

Recurrent oral ulcers and hyperactive blood neutrophils are characteristic of Behҫet’s Disease (BD). High levels of enzymatically active neutrophil elastase (NE) were measured in saliva of BD patients without ulcers (BDq), which can lead to extracellular matrix degradation and mucosal instability. BD oral epithelial cells expressed high levels of secretory leukocyte protease inhibitor (SLPI) mRNA, however, salivary SLPI protein was depleted. Although high levels of salivary alpha‐1 antitrypsin (α1AT) were detected, NE was not fully inhibited. We propose that BDq individuals experience a protease‐anti‐protease imbalance in favour of NE which may contribute to their recurrent oral ulceration episodes.
Keywords: autoinflammatory disease, human, inflammation, mucosa, neutrophils
Summary
Behçet’s disease (BD) is a vasculitis of unknown aetiology typified by chronic recurrent oral ulcers and systemic inflammatory manifestations. Neutrophils, and specifically their protease neutrophil elastase (NE), have been implicated in its pathology. Although NE is an effective anti‐microbial, excessive NE can damage host tissue. Recurrent oral ulceration is a primary BD symptom, therefore we hypothesized that excessive neutrophil infiltration evidenced by increased NE and a reduction in specific endogenous inhibitors, secretory leucocyte protease inhibitor (SLPI) and alpha1‐anti‐trypsin (α1AT) contributes to BD mucosal instability. NE, SLPI and α1AT were quantified in saliva from BD patients with active oral ulcers (BDa) and quiet without ulcers (BDq), recurrent aphthous stomatitis (RASa; RASq) and healthy controls (HC). Although BDq saliva had marginally higher median NE levels (1112 ng/ml) compared to both RASq (1043 ng/ml) and HC (999 ng/ml), SLPI was significantly reduced in BDq (P < 0·01). Despite decreased SLPI protein, mRNA expression was significantly increased in BDq buccal epithelial swabs compared to RASq and HC (P < 0·05, P < 0·001). NE remained enzymatically active, although α1AT levels were at least eight times higher than SLPI in all groups, suggesting that α1AT does not have a primary role in counteracting NE in saliva. Furthermore, NE levels in BDa patients medicated with both azathioprine (AZA) and colchicine (COLC) were significantly lower than those on COLC (P = 0·0008) or neither (P = 0·02), indicating that combining AZA + COLC may help to regulate excessive NE during ulceration. This study showed that enzymatically active NE coupled with reduced SLPI in BD saliva may contribute to recurrent oral ulcerations.
Introduction
Behçet’s disease (BD) is a chronic, multi‐systemic disorder with evidence of both autoinflammatory and autoimmune characteristics typified by recurrent oral and genital mucocutaneous lesions [1, 2]. Individuals may also experience arthritis, eye lesions, gastrointestinal issues, vascular inflammation and/or neurological disorders. Although the mechanism of pathogenesis is unknown, there is some evidence of autoimmunity by way of anti‐endothelial cell antibodies as well as immune dysfunction with a genetic predisposition to the human leucocyte antigen complex, HLA‐B51 [3, 4, 5]. Oral ulceration occurs in all BD patients, typically preceding systemic symptoms, suggesting that these ulcers are a key event in this disease [6]. Morphologically, BD ulcers are similar to recurrent aphthous stomatitis (RAS), with a few differentiating characteristics [7]. In BD, a large swollen peri‐ulcer with erythema is visible. Majors ulcers tend to be destructive, with scars distorting the oral mucosa [8]. In RAS, peri‐ulcer erythema is reduced and quickly diminishes as the ulcer heals from the base. During quiet episodes, BD patients frequently still have erythematous mucosal areas. Histologically, BD and RAS ulcers share similar features [9]. Biopsies show infiltration of neutrophils, macrophages, T cells and proinflammatory cytokines [1, 10]. However, interleukin (IL)‐4 has only been detected in BD ulcers [11]. The dichotomy of IL‐4 is that it has been shown to down‐regulate neutrophil migration [12], but can also delay neutrophil apoptosis and induce IL‐8, a potent neutrophil chemoattractant [13]. There is a lack of research regarding the possible mechanisms that are involved in the induction of BD oral manifestations despite the fact that the initial onset of oral ulceration precedes all other symptoms.
Neutrophil chemotaxis and infiltration is evident in BD oral, ocular and cutaneous lesions and has been implicated in the disease’s progression and pathology [14, 15, 16]. Gene HLA‐B51, having the strongest association with BD [5], was also found to be associated with neutrophil hyperactivity [17]. Treatment is aimed at preventing systemic reactivation. Immunomodulatory medication, such as colchicine (COLC), is used in patients with recurrent mucocutaneous BD to reduce neutrophil chemotactic activity [18].
Neutrophil elastase (NE) is a serine protease produced mainly by promyelocytes [19]. NE is contained in azurophilic granules alongside cathepsin G (CatG) and proteinase‐3 in a ready‐to‐use state. The controlled mechanism of NE delivery consists of either the release of granules into a phagolysosome where microbes are degraded or exocytosis of the azurophil into extracellular spaces [20]. Uncontrolled release of NE can also be due to necrosis and/or NET activation and release (NETosis) [20, 21]. Neutrophil degranulation can be triggered by various inflammatory factors or direct contact with the extracellular matrix (ECM) [22], and increased neutrophil recruitment can extend the duration of inflammation [23]. High extracellular NE levels in chronically inflamed tissue of individuals with chronic obstructive pulmonary disease (COPD) [24] and inflammatory bowel disease has been implicated as a cause of tissue damage [23]. Being kinetically faster than CatG and proteinase‐3 [25], NE is capable of quickly degrading ECM components such as elastin, collagen and fibronectin [23]. Therefore, NE regulation is vital.
