs pre-TBS p = 0.30, paired t test; n = 4/4 mice; Fig. 5E). Similar effects were seen in slices from older animals (P35+) also where 50 Hz TBS led to potentiation (aCSF 50 Hz: 131.41 ± 14.97%, vs pre-TBS p = 0.025, paired t test; n = 4/4 mice), which was blocked by the addition of histamine to the aCSF (histamine 50 Hz: 106.33 ± 3.51%, vs pre-TBS p = 0.15, paired t test; n = 3/3 mice). The LTP at P21–P35 observed in aCSF after 50 Hz TBS was blocked by the addition of the NMDA receptor antagonist D-AP5 (50 μm) to the recording aCSF (97.61 ± 7.39%, aCSF vs aCSF+D-AP5: p = 0.0036, paired t test; n = 5/5 mice and n = 10/5 mice; Fig. 5E), and the addition of the H3 receptor antagonist thioperamide(10 μm) to aCSF-containing histamine could rescue the LTP (130.86 ± 8.60%, vs pre-TBS p = 0.0025, paired t test; n = 9/3 mice; Fig. 5E).
In conclusion, we find that TBS stimulation at both 10 and 50 Hz frequency did not lead to plastic changes at corticostriatal synapses in the first postnatal week. However, in the second postnatal week, brief bursts of activity at 50 Hz are able to induce a long-term increase in the synaptic strength of corticostriatal synapses, but only in the presence of histamine and dependent on both H3 histamine receptors and NMDA receptors. In contrast, in the fourth postnatal week, older brief bursts of activity at 50 Hz reliably induced long-term increases in synaptic efficacy; but during this period, this is blocked by the addition of histamine and also dependent on H3 histamine receptors and NMDA receptors.
Discussion
The findings of the present study demonstrate that the mouse striatum receives histaminergic innervation and contains transcripts for the H1, H2, and H3 histamine receptors from the first postnatal weeks onwards. These receptors are functional as superfusion of histamine has acute effects on both the intrinsic electrical properties of striatal SPNs, as well as on excitatory transmission at corticostriatal synapses. First, we find that histamine modulates diverse electrical characteristics of SPNs, but during the latter stages of postnatal development mainly leads to their depolarization and an overall reduction in their action potential output. Second, histamine is able to negatively modulate corticostriatal transmission by acting at H3 receptors, and it does this at all developmental ages studied. Importantly, we find that histamine is able to control longer-term changes at corticostriatal synapses, as it facilitates the induction of NMDA receptor-dependent long-term synaptic plasticity during the second postnatal week, but inhibits the induction of long-term synaptic plasticity at later developmental stages, with both processes dependent on H3 histamine receptors. Together, these results demonstrate that histamine is an active neuromodulator during early striatal development, and suggests that alterations in the levelsof histamine can significantly affect developing corticostriatal circuits.
The striatum is the main input nucleus of the basal ganglia and in adulthood is innervated by neuromodulator-containing afferents, such as dopamine (Surmeier et al., 2011), serotonin (Steinbusch, 1981), noradrenaline (Aston-Jones and Bloom, 1981), and histamine (Haas and Panula, 2003). Here we corroborate previous observations (Auvinen and Panula, 1988; Panula et al., 2014) that histamine-containing afferents are found in the striatum from the second postnatal week onwards, with little to no detectable innervation during earlier periods, suggesting that afferents are still maturing and have not yet reached dorsal striatum, in contrast to dopaminergic afferents (Specht et al., 1981). Overall, the density of histaminergic afferents is low at all developmental periods studied, which together with the observation that varicosities only infrequently make synapses in adulthood (Takagi et al., 1986), suggests that histamine might diffuse far and act at a distance from its release sites. Indeed, although low-affinity organic cation transporters likely play a role in the uptake of histamine (Amphoux et al., 2006), to date no specific histamine transporter or uptake mechanism has been described in the mammalian CNS, but see in Drosophila (Borycz et al., 2002; Chaturvedi et al., 2014; Stenesen et al., 2015; Xu et al., 2015), which suggests that histamine might be degraded in the extracellular space (Schwartz et al., 1991; Haas and Panula, 2003) with a possible half-life ranging in minutes (Schwartz et al., 1991). Interestingly, we found transcripts for all the three major histamine receptors (H1, H2, and H3) in the striatum from the first postnatal days onwards, extending on previous in situ work demonstrating early expression of histamine receptors in the brain (Kinnunen et al., 1998; Heron et al., 2001; Karlstedt et al., 2001), and suggesting that released histamine could act at these receptors from birth regulating many developmental processes. Although the expression levels of the H1 and H2 receptors remain comparatively constant, the H3 receptor exhibited a rapid developmental increase, suggesting an increased ability of histamine in modulating H3 receptor-dependent processes (Haas and Panula, 2003), including, for example, the synaptic plasticity found in this study and discussed further below. Last, although the extent to which the H4 histamine receptor is expressed in the CNS is debated (Schneider and Seifert, 2016), we did not find evidence for striatal expression of these receptors as transcripts at any of the developmental age ranges studied.
To investigate whether histamine modulates developing striatal neurons and synapses, two different electrophysiological approaches were used. First, we performed whole-cell patch-clamp recordings of developing striatal SPNs in combination with histamine superfusion and observed many and diverse modulatory effects on their intrinsic electrical properties. In the first postnatal week, histamine superfusion led to a significant reduction in input resistance, together with a pronounced hyperpolarization, suggestive of activation of potassium leak currents (Nisenbaum et al., 1996; Weiger et al., 1997; Shen et al., 2007). Similar hyperpolarization has been observed in thalamic neurons and was dependent on activity at H1 receptors (Sittig and Davidowa, 2001). In the second postnatal week and later, we do not observe changes in input resistance, possible through developmental changes in ion channel expression (Tepper et al., 1998), and instead find that histamine leads to a pronounced depolarization of SPNs (Haas and Konnerth, 1983; Ellender et al., 2011). Indeed, developmental changes in ion channel expression also likely underpin our observations of a progressively decreasing input resistance and membrane time constant across the age ranges studied, which will affect the temporal responsiveness and precision to synaptic inputs of SPNs. The histamine-mediated depolarization of striatal neurons was also observed in other studies and relied on activity at H2 receptors (Ellender et al., 2011), or a combination of H1 and H2 receptors (Munakata and Akaike, 1994; Zhuang et al., 2018). Contrary to expectations, the histamine-induced depolarized state did not lead to an increase in action potential frequency on positive current injections, which instead were significantly lowered. Similar decreases in action potential frequency have been observed in cortical interneurons and were a reflection of histamine modulating Kv3.2-containing K+ channels (Atzori et al., 2000), but the overall low rates of action potential firing of striatal SPNs suggest modulation through other channels mediating a prolongation of ISIs (Nisenbaum et al., 1994; Baranauskas et al., 2003; Shen et al., 2004, 2005). A recent elegant study demonstrated that whole-cell recordings of SPNs can lead to significant dialysis of cytosolic components altering their intrinsic firing properties and critically affecting their response to dopaminergic signaling (Lahiri and Bevan, 2020). Interestingly, clear postsynaptic effects of histamine were seen in our whole-cell patch-clamp configurations at all age ranges studied, which might have resulted from use of high-resistance pipettes, although the variance in responses could have resulted from our recording configuration. Indeed, perforated patch clamp would be the ideal recording configuration when studying G-protein-coupled receptor physiology, especially during recordings of longer duration, and concerns about dialysis of cytosolic components was one of the reasons to opt for fEPSP recordings for the long-term plasticity experiments.
Our second electrophysiological approach consisted of field recordings in acute brain slices to investigate the effect of histamine on cortically evoked glutamatergic transmission in the striatum. Histamine was able to negatively modulate the amplitude of the cortically evoked fEPSP from the first postnatal days onwards, which was dependent on H3 receptors. Thus, the ability of histamine to modulate corticostriatal synapses (Doreulee et al., 2001; Ellender et al., 2011) is already established during the first postnatal days, and the rapid increase in expression of the H3 histamine receptor is likely a reflection of both an increase in the number of striatal synapses and the ability of histamine to modulate these synapses (Ellender et al., 2011). It is important to note that further changes in the physiological function of histamine might occur beyond the age ranges investigated here. Indeed, precedent can be found in both the neuromodulators dopamine and acetylcholine (Teicher et al., 1995; Lieberman et al., 2018; McGuirt et al., 2020), where continued dynamic changes in their function are observed at 120 d. Last, the use of field recordings masks possible differential effects of histamine on the two populations of striatal SPNs: the D1-expressing direct pathway SPNs and the D2-expressing indirect pathway SPNs (Day et al., 2008; Gertler et al., 2008). Although we cannot exclude that histamine might affect these two populations differentially during particular periods of development, previous work in more mature striatal SPNs would suggest that histamine affects both SPNs in similar ways. Indeed, they have been shown to both depolarize in response to histamine acting at H1 and/or H2 receptors (Ellender et al., 2011; Zhuang et al., 2018), with a similar IC50 (Zhuang et al., 2018), both cortical and thalamic excitatory glutamatergic synaptic transmission are equally depressed by histamine acting at presynaptic H3 receptors (Ellender et al., 2011; Zhuang et al., 2018), qRT-PCR experiments revealed comparable levels of expression of transcripts for the H1 and H2 receptors in D1 and D2 SPNs (Zhuang et al., 2018) and both D1 and D2 SPNs express H3 receptors (Ryu et al., 1994; Pillot et al., 2002). These results support the idea that, at least at more mature stages, both presynaptic and postsynaptic sites at D1 and D2 SPNs express a complement of receptors to similarly respond to histamine.