Secretory leucocyte protease inhibitor (SLPI) has been reported as one of the primary regulatory inhibitors of NE in COPD patients’ sputum [26]. SLPI is a stable innate inhibitory protein which has broad‐spectrum anti‐microbial activity and protects mucosal epithelial surfaces from proteolytic enzymes such as NE, CatG and trypsin [27]. SLPI is produced in many cell types, including saliva gland acinar cells and mucosal epithelial cells [28], and has been detected in various secretions including bronchial fluid [29] and saliva [30].
Alpha1‐anti‐trypsin (α1AT), a systemically circulating serine protease produced mainly in the liver, is a well‐known inhibitor of NE in the lung. It has been measured in saliva [31] and gingival crevicular fluid in periodontal lesions [32]. Notably, both SLPI and α1AT have also been identified in neutrophil cytoplasm separated from the enclosed NE granule, suggesting that neutrophils have evolved to protect themselves from their own potentially damaging enzymes [27, 33].
Neutrophil infiltration is evident in BD oral ulcers and, uniquely, IL‐4, which could delay neutrophil apoptosis. Degranulation is triggered by various inflammatory factors such as tumour necrosis factor (TNF)‐α, IL‐8, C5a and lipopolysaccharide (LPS) [34] releasing excessive NE into the oral environment, which can quickly degrade epithelial tissue [35]. The aim of this study was to clarify the role of NE and its endogenous inhibitors in the oral cavity which may help to elucidate the mechanisms by which mucosal ulceration occurs in BD. It was hypothesized that BD patients have insufficient levels of salivary SLPI and α1AT, enabling excessive extracellular NE to damage the oral epithelial mucosa leading to cellular instability and recurrent ulcerations. The information deduced from this research may help to improve targeted drug therapy to decrease recurrent episodes, increase the healing rate and ultimately to prevent ulceration.
Materials and methods
Patients and saliva sample collection
This study was approved by the local research ethics service of the City and East London (P/03/122). All subjects gave written consent and all samples were collected between January 2009 and December 2012. STROBE cohort guidelines were followed. BD patients fulfilled the International Study Group Criteria [36]. Although BD is a multi‐system disorder with a broad range of clinical manifestations, we focused upon the oral environment, and therefore refer in the manuscript to an active oral episode (BDa) as the presence of at least one oral ulcer at the time of sampling regardless of size, duration or other clinical symptoms. BD and RAS ulcers can resolve and re‐occur intermittently. If no oral ulcer was present at the time of sample collection, regardless of other systemic symptoms, the individual was deemed orally quiet, or BDq. A separate questionnaire for BD patients’ systemic symptoms was recorded on the day of their clinic visit. Evidence of disease activity at different sites, such as their skin, genitals or joints, was recorded separately as quiet or active (Supporting information, Table S1).
Saliva was collected from BD patients with active oral ulcers (BDa, n = 93) and those without oral ulcers deemed BD quiet (BDq, n = 103) at monthly‐held Behçet’s daytime clinics at the Royal London Hospital. RAS subjects with active oral ulcers (RASa, n = 19), RAS without ulcers (RASq, n = 16) and healthy controls (HC, n = 82) were recruited using local flyers. Participants rinsed with 5 ml of water for 10 s and expelled. Passive drooling of 1–5 ml of unstimulated saliva was collected into a sterile container, transferred to the laboratory on ice and centrifuged at 3500 g for 15 min at 4°C. Supernatants were aliquoted and stored at −80°C until further analysis.
Salivary levels of SLPI, NE and α1AT
Enzyme‐linked immunosorbent assays (ELISA) were used to measure human SLPI (Quantikine® ELISA; R&D Systems, Abingdon, UK), NE (HK319; HyCult Biotech, Uden, The Netherlands) and α1AT (Genway Biotech, Inc., San Diego, CA, USA) in saliva following the manufacturers’ protocols. Table 1 summarizes the number of BD, RAS and HC saliva samples tested per assay. Saliva was diluted 1 : 200 in phosphate‐buffered saline (PBS) and run in duplicate alongside standards. Initial validation experiments were carried out for each ELISA. This involved adding recombinant or purified target protein to ELISA wells alone or in the presence of BD saliva in order to assess the accuracy of the assays. Each ELISA successfully recovered the spiked amount at a 1 : 200 saliva dilution (acceptable recovery being between 80 and 120%).
Table 1.