The first postnatal weeks in the striatum are defined by a rapid increase in the number and strength of excitatory synaptic connections (Tepper et al., 1998; Kozorovitskiy et al., 2012; Peixoto et al., 2016, 2019; Krajeski et al., 2019). It has been shown that neural activity is critical in the establishment of these synaptic connections (Kozorovitskiy et al., 2012) and this likely involves a form of synaptic plasticity (Calabresi et al., 1992; Kreitzer and Malenka, 2008), but the exact rules that govern these processes at early periods of postnatal development are unknown. We explored the ability of brief bursts of cortical activity, mimicking naturalistic neural activity patterns (Khazipov et al., 2004; Hanganu et al., 2006; Yang et al., 2009), to induce long-term changes in the strength of developing corticostriatal synapses. We discovered that corticostriatal synapses exhibited NMDA receptor-dependent synaptic plasticity from the second postnatal week onwards induced by brief 50 Hz bursts of activity. Such NMDA receptor-dependent synaptic plasticity has also been observed with sustained higher-frequency stimulation at later stages of striatal development (Partridge et al., 2000). Interestingly, this form of synaptic plasticity at corticostriatal synapses was enabled by histamine at P9–P12, possibly in part through the histamine-mediated dendritic depolarization and a facilitated release of the NMDA receptor Mg2+ block (Calabresi et al., 1992), and inhibited by histamine at older ages. Unexpectedly, both processes were dependent on H3 histamine receptors. Whereas classically the H3 receptor is thought to regulate Gi/o pathways leading to reduced levels of cAMP and PKA (Haas and Panula, 2003), there is evidence to suggest that in adulthood alternative isoforms of H3 receptors exist, which are each differentially coupled to other intracellular pathways (e.g., MAPK pathways) (Drutel et al., 2001; Nieto-Alamilla et al., 2016; Rapanelli et al., 2016). This, in combination with observations of the existence of H3 heteromeric channels with other G-protein-coupled receptors (Ferrada et al., 2009; Moreno et al., 2011; Márquez-Gómez et al., 2018), suggests a great complexity by which histamine acting at H3 receptors can signal to neurons and might explain the opposing effects of histamine on the observed synaptic plasticity depending on the developmental period studied. Indeed, H3 receptors can form heteromers with D1 and D2 dopamine receptors, as well as A2A adenosine receptors, at least in isolated cellular systems (Ferrada et al., 2008; Moreno et al., 2011; Márquez-Gómez et al., 2018), which can result in complex and difficult to predict interactions and responses to histamine, dopamine, and adenosine in the D1 direct pathway SPNs and the D2 indirect pathway SPNs, not least as H3 noncanonical pathways (MAPK and GSK3β) also differ between D1 and D2 SPNs (Rapanelli et al., 2016), and these together might affect processes, such as long-term plasticity (Shen et al., 2008). Furthermore, despite careful placement of stimulating electrodes and use of low levels of stimulation, we cannot exclude the possibility of indirect recruitment of cholinergic interneurons anddopaminergic afferents with potential interactions between neuromodulators affecting the parameters investigated (Sulzer et al., 2016; Condon et al., 2019). Last, we cannot exclude the possibility that histamine might also have some action directly at NMDA receptors (Brown et al., 1995; Haas and Panula, 2003).
In conclusion, our results show that histamine is an active neuromodulator during early postnatal development and exhibits many actions at both striatal SPNs and striatal synapses. Our results are in line with observations regarding other neuromodulators and brain development, such as serotonin (Gaspar et al., 2003), acetylcholine (Role and Berg, 1996), and dopamine (Kozorovitskiy et al., 2015; Lieberman et al., 2018), and can provide insight into the possible etiology of neurodevelopmental disorders resulting from changes in brain histamine levels. Importantly, our results show that histamine is key in gating synaptic plasticity at developing corticostriatal synapses at an early critical period when synapses are actively remodeling (Tepper et al., 1998; Kozorovitskiy et al., 2015; Peixoto et al., 2016, 2019; Mowery et al., 2017; Krajeski et al., 2019), and this period seems to precede later critical periods important for modulation of the intrinsic electrical properties of SPNs (Lieberman et al., 2018).
Footnotes
T.E. was supported by Medical Research Council Career Development Award MR/M009599/1. S.H.was supported by the Korean Government Scholarship Program for Study Overseas, Dr Joanna Chor Ying Ho Scholarship in Pharmacology at Lady Margaret Hall, Mogam Science Scholarship Foundation, Oak Han-Heum Scholarship, Edward Kwanhun Rim, Jong Koo Lee, and Jong Bum Park. R.M.-G. was supported by a Royal Society Newton International Fellowship. We thank Colin Akerman and Fran Platt for generously providing access to equipment; Dawn Shepherd and Judith Schweimer for providing technical assistance; and Paul Bolam for advice and comments.
The authors declare no competing financial interests.
References
- Abdurakhmanova S, Chary K, Kettunen M, Sierra A, Panula P (2017) Behavioral and stereological characterization of Hdc KO mice: relation to Tourette syndrome. J Comp Neurol 525:3476–3487. 10.1002/cne.24279 [DOI] [PubMed] [Google Scholar]
- Albin RL. (2006) Neurobiology of basal ganglia and Tourette syndrome: striatal and dopamine function. Adv Neurol 99:99–106. [PubMed] [Google Scholar]
- Amphoux A, Vialou V, Drescher E, Brüss M, Mannoury La Cour C, Rochat C, Millan MJ, Giros B, Bönisch H, Gautron S (2006) Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain. Neuropharmacology 50:941–952. 10.1016/j.neuropharm.2006.01.005 [DOI] [PubMed] [Google Scholar]
- Arias-Montano JA, Floran B, Garcia M, Aceves J, Young JM (2001) Histamine H(3) receptor-mediated inhibition of depolarization-induced, dopamine D(1) receptor-dependent release of [(3)H]-gamma-aminobutryic acid from rat striatal slices. Br J Pharmacol 133:165–171. 10.1038/sj.bjp.0704053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Lancelot JC, Lecomte JM, Pollard H, Robba M, Schunack W, Schwartz JC (1987) Highly potent and selective ligands for histamine H3-receptors. Nature 327:117–123. 10.1038/327117a0 [DOI] [PubMed] [Google Scholar]
- Arrang JM, Drutel G, Schwartz JC (1995) Characterization of histamine H3 receptors regulating acetylcholine release in rat entorhinal cortex. Br J Pharmacol 114:1518–1522. 10.1111/j.1476-5381.1995.tb13379.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Bloom FE (1981) Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci 1:876–886. 10.1523/JNEUROSCI.01-08-00876.1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzori M, Lau D, Tansey EP, Chow A, Ozaita A, Rudy B, McBain CJ (2000) H2 histamine receptor-phosphorylation of Kv3.2 modulates interneuron fast spiking. Nat Neurosci 3:791–798. 10.1038/77693 [DOI] [PubMed] [Google Scholar]
- Auvinen S, Panula P (1988) Development of histamine-immunoreactive neurons in the rat brain. J Comp Neurol 276:289–303. 10.1002/cne.902760211 [DOI] [PubMed] [Google Scholar]
- Baranauskas G, Tkatch T, Nagata K, Yeh JZ, Surmeier DJ (2003) Kv3.4 subunits enhance the repolarizing efficiency of Kv3.1 channels in fast-spiking neurons. Nat Neurosci 6:258–266. 10.1038/nn1019 [DOI] [PubMed] [Google Scholar]
- Bolam JP, Ellender TJ (2016) Histamine and the striatum. Neuropharmacology 106:74–84. 10.1016/j.neuropharm.2015.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borycz J, Borycz JA, Loubani M, Meinertzhagen IA (2002) tan and ebony genes regulate a novel pathway for transmitter metabolism at fly photoreceptor terminals. J Neurosci 22:10549–10557. 10.1523/JNEUROSCI.22-24-10549.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Haas HL (1999) On the mechanism of histaminergic inhibition of glutamate release in the rat dentate gyrus. J Physiol 515: 777–786. 10.1111/j.1469-7793.1999.777ab.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Fedorov NB, Haas HL, Reymann KG (1995) Histaminergic modulation of synaptic plasticity in area CA1 of rat hippocampal slices. Neuropharmacology 34:181–190. 10.1016/0028-3908(94)00138-I [DOI] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G (1992) Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. Eur J Neurosci 4:929–935. 10.1111/j.1460-568.1992.tb00119.x [DOI] [PubMed] [Google Scholar]
- Castellan Baldan L, Williams KA, Gallezot JD, Pogorelov V, Rapanelli M, Crowley M, Anderson GM, Loring E, Gorczyca R, Billingslea E, Wasylink S, Panza KE, Ercan-Sencicek AG, Krusong K, Leventhal BL, Ohtsu H, Bloch MH, Hughes ZA, Krystal JH, Mayes L, et al. (2014) Histidine decarboxylase deficiency causes Tourette syndrome: parallel findings in humans and mice. Neuron 81:77–90. 10.1016/j.neuron.2013.10.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi R, Reddig K, Li HS (2014) Long-distance mechanism of neurotransmitter recycling mediated by glial network facilitates visual function in Drosophila. Proc Natl Acad Sci USA 111:2812–2817. 10.1073/pnas.1323714111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon MD, Platt NJ, Zhang YF, Roberts BM, Clements MA, Vietti-Michelina S, Tseu MY, Brimblecombe KR, Threlfell S, Mann EO, Cragg SJ (2019) Plasticity in striatal dopamine release is governed by release-independent depression and the dopamine transporter. Nat Commun 10:4263. 10.1038/s41467-019-12264-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Wokosin D, Plotkin JL, Tian X, Surmeier DJ (2008) Differential excitability and modulation of striatal medium spiny neuron dendrites. J Neurosci 28:11603–11614. 10.1523/JNEUROSCI.1840-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehorter N, Michel FJ, Marissal T, Rotrou Y, Matrot B, Lopez C, Humphries MD, Hammond C (2011) Onset of pup locomotion coincides with loss of NR2C/D-mediated cortico-striatal EPSCs and dampening of striatal network immature activity. Front Cell Neurosci 5:24. 10.3389/fncel.2011.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doreulee N, Yanovsky Y, Flagmeyer I, Stevens DR, Haas HL, Brown RE (2001) Histamine H(3) receptors depress synaptic transmission in the corticostriatal pathway. Neuropharmacology 40:106–113. 10.1016/S0028-3908(00)00101-5 [DOI] [PubMed] [Google Scholar]