Subjects’ Demographics at the Time of Sample Collection
| ELISA | Subject group | Total n | Age in years (median, IQR) | Male (%) |
|---|---|---|---|---|
| NE ELISA | ||||
| HC* | 54 | 33 (29, 50) | 33·3% | |
| BDq | 59 | 38 (31, 49) | 30·5% | |
| BDa | 49 | 39 (29, 44) | 36·7% | |
| RASq | 10 | 42 (24, 45) | 40% | |
| RASa | 12 | 48 (40, 49) | 58·3% | |
| SLPI ELISA | ||||
| HC* | 49 | 32 (29, 48) | 34·7% | |
| BDq | 56 | 37 (31, 49) | 33·9% | |
| BDa | 52 | 39 (28, 47) | 36·5% | |
| RASq | 11 | 40 (27, 45) | 36·4% | |
| RASa | 15 | 47 (45, 49) | 40·0% | |
| α1AT ELISA | ||||
| HC* | 19 | 30 (27, 31) | 36·8% | |
| BDq | 29 | 43 (35, 49) | 34·5% | |
| BDa | 32 | 42 (29, 50) | 31·3% | |
| RASq | 5 | 35 (24, 47) | 37·5% | |
| RASa | 4 | 38 (27, 45) | 25·0% |
HC NE ELISA had n = 17 anonymous donors, SLPI n = 10, α1AT n = 2.
IQR = interquartile range; ELISA = enzyme‐linked immunosorbent assay; NE = neutrophil elastase; SLPI = secretory leucocyte protease inhibitor; α1AT = alpha1‐anti‐trypsin; HC = healthy controls; BDq = Behçet’s disease quiet patients without oral ulcers; BDa = BD patients with active oral ulcers; RASa = recurrent aphthous stomatitis patients with active oral ulcers; RASq = recurrent aphthous stomatitis quiet patients without oral ulcers.
Elastase activity in saliva samples
Total protease activity as well as NE‐specific enzymatic activity in saliva was measured using the EnzChek® Elastase Assay Kit (Molecular Probes/Life Technologies, Paisley, UK), as per the manufacturer’s instructions. The following patients’ saliva samples were tested: BDa, n = 7; BDq, n = 6; RASa, n = 4; RASq, n = 5; and HC, n = 8. First, BDa and RASa saliva samples were diluted 1 : 20 using the 1× reaction buffer, and those from BDq, RASq and HCs were diluted to 1 : 10. An aliquot of 100 µl of these diluted samples was added in duplicate to a 96‐well plate containing 50 µl of 800 µM NE inhibitor (N‐methoxysuccinyl‐Ala‐Ala‐Pro‐Val‐chloromethyl ketone) or 50 µl of 1× reaction buffer for the non‐inhibited reaction. Next, 50 µl of 100 µg/ml of the DQTM elastin conjugated with Bodipy® FL dye was added to each well to bring the final volume to 200 µl. Porcine elastase was provided as the protease activity positive control. Its reaction consisted of the same volume of DQTM elastin with and without inhibitor. A negative control with only reaction buffer and DQTM elastin was also included. With an excitation wavelength of 480 nm, emitted fluorescence at a wavelength of 520 nm was detected on an Optima FLUOStar spectrophotometer (BMG Labtech, Ortenberg, Germany) and recorded every 5 min for a total of 95 min at 37°C. An increase in fluorescence during 95 min indicated high elastase activity in the saliva. After adjusting for sample dilutions, the difference between neutrophil‐specific elastase activity and total elastase activity (i.e. with and without inhibitor) was calculated in order to determine the NE‐specific activity in each saliva sample.
Oral buccal swab sample collection
A swab of oral buccal epithelial cells was collected from all participants in order to determine the extent of the local expression of SLPI mRNA other than from the saliva glands’ acini. Therefore, after saliva was collected, oral buccal swabs (OBS) were collected from HC, RAS and BD patients by brushing the inside of the cheek, sweeping up and down 10 times using a nylon cytology brush (Deltalab, Barcelona, Spain). If sloughed from an ulcerated site, the sample was denoted as ‘U’ and if from a non‐ulcerated site as ‘NU’. Gentle pressure was used for obtaining swabs from ulcerated sites in order to minimize discomfort to patients. The brush was then vigorously washed in 400 μl of RLT Buffer (Qiagen, Manchester, UK). The samples were frozen on dry ice and transferred to −80°C.
Extraction, purification and reverse transcription of RNA
OBS were defrosted on ice and RNA extracted using the RNeasy Mini Kit (Qiagen), as described by the supplier’s protocol, followed by DNase enzyme digestion using TURBO™ DNA‐free kit (Ambion, Chipping Norton, UK). RNA quantity and quality were assessed using Nanodrop™ 1000 (ThermoScientific, Loughborough, UK). A ratio of 1·7–2·0 indicated good‐quality RNA. Reverse transcription (RT) was performed on ≤ 2 µg per 20 µl reactions using the high‐capacity cDNA reverse transcription kit with RNase inhibitor (Applied Biosystems, Warrington, UK), as per the manufacturer’s instructions. Each 0·2 ml reaction tube was placed into a Veriti Thermocycler (ThermoScientific) with the following incubation times and temperatures: 25°C for 10 min, 37°C for 120 min and 85°C for 5 min to deactivate the RT enzyme, and cooled to 4°C. The new complementary (c)DNA was aliquoted and kept at −20°C.
OBS reference gene panel
The real‐time ready Human Reference Gene 384‐well Panel (Roche, Mannheim, Germany) was used to test a panel of 19 potential reference genes in a total of 48 OBS from BD, RAS and HC subjects. The manufacturer’s protocol was followed. For our plate set‐up, each cDNA sample was first diluted 1 : 10 in RNase‐free water to a final volume of 125 µl. To this, 125 µl of 2× LightCycler® 480 Probes Master reaction mix (Roche) was added. After mixing, 10 µl was pipetted into each well across an individual row (columns 1–23), i.e. each row consisted of one patient sample. A total of 16 patient RNA samples were applied to one plate. Columns 23 and 24 (the RT‐negative controls) each had 1 µl of the previously set aside non‐transcribed OBS RNA sample, 4 µl of water and 5 µl of the 2× LightCycler® 480 Probes Master reaction mix. Plates were sealed, centrifuged briefly and run on the LightCycler® 480 System I instrument (Roche) by uploading the company‐provided macros program.