- Drutel G, Peitsaro N, Karlstedt K, Wieland K, Smit MJ, Timmerman H, Panula P, Leurs R (2001) Identification of rat H3 receptor isoforms with different brain expression and signaling properties. Mol Pharmacol 59:1–8. 10.1124/mol.59.1.1 [DOI] [PubMed] [Google Scholar]
- Ellender TJ, Huerta-Ocampo I, Deisseroth K, Capogna M, Bolam JP (2011) Differential modulation of excitatory and inhibitory striatal synaptic transmission by histamine. J Neurosci 31:15340–15351. 10.1523/JNEUROSCI.3144-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan-Sencicek AG, Stillman AA, Ghosh AK, Bilguvar K, O'Roak BJ, Mason CE, Abbott T, Gupta A, King RA, Pauls DL, Tischfield JA, Heiman GA, Singer HS, Gilbert DL, Hoekstra PJ, Morgan TM, Loring E, Yasuno K, Fernandez T, Sanders S, et al. (2010) L-histidine decarboxylase and Tourette's syndrome. N Engl J Med 362:1901–1908. 10.1056/NEJMoa0907006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felling RJ, Singer HS (2011) Neurobiology of Tourette syndrome: current status and need for further investigation. J Neurosci 31:12387–12395. 10.1523/JNEUROSCI.0150-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrada C, Ferre S, Casado V, Cortes A, Justinova Z, Barnes C, Canela EI, Goldberg SR, Leurs R, Lluis C, Franco R (2008) Interactions between histamine H3 and dopamine D2 receptors and the implications for striatal function. Neuropharmacology 55:190–197. 10.1016/j.neuropharm.2008.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrada C, Moreno E, Casado V, Bongers G, Cortes A, Mallol J, Canela EI, Leurs R, Ferre S, Lluis C, Franco R (2009) Marked changes in signal transduction upon heteromerization of dopamine D1 and histamine H3 receptors. Br J Pharmacol 157:64–75. 10.1111/j.1476-5381.2009.00152.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L (2005) Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci 25:11279–11287. 10.1523/JNEUROSCI.4476-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SD, Robertson PB, Black MJ, Redgrave P, Sagar MA, Abraham WC, Reynolds JN (2017) Reinforcement determines the timing dependence of corticostriatal synaptic plasticity in vivo. Nat Commun 8:334. 10.1038/s41467-017-00394-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagmeyer I, Haas HL, Stevens DR (1997) Adenosine A1 receptor-mediated depression of corticostriatal and thalamostriatal glutamatergic synaptic potentials in vitro. Brain Res 778:178–185. 10.1016/S0006-8993(97)01060-3 [DOI] [PubMed] [Google Scholar]
- Foster HL, Small JD, Fox JG (1983) The mouse in biomedical research. In: Normative biology, immunology, and husbandry. San Diego: Academic. [Google Scholar]
- Gaspar P, Cases O, Maroteaux L (2003) The developmental role of serotonin: news from mouse molecular genetics. Nat Rev Neurosci 4:1002–1012. 10.1038/nrn1256 [DOI] [PubMed] [Google Scholar]
- Gbahou F, Vincent L, Humbert-Claude M, Tardivel-Lacombe J, Chabret C, Arrang JM (2006) Compared pharmacology of human histamine H3 and H4 receptors: structure-activity relationships of histamine derivatives. Br J Pharmacol 147:744–754. 10.1038/sj.bjp.0706666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler TS, Chan CS, Surmeier DJ (2008) Dichotomous anatomical properties of adult striatal medium spiny neurons. J Neurosci 28:10814–10824. 10.1523/JNEUROSCI.2660-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty AW, Kimura M (1994) The basal ganglia and adaptive motor control. Science 265:1826–1831. 10.1126/science.8091209 [DOI] [PubMed] [Google Scholar]
- Grillner S, Hellgren J, Menard A, Saitoh K, Wikstrom MA (2005) Mechanisms for selection of basic motor programs: roles for the striatum and pallidum. Trends Neurosci 28:364–370. 10.1016/j.tins.2005.05.004 [DOI] [PubMed] [Google Scholar]
- Haas HL, Konnerth A (1983) Histamine and noradrenaline decrease calcium-activated potassium conductance in hippocampal pyramidal cells. Nature 302:432–434. 10.1038/302432a0 [DOI] [PubMed] [Google Scholar]
- Haas HL, Panula P (2003) The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev Neurosci 4:121–130. 10.1038/nrn1034 [DOI] [PubMed] [Google Scholar]
- Haas HL, Schaerer B, Vosmansky M (1979) A simple perfusion chamber for the study of nervous tissue slices in vitro. J Neurosci Methods 1:323–325. 10.1016/0165-0270(79)90021-9 [DOI] [PubMed] [Google Scholar]
- Hanganu IL, Ben-Ari Y, Khazipov R (2006) Retinal waves trigger spindle bursts in the neonatal rat visual cortex. J Neurosci 26:6728–6736. 10.1523/JNEUROSCI.0752-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawes SL, Gillani F, Evans RC, Benkert EA, Blackwell KT (2013) Sensitivity to theta-burst timing permits LTP in dorsal striatal adult brain slice. J Neurophysiol 110:2027–2036. 10.1152/jn.00115.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron A, Rouleau A, Cochois V, Pillot C, Schwartz JC, Arrang JM (2001) Expression analysis of the histamine H(3) receptor in developing rat tissues. Mech Dev 105:167–173. 10.1016/S0925-4773(01)00389-6 [DOI] [PubMed] [Google Scholar]
- Hew RW, Hodgkinson CR, Hill SJ (1990) Characterization of histamine H3-receptors in guinea-pig ileum with H3-selective ligands. Br J Pharmacol 101:621–624. 10.1111/j.1476-5381.1990.tb14130.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Yamatodani A, Ando-Yamamoto M, Tohyama M, Watanabe T, Wada H (1988) Organization of histaminergic fibers in the rat brain. J Comp Neurol 273:283–300. 10.1002/cne.902730302 [DOI] [PubMed] [Google Scholar]
- Jafri MS, Moore KA, Taylor GE, Weinreich D (1997) Histamine H1 receptor activation blocks two classes of potassium current, IK(rest) and IAHP, to excite ferret vagal afferents. J Physiol 503:533–546. 10.1111/j.1469-7793.1997.533bg.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlstedt K, Senkas A, Ahman M, Panula P (2001) Regional expression of the histamine H(2) receptor in adult and developing rat brain. Neuroscience 102:201–208. 10.1016/S0306-4522(00)00464-4 [DOI] [PubMed] [Google Scholar]
- Khazipov R, Sirota A, Leinekugel X, Holmes GL, Ben-Ari Y, Buzsáki G (2004) Early motor activity drives spindle bursts in the developing somatosensory cortex. Nature 432:758–761. 10.1038/nature03132 [DOI] [PubMed] [Google Scholar]
- Kinnunen A, Lintunen M, Karlstedt K, Fukui H, Panula P (1998) In situ detection of H1-receptor mRNA and absence of apoptosis in the transient histamine system of the embryonic rat brain. J Comp Neurol 394:127–137. [DOI] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, Sabatini BL (2012) Recurrent network activity drives striatal synaptogenesis. Nature 485:646–650. 10.1038/nature11052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Peixoto R, Wang W, Saunders A, Sabatini BL (2015) Neuromodulation of excitatory synaptogenesis in striatal development. Elife 4:e10111 10.7554/eLife.10111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajeski RN, Macey-Dare A, van Heusden F, Ebrahimjee F, Ellender TJ (2019) Dynamic postnatal development of the cellular and circuit properties of striatal D1 and D2 spiny projection neurons. J Physiol 597:5265–5293. 10.1113/JP278416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC (2008) Striatal plasticity and basal ganglia circuit function. Neuron 60:543–554. 10.1016/j.neuron.2008.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri AK, Bevan MD (2020) Dopaminergic transmission rapidly and persistently enhances excitability of D1 receptor-expressing striatal projection neurons. Neuron 106:277–290.e6. 10.1016/j.neuron.2020.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Wong D, Lynch G (1986) Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res 368:347–350. 10.1016/0006-8993(86)90579-2 [DOI] [PubMed] [Google Scholar]
- Lieberman OJ, McGuirt AF, Mosharov EV, Pigulevskiy I, Hobson BD, Choi S, Frier MD, Santini E, Borgkvist A, Sulzer D (2018) Dopamine triggers the maturation of striatal spiny projection neuron excitability during a critical period. Neuron 99:540–554.e544. 10.1016/j.neuron.2018.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Kocsis JD (1988) Presynaptic actions of carbachol and adenosine on corticostriatal synaptic transmission studied in vitro. J Neurosci 8:3750–3756. 10.1523/JNEUROSCI.08-10-03750.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márquez-Gómez R, Robins MT, Gutiérrez-Rodelo C, Arias JM, Olivares-Reyes JA, van Rijn RM, Arias-Montaño JA (2018) Functional histamine H3 and adenosine A2A receptor heteromers in recombinant cells and rat striatum. Pharmacol Res 129:515–525. 10.1016/j.phrs.2017.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuirt A, Lieberman O, Post M, Pigulevskiy I, Sulzer D (2020) Coordinated postnatal maturation of striatal cholinergic interneurons and dopamine release dynamics in mice. bioRxiv 022152. doi: 10.1101/2020.04.02.022152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaught KS, Mink JW (2011) Advances in understanding and treatment of Tourette syndrome. Nat Rev Neurol 7:667–676. 10.1038/nrneurol.2011.167 [DOI] [PubMed] [Google Scholar]
- Mink JW. (2001) Neurobiology of basal ganglia circuits in Tourette syndrome: faulty inhibition of unwanted motor patterns? Adv Neurol 85:113–122. [PubMed] [Google Scholar]
- Molina-Hernández A, Nuñez A, Sierra JJ, Arias-Montaño JA (2001) Histamine H3 receptor activation inhibits glutamate release from rat striatal synaptosomes. Neuropharmacology 41:928–934. 10.1016/S0028-3908(01)00144-7 [DOI] [PubMed] [Google Scholar]
- Moreno E, Hoffmann H, Gonzalez-Sepulveda M, Navarro G, Casado V, Cortes A, Mallol J, Vignes M, McCormick PJ, Canela EI, Lluis C, Moratalla R, Ferre S, Ortiz J, Franco R (2011) Dopamine D1-histamine H3 receptor heteromers provide a selective link to MAPK signaling in GABAergic neurons of the direct striatal pathway. J Biol Chem 286:5846–5854. 10.1074/jbc.M110.161489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisset S, Rouleau A, Ligneau X, Gbahou F, Tardivel-Lacombe J, Stark H, Schunack W, Ganellin CR, Schwartz JC, Arrang JM (2000) High constitutive activity of native H3 receptors regulates histamine neurons in brain. Nature 408:860–864. 10.1038/35048583 [DOI] [PubMed] [Google Scholar]