SLPI and peptidylprolyl isomerase A (PPIA) qPCR optimization
All primers were designed based on human mRNA database sequences, taking into account any multiple transcript variations. SLPI (Accession no. NM003064.2) primers were designed by the Universal Probe Library program (Roche Applied Science, Nottingham, UK): SLPI forward primer: 5′‐CTGTGGAAGGCTCTGGAAAG and SLPI reverse primer 5′‐GGCACTGGGCAGATTTCTTA. Primer design for one of the most stable reference genes, peptidylprolyl isomerase A (PPIA, Accession no. NM021130), was selected from RTPrimerDB: (forward primer 5′‐TCATCTGCACTGCCAAGACTG, reverse primer 5′‐CATGCCTTCTTTCACTTTGCC). Primers were manufactured by Sigma‐Aldrich (Poole, UK).
During quantitative (q)PCR assay optimization, primers underwent an initial reaction with 2× Sybr Green I Mastermix (Roche) and at least two different cDNA OBS samples to ensure amplification of one product. In order to assess this, the melt temperature (Tm) of the amplicon was completed and water (negative) controls were run every time and monitored for any amplification. Single amplicon product sizes, 66 base pairs (bp) for SLPI and 71 bp for PPIA, were confirmed by DNA electrophoresis on a 1% agarose gel. Purified cDNA was subsequently used for standards. Because a pre‐mixed 2X Sybr Green I Mastermix was used for all reactions, dNTPs, Taq polymerase and MgCl2 concentrations were all predetermined by the manufacturer, and therefore special consideration was spent on forward and reverse primer concentration as well as annealing temperature and time in order to optimize the individual assays.
The final qPCR assays for each target consisted of 400 nM forward and reverse primers with 2× Sybr Green I Mastermix (Roche) and 5 µl of cDNA in a final 12·5 µl reaction. Both assays were optimized to run at the same temperature cycles, which included initial activation of Taq polymerase at 95°C for 5 min followed by 45 cycles of 95°C for 10 s, 65°C for 10 s and 72°C for 10 s on a LightCycler® 480 System I instrument (Roche). A total of 86 OBS samples were analyzed for SLPI/PPIA mRNA (HC, n = 16; BDq, n = 27; BDa NU, n = 20; BDa U, n = 6; RASq, n = 11; RASa NU, n = 4; RASa U, n = 2).
Statistical analysis
IBM spss version 23 was used to analyze ELISA and enzyme kinetics data using the non‐parametric Mann–Whitney U‐test. Significance was based on two‐tailed, 95% confidence intervals (CI) where *P < 0·05, **P < 0·01, ***P ≤ 0·001. Spearman’s rho with two‐tailed significance was used for correlation analysis. QBasePlus software version 2.1 (Biogazelle, Gent, Belgium) was used for normalization of qPCR data followed by analysis using an unpaired t‐test with Welch’s correction on GraphPad version 7.03.
Results
Salivary SLPI, NE and α1AT levels
BD and RAS patients with ulcers had significantly higher NE levels compared to those without ulcers. Figure 1a shows that BDa saliva (1794 ng/ml, median) and RASa saliva (3080·7 ng/ml) were significantly higher than all other groups without ulcers: BDq (1111·7 ng/ml, P < 0·05), RASq (1043·4 ng/ml, P < 0·05) and HC (998·9 ng/ml, P < 0·01). The median NE level in the BDq group was higher than HC and RASq, but not significantly so.
Fig. 1.
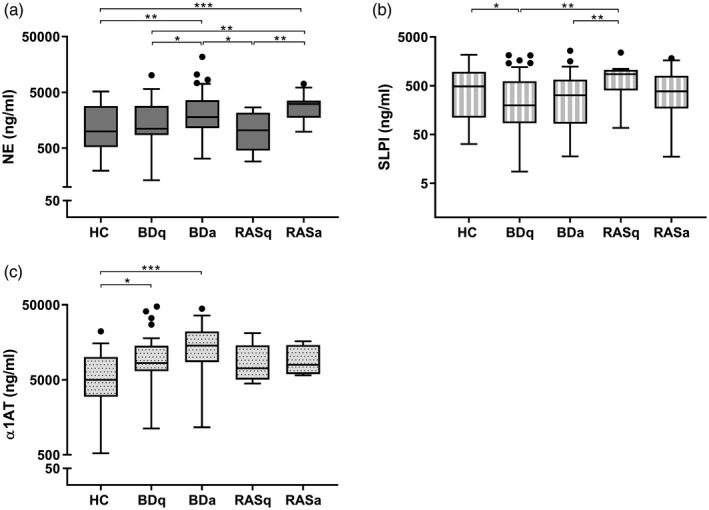
Saliva protein levels among patient groups. Tukey box‐plots show (a) neutrophil elastase (NE), (b) secretory leucocyte protease inhibitor (SLPI) and (c) alpha1‐anti‐trypsin (α1AT) levels in healthy controls (HC), recurrent aphthous stomatitis (RAS) and Behçet’s disease (BD) patients; q = quiet episode/no ulcers; a = at least one active mouth ulcer regardless of other systemic symptoms. Each saliva sample was measured in duplicate. The Mann–Whitney U‐test was used to assess differences between two groups at a time. Significance based on two‐tailed 95% confidence interval (CI). *P < 0·05, **P < 0·01, ***P < 0·001.