- Mowery TM, Penikis KB, Young SK, Ferrer CE, Kotak VC, Sanes DH (2017) The sensory striatum is permanently impaired by transient developmental deprivation. Cell Rep 19:2462–2468. 10.1016/j.celrep.2017.05.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munakata M, Akaike N (1994) Regulation of K+ conductance by histamine H1 and H2 receptors in neurones dissociated from rat neostriatum. J Physiol 480:233–245. 10.1113/jphysiol.1994.sp020356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto-Alamilla G, Márquez-Gómez R, García-Gálvez AM, Morales-Figueroa GE, Arias-Montaño JA (2016) The histamine H3 receptor: structure. Mol Pharmacol 90:649–673. 10.1124/mol.116.104752 [DOI] [PubMed] [Google Scholar]
- Nisenbaum ES, Xu ZC, Wilson CJ (1994) Contribution of a slowly inactivating potassium current to the transition to firing of neostriatal spiny projection neurons. J Neurophysiol 71:1174–1189. 10.1152/jn.1994.71.3.1174 [DOI] [PubMed] [Google Scholar]
- Nisenbaum ES, Wilson CJ, Foehring RC, Surmeier DJ (1996) Isolation and characterization of a persistent potassium current in neostriatal neurons. J Neurophysiol 76:1180–1194. 10.1152/jn.1996.76.2.1180 [DOI] [PubMed] [Google Scholar]
- Nissinen MJ, Panula P (1995) Developmental patterns of histamine-like immunoreactivity in the mouse. J Histochem Cytochem 43:211–227. 10.1177/43.2.7822777 [DOI] [PubMed] [Google Scholar]
- Panula P, Nuutinen S (2013) The histaminergic network in the brain: basic organization and role in disease. Nat Rev Neurosci 14:472–487. 10.1038/nrn3526 [DOI] [PubMed] [Google Scholar]
- Panula P, Sundvik M, Karlstedt K (2014) Developmental roles of brain histamine. Trends Neurosci 37:159–168. 10.1016/j.tins.2014.01.001 [DOI] [PubMed] [Google Scholar]
- Partridge JG, Tang KC, Lovinger DM (2000) Regional and postnatal heterogeneity of activity-dependent long-term changes in synaptic efficacy in the dorsal striatum. J Neurophysiol 84:1422–1429. 10.1152/jn.2000.84.3.1422 [DOI] [PubMed] [Google Scholar]
- Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, Sabatini BL (2016) Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(–/–) mice. Nat Neurosci 19:716–724. 10.1038/nn.4260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto RT, Chantranupong L, Hakim R, Levasseur J, Wang W, Merchant T, Gorman K, Budnik B, Sabatini BL (2019) Abnormal striatal development underlies the early onset of behavioral deficits in Shank3B(–/–) mice. Cell Rep 29:2016–2027.e2014. 10.1016/j.celrep.2019.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillot C, Heron A, Cochois V, Tardivel-Lacombe J, Ligneau X, Schwartz JC, Arrang JM (2002) A detailed mapping of the histamine H(3) receptor and its gene transcripts in rat brain. Neuroscience 114:173–193. 10.1016/S0306-4522(02)00135-5 [DOI] [PubMed] [Google Scholar]
- Rapanelli M, Frick LR, Horn KD, Schwarcz RC, Pogorelov V, Nairn AC, Pittenger C (2016) The histamine H3 receptor differentially modulates mitogen-activated protein kinase (MAPK) and Akt signaling in striatonigral and striatopallidal neurons. J Biol Chem 291:21042–21052. 10.1074/jbc.M116.731406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapanelli M, Frick L, Bito H, Pittenger C (2017a) Histamine modulation of the basal ganglia circuitry in the development of pathological grooming. Proc Natl Acad Sci USA 114:6599–6604. 10.1073/pnas.1704547114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapanelli M, Frick L, Pogorelov V, Ohtsu H, Bito H, Pittenger C (2017b) Histamine H3R receptor activation in the dorsal striatum triggers stereotypies in a mouse model of tic disorders. Transl Psychiatry 7:e1013. 10.1038/tp.2016.290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Role LW, Berg DK (1996) Nicotinic receptors in the development and modulation of CNS synapses. Neuron 16:1077–1085. 10.1016/S0896-6273(00)80134-8 [DOI] [PubMed] [Google Scholar]
- Ryu JH, Yanai K, Iwata R, Ido T, Watanabe T (1994) Heterogeneous distributions of histamine H3, dopamine D1 and D2 receptors in rat brain. Neuroreport 5:621–624. 10.1097/00001756-199401000-00022 [DOI] [PubMed] [Google Scholar]
- Schneider EH, Seifert R (2016) The histamine H4-receptor and the central and peripheral nervous system: a critical analysis of the literature. Neuropharmacology 106:116–128. 10.1016/j.neuropharm.2015.05.004 [DOI] [PubMed] [Google Scholar]
- Schwartz JC, Arrang JM, Garbarg M, Pollard H, Ruat M (1991) Histaminergic transmission in the mammalian brain. Physiol Rev 71:1–51. 10.1152/physrev.1991.71.1.1 [DOI] [PubMed] [Google Scholar]
- Shen W, Hernandez-Lopez S, Tkatch T, Held JE, Surmeier DJ (2004) Kv1.2-containing K+ channels regulate subthreshold excitability of striatal medium spiny neurons. J Neurophysiol 91:1337–1349. 10.1152/jn.00414.2003 [DOI] [PubMed] [Google Scholar]