However, SLPI was significantly reduced in BDq patient’s saliva (198·7 ng/ml, P < 0·05) compared to HC and RAS and had the lowest median salivary SLPI levels among all groups (Fig. 1b). BDa and RASa had similar levels of SLPI at 320·6 and 383 ng/ml, respectively; however, only BDa saliva was significantly lower than RASq (P < 0·01). SLPI levels were highest in HCs (487·3 ng/ml) and RASq (860·1 ng/ml) patients. Spearman’s rho correlation analysis of n = 37 BDq patients whose saliva was tested for both SLPI and NE showed a diminished inverse relationship (r = −0·195, P = 0·25) compared to HC (r = −0·419, P = 0·02) and BDa (r = −0·299, P = 0·09) (see Supporting information, Fig. S1).
Salivary α1AT levels were significantly higher in BDq (8395·3 ng/ml) compared to HC (4995 ng/ml, P < 0·05) (Fig 1c). BDa α1AT (14 326·1 ng/ml) was nearly double the quantity of BDq and significantly higher than HC (P < 0·001), but not BDq. RASa and RASq had higher α1AT levels than HC, but neither was significant.
Although the focus was upon investigating NE, SLPI and α1AT levels in saliva, BD patients were also clinically assessed for other systemic symptoms (Supporting information, Table S1). BDq patients whose saliva was tested for NE, SLPI and α1AT also commonly had painful joints (19, 21, 38%) and folliculitis (12, 14, 14%), respectively. The median saliva NE level for BDq patients experiencing joint pain and folliculitis were both 1843.6 ng/ml. There was no significant difference in the BDq salivary NE concentration when these two sites were recorded as clinically active (Supporting information, Fig. S2a). The median SLPI saliva concentration for BDq with joint pain was 147·3 ng/ml, while those with folliculitis was 367·7 ng/ml. Again, there was no significant difference in the saliva levels when these two most common sites were active (Supporting information, Fig. S2b). However, when BDq had active joints (n = 11), the α1AT in their saliva was significantly higher (P = 0·02) (Supporting information, Fig. S2c). For BD patients with active oral ulcers the most frequent ailments were follicultis (34–46%), followed closely by joint pain (33–44%) and genital ulcers (25–33%) (Supporting information, Table S1). The NE, SLPI and α1AT saliva levels in BDa patients were not significantly higher when they were experiencing folliculitis or joint pain (Supporting information, Fig. S2d–f).
Salivary NE levels and patient medications
While no systemic medications were reported for HC or RAS groups, similar numbers of BDa and BDq patients were receiving immunomodulatory medications at the time of sample collection (Supporting information, Table S2). Saliva samples were chosen from this cohort based on active or quiet ulcer status. The medications were analysed as a secondary variable.
Salivary NE levels were measured in both BDa and BDq patients taking COLC or azathioprine (AZA), both, or neither COLC or AZA (Fig. 2). Although there was no significant difference in NE levels among BDq patients, those who were taking both COLC and AZA tended to have increased NE (2309 ng/ml). However, the NE levels in BDa patients on both COLC and AZA were significantly lower (1131 ng/ml) compared to those taking only COLC (1816 ng/ml, P = 0·0008) or those on neither COLC or AZA (2141 ng/ml, P = 0·02). This group of BDa patients on COLC and AZA had similar NE levels to healthy controls (999 ng/ml, Fig. 2a) and although this group consisted of a small number of patients (n = 7), they exhibited a narrow range of NE levels (490–1347 ng/ml).
Fig. 2.
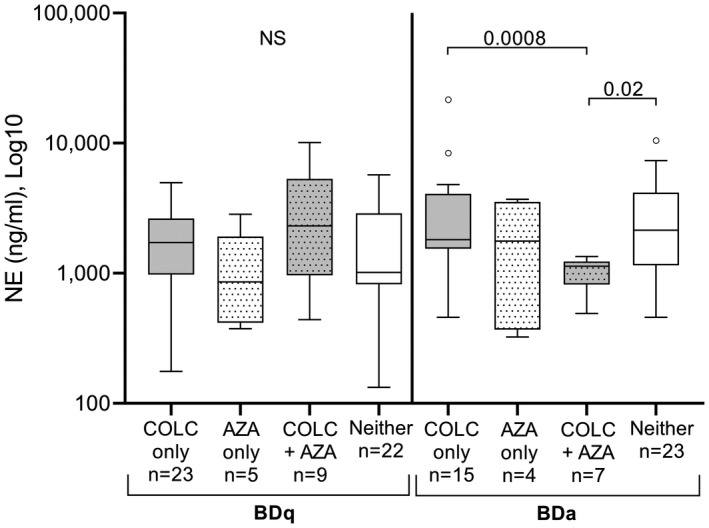
Saliva neutrophil elastase (NE) levels of Behçet’s disease (BD) patients taking systemic colchicine (COLC) and/or azathioprine (AZA) during quiet or active oral ulceration. The Tukey box‐plots show enzyme‐linked immunosorbent assay (ELISA) NE levels in the following groups: Behçet’s disease (BD)q patients with no oral ulceration at the time of sampling and BDa patients who had oral ulcer(s). Patients were identified as taking COLC or AZA, both, or neither AZA or COLC. ‘COLC only’ refers to the exclusion of AZA and vice versa; however, this does not exclude other medications that the patient may have been taking. Mann–Whitney U‐test, significance based on two‐tailed 95% confidence interval (CI). Exact P‐values are indicated; n.s. = not significant.
Enzymatically active NE in saliva
Elastin‐specific proteases were measured, and HC saliva had the lowest overall protease activity compared to all other study groups (Fig. 3a) based on an equal amount of elastin substrate being added to each saliva sample. Within 95 min of exposure to the substrate, the total protease and NE enzyme activity was significantly higher in both BDa and BDq patients compared to HC (P < 0·05). When comparing each group’s NE activity relative to their total protease activity, there were no significant differences (Fig. 3b). This was determined by dividing the median fluorescence of the NE‐specific activity by the total protease activity fluorescence for each group and multiplying by 100. The NE in HC saliva comprised ~78% of the total protease activity. Similarly, RAS and BD saliva samples also showed that 79·6 and 82·8% of their total protease activity, respectively, was due to NE.
Fig. 3.
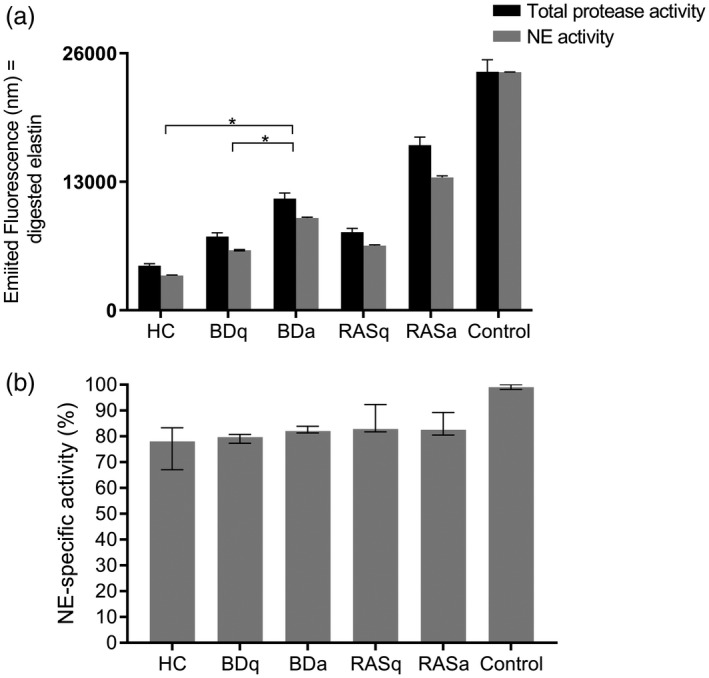
Although the quantity of neutrophil elastase (NE) differs among patient groups, it makes up the majority of elastin‐targeting proteases in saliva. The total protease and NE‐specific activity were measured in saliva samples based on the breakdown and release of fluorescent‐conjugated elastin. Saliva samples were tested in duplicate from each patient group: healthy controls (HC) n = 8, BDq (quiet, no ulcer) n = 6, BDa (active oral ulcer) n = 7, recurrent aphthous stomatitis (RAS)q, n = 5; RASa, n = 4. (a) The black bars represent each group’s total protease activity based on the median fluorescence intensity. The grey bars represent the level of NE‐specific activity for the same saliva samples. Error bars are 5% of the group median. Porcine elastase was used as a positive control. Mann–Whitney U‐test, *P < 0·05. Only significant differences are indicated. (b) Percentage of NE activity relative to the total protease activity (group median values). Error bars indicate the interquartile region. The enzymatic ability of NE to digest elastin was similar in all groups, regardless of ulceration.
Stable reference genes in OBS
Duplicate SLPI cycle quantification (Cq) values were averaged and normalized (calibrated normalized relative quantity) using the qBasePlus (Biogazelle software) against the Cq values for the PPIA reference gene. Of 48 OBS, 13 samples were excluded from analysis due to a final Cq value > 40. RASa U samples were completed but not included in analysis due to the small sample size, only n = 2. None of the RT‐negative samples amplified, and were therefore clear of any gDNA contamination. The four most stable reference genes validated for BD, RAS and HC OBS were PPIA, glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH), phosphoglycerate kinase 1 (PGK1) and beta‐actin (ACTB) (Supporting information, Fig. S3). PPIA primers performed the most efficiently, and due to the limited amount of material for multiple assays this reference gene was used to normalize the qPCR results for SLPI mRNA expression.
SLPI mRNA expression
SLPI mRNA was significantly increased in BDa, NU (P ≤ 0·01) and BDa, U (P ≤ 0·05) when compared to HC (Fig. 4). Also, SLPI mRNA was significantly up‐regulated in BDq (P ≤ 0·001) compared to HC and RASq (P ≤ 0·05). Unfortunately, the only two RASa U samples available showed a wide variable range, and were therefore excluded from the final analysis.
Fig. 4.