- Shen W, Hamilton SE, Nathanson NM, Surmeier DJ (2005) Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J Neurosci 25:7449–7458. 10.1523/JNEUROSCI.1381-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Tian X, Day M, Ulrich S, Tkatch T, Nathanson NM, Surmeier DJ (2007) Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat Neurosci 10:1458–1466. 10.1038/nn1972 [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ (2008) Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321:848–851. 10.1126/science.1160575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittig N, Davidowa H (2001) Histamine reduces firing and bursting of anterior and intralaminar thalamic neurons and activates striatal cells in anesthetized rats. Behav Brain Res 124:137–143. 10.1016/S0166-4328(01)00223-6 [DOI] [PubMed] [Google Scholar]
- Specht LA, Pickel VM, Joh TH, Reis DJ (1981) Light-microscopic immunocytochemical localization of tyrosine hydroxylase in prenatal rat brain: I. Early ontogeny. J Comp Neurol 199:233–253. 10.1002/cne.901990207 [DOI] [PubMed] [Google Scholar]
- Steinbusch HW. (1981) Distribution of serotonin-immunoreactivity in the central nervous system of the rat-cell bodies and terminals. Neuroscience 6:557–618. 10.1016/0306-4522(81)90146-9 [DOI] [PubMed] [Google Scholar]
- Stenesen D, Moehlman AT, Kramer H (2015) The carcinine transporter CarT is required in Drosophila photoreceptor neurons to sustain histamine recycling. Elife 4:e10972. 10.7554/eLife.10972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Cragg SJ, Rice ME (2016) Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia 6:123–148. 10.1016/j.baga.2016.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Carrillo-Reid L, Bargas J (2011) Dopaminergic modulation of striatal neurons, circuits, and assemblies. Neuroscience 198:3–18. 10.1016/j.neuroscience.2011.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi H, Morishima Y, Matsuyama T, Hayashi H, Watanabe T, Wada H (1986) Histaminergic axons in the neostriatum and cerebral cortex of the rat: a correlated light and electron microscopic immunocytochemical study using histidine decarboxylase as a marker. Brain Res 364:114–123. 10.1016/0006-8993(86)90992-3 [DOI] [PubMed] [Google Scholar]
- Teicher MH, Andersen SL, Hostetter JC Jr (1995) Evidence for dopamine receptor pruning between adolescence and adulthood in striatum but not nucleus accumbens. Brain Res Dev Brain Res 89:167–172. 10.1016/0165-3806(95)00109-Q [DOI] [PubMed] [Google Scholar]
- Tepper JM, Sharpe NA, Koos TZ, Trent F (1998) Postnatal development of the rat neostriatum: electrophysiological, light- and electron-microscopic studies. Dev Neurosci 20:125–145. 10.1159/000017308 [DOI] [PubMed] [Google Scholar]
- Tort AB, Kramer MA, Thorn C, Gibson DJ, Kubota Y, Graybiel AM, Kopell NJ (2008) Dynamic cross-frequency couplings of local field potential oscillations in rat striatum and hippocampus during performance of a T-maze task. Proc Natl Acad Sci USA 105:20517–20522. 10.1073/pnas.0810524105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota H, Dugovic C, Koehl M, Laposky AD, Weber C, Ngo K, Wu Y, Lee DH, Yanai K, Sakurai E, Watanabe T, Liu C, Chen J, Barbier AJ, Turek FW, Fung-Leung WP, Lovenberg TW (2002) Behavioral characterization of mice lacking histamine H(3) receptors. Mol Pharmacol 62:389–397. 10.1124/mol.62.2.389 [DOI] [PubMed] [Google Scholar]
- Vanhala A, Yamatodani A, Panula P (1994) Distribution of histamine-, 5-hydroxytryptamine-, and tyrosine hydroxylase-immunoreactive neurons and nerve fibers in developing rat brain. J Comp Neurol 347:101–114. 10.1002/cne.903470108 [DOI] [PubMed] [Google Scholar]
- Weiger T, Stevens DR, Wunder L, Haas HL (1997) Histamine H1 receptors in C6 glial cells are coupled to calcium-dependent potassium channels via release of calcium from internal stores. Naunyn Schmiedebergs Arch Pharmacol 355:559–565. 10.1007/pl00004983 [DOI] [PubMed] [Google Scholar]
- Xu Y, An F, Borycz JA, Borycz J, Meinertzhagen IA, Wang T (2015) Histamine recycling is mediated by CarT, a carcinine transporter in Drosophila photoreceptors. PLoS Genet 11:e1005764. 10.1371/journal.pgen.1005764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yael D, Vinner E, Bar-Gad I (2015) Pathophysiology of tic disorders. Mov Disord 30:1171–1178. 10.1002/mds.26304 [DOI] [PubMed] [Google Scholar]
- Yang JW, Hanganu-Opatz IL, Sun JJ, Luhmann HJ (2009) Three patterns of oscillatory activity differentially synchronize developing neocortical networks in vivo. J Neurosci 29:9011–9025. 10.1523/JNEUROSCI.5646-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YC, Satoh H, Wu SM, Marshak DW (2009) Histamine enhances voltage-gated potassium currents of ON bipolar cells in macaque retina. Invest Ophthalmol Vis Sci 50:959–965. 10.1167/iovs.08-2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecharia AY, Yu X, Gotz T, Ye Z, Carr DR, Wulff P, Bettler B, Vyssotski AL, Brickley SG, Franks NP, Wisden W (2012) GABAergic inhibition of histaminergic neurons regulates active waking but not the sleep-wake switch or propofol-induced loss of consciousness. J Neurosci 32:13062–13075. 10.1523/JNEUROSCI.2931-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang QX, Xu HT, Lu XJ, Li B, Yung WH, Wang JJ, Zhu JN (2018) Histamine excites striatal dopamine D1 and D2 receptor-expressing neurons via postsynaptic H1 and H2 receptors. Mol Neurobiol 55:8059–8070. 10.1007/s12035-018-0976-1 [DOI] [PubMed] [Google Scholar]