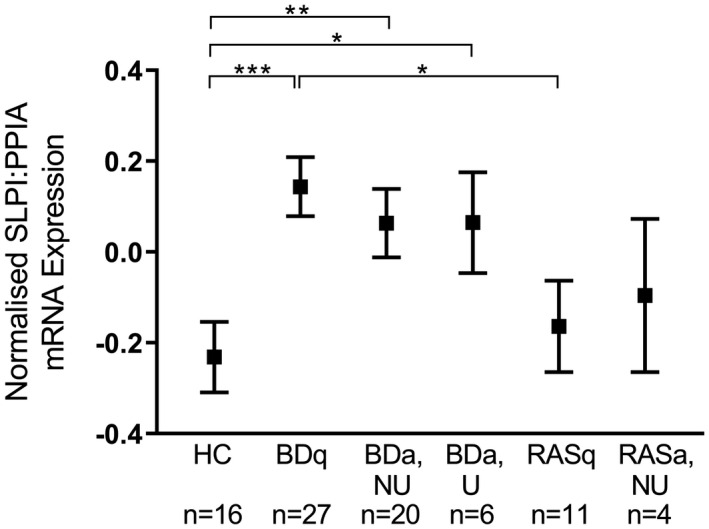
Mean secretory leucocyte protease inhibitor (SLPI) mRNA expression in oral buccal swabs among different patient groups. SLPI cycle quantification (Cq) values normalized using peptidylprolyl isomerase A (PPIA) as the reference gene and compared among healthy controls (HC), recurrent aphthous stomatitis (RAS), Behçet’s disease (BD); q = quiet, no ulcers; a = at least one active mouth ulcer regardless of other systemic symptoms. Non‐ulcerated site (NU) = swab taken from the buccal mucosa away from the oral ulcer; U = swab taken directly from the ulcer. Data analysed using unpaired t‐test with Welch’s correction (two‐tailed, 95% confidence interval). *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001.
Discussion
Increased circulating neutrophils and their degranulated enzyme, NE, have previously been implicated in the systemic chronic inflammatory response in BD [14, 15]. The recurrent oral ulcers that accompany BD may also result from unregulated NE‐stimulated inflammation. As observed in a previous study, it was thought that protective salivary SLPI would increase during oral ulceration, as it has been shown to aid wound healing [37]. We report here for the first time, to our knowledge, that during quiet oral episodes in which BD mucosa is not ulcerated, individuals with BD have active proteolytic NE in their saliva in quantities similar to HCs and RASq subjects; however, levels of SLPI in BDq are significantly lower. Taken together, these results indicate that the presence of active NE and reduced SLPI may be contributory factors to initiation of BD oral ulceration (Fig. 5). Another endogenous inhibitor, α1AT, despite its abundance in saliva, does not appear to affect NE activity. The results suggest that there is an association between high levels of proteolytic enzymes and oral mucosal instability leading to oral ulceration. Furthermore, our data show that BDa patients medicated with both AZA and COLC have reduced salivary NE.
Fig. 5.
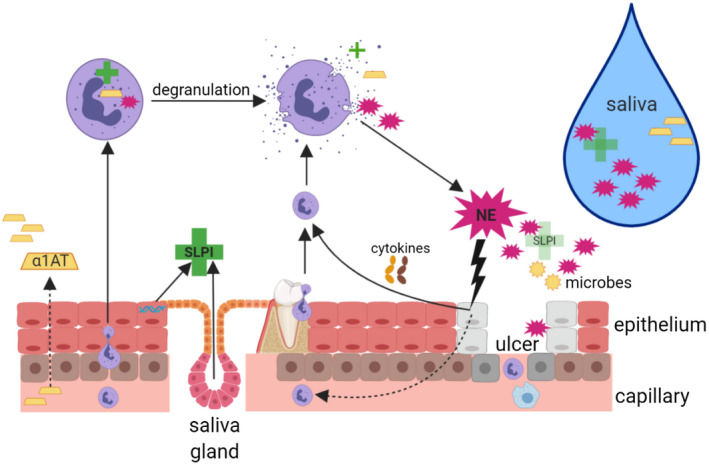
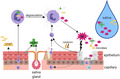
Proposed working model of Behҫet’s disease (BD) oral environment: summary of the interaction of the epithelium, neutrophil elastase (NE), secretory leucocyte protease inhibitor (SLPI) and alpha1‐anti‐trypsin (α1AT). Individuals with BD have an increased presence of neutrophils in their vasculature which are recruited to the oral cavity due to hypersensitivity to ever‐present microbes and proinflammatory cytokines [tumour necrosis factor (TNF)‐α, interleukin (IL‐8)] produced by oral epithelial keratinocytes and immune cells. Fibroblasts and leucocytes further contribute IL‐1β, which induces additional IL‐8 production – a neutrophil chemoattractant. Neutrophils continue to migrate through the epithelium towards the site of accumulating inflammatory signals. Neutrophils phagocytose foreign entities or degranulate releasing NE into the extracellular environment. Activated neutrophils also release IL‐8, thus reinforcing a cycle of inflammation. Neutrophils may apoptose shortly after controlled degranulation, but may become necrotic or undergo NET activation and release (NETosis), which releases NE non‐discriminately. Salivary glands and epithelial cells work to produce SLPI to counteract the excessive NE, but SLPI is steadily used up. Although serum‐derived α1AT is carried to the oral cavity, there is no evidence that it can reduce NE activity. Neutrophils also have intracellular α1AT and SLPI reserves, but the extent of their external inhibitory contribution is not known. Excessive, prolonged exposure to NE can directly damage epithelial cells and the integrity of the mucosal lining by breaking down extracellular matrix (ECM), resulting in an ulcer. The injured tissue reinforces the inflammatory cycle and recruitment of neutrophils. Fluctuations of these inflammatory elements and reduced healing components may contribute to the recurrent ulcers seen in this disease (created using BioRender).
The inverse relationship of NE and SLPI
All individuals had quantifiable NE and SLPI in their saliva. Our data show a ratio of 2 : 1, NE : SLPI concentration based on collective HC median values using an ELISA with monoclonal antibody detection of each protein. While NE increased during oral ulceration in RAS and BD patients, BDq patients had only slightly higher NE levels than HCs, but showed a significant reduction of SLPI (the lowest of all groups), giving a concentration ratio of 5 : 1, NE : SLPI – precisely the same NE : SLPI ratio as BDa levels. This suggests that SLPI is depleted in BDq patients despite being orally asymptomatic. A similar inverse NE/SLPI dynamic has been previously reported in active periodontitis saliva [38], and reduced SLPI has been associated with periodontal disease progression [39].
SLPI has two structural domains allowing the binding of two different targets [28]. We therefore postulated that as SLPI derived from whole saliva could simultaneously bind to both NE and a different second target (such as a microbe), this could block SLPI detection by ELISA antibody. To further validate and scrutinize the reduction of SLPI in our BDq cohort, we conducted a spike‐recovery ELISA experiment. After adding known concentrations of recombinant (r)SLPI into BDq saliva in which SLPI had previously been measured, the detection recovery of rSLPI was between 89 and 99%. This provided us with evidence that the ELISA detection was accurate despite the possibility of SLPI to be masked while exposed to multiple targets in saliva.
Other reasons for the depletion of SLPI in saliva may involve cysteine proteases such as cathepsins or chymase, a serine protease from mast cells, which can cleave and inactivate SLPI [38]. NE has also been shown to directly degrade SLPI, which could explain the inverse concentration levels [40]. Another reason for the lower SLPI concentration could be due to its direct attachment to oral microbes. There is strong evidence that SLPI has broad‐spectrum anti‐microbial activity [28]. In a study by Seoudi et al., the oral bacterial profile of BD and RAS patients with and without ulcers and HCs revealed that the different subject groups had different bacterial species colonization [41]. Microbial flora have been previously implicated in BD pathogenesis [42], which may further explain their decreased levels of SLPI.
Increased SLPI mRNA but decreased SLPI protein is characteristic during BDq
Reduced SLPI levels could also indicate that sufficient levels of SLPI were not available in the first place. This would be especially detrimental in an inflammatory condition such as BD, in which regulation of proinflammatory factors is vital to reduce symptoms. This prompted us to investigate local SLPI mRNA expression. Both saliva gland acini and epithelial cells contribute to salivary SLPI levels. Excising patient gland cells was unrealistic so, instead, we investigated SLPI mRNA expression in the oral epithelium using buccal swabs. The vast majority of studies investigating oral buccal epithelial cell RNA involves oral squamous cell carcinoma and many do not mention validation of their reference genes. To date, this is the first study to report stable RNA reference genes in OBS from HC as well as BD and RAS patients with and without oral ulcers and is explored in depth in the Supplementary information, Part II.
The expectation was that SLPI mRNA would be reduced, reflecting the low salivary levels. Surprisingly, the results indicated that SLPI mRNA was significantly up‐regulated in BDq patients’ buccal epithelial cells. A similar paradox was reported in an in‐vitro experiment by Sallenave et al., in which NE was used to stimulate A549 epithelial cells. After 24 h, SLPI protein levels were decreased by 94% while SLPI mRNA was up‐regulated [43].The rate of translation (mRNA to protein), production and cellular exocytosis of SLPI is unknown for buccal epithelial cells; however, ex‐vivo human upper respiratory tract submucosal cells have shown a delay of SLPI exocytosis after NE exposure [44]. We speculate that once SLPI is available in the extracellular space in the presence of high levels of NE it is rapidly utilized.
In BDa patients, the up‐regulation of SLPI mRNA could be directly stimulated by high levels of salivary NE. However, what is decisively characteristic is that BDq patients with no oral ulceration have the highest SLPI mRNA expression, lowest salivary SLPI and NE levels, on par with HC and RASq. This suggests that BDq patients are probably more sensitive to NE stimulation in comparison to HC or RAS patients. Also, the SLPI mRNA data provides compelling evidence of a differentiation between the oral mucosal environment of BDq and RASq patients.
A small proportion of the BDq patients in this study presented with other systemic clinical manifestations, such as joint pain, skin lesions and genital ulcers at the time of their saliva donation, demonstrating their underlying systemic inflammatory activity. Although the numbers were small, it was found that only salivary α1AT levels were significantly increased in BDq patients with active joint pain (P = 0·02). This could be due to increased α1AT production by the liver and vascular dissemination. None of the BDa patients showed any significant difference in their NE, SLPI or α1AT saliva levels when assessing their most common systemic symptoms. Our data would suggest that NE and SLPI in saliva do not necessarily reflect other site‐specific systemic activity. Importantly, therefore, when assessing SLPI as a biomarker in BDq patients, other systemic symptoms should not greatly influence the oral concentration.
Even though our data did not show significantly higher NE saliva levels during other active systemic symptoms, increased circulating peripheral blood neutrophils are likely to influence tissue and oral infiltration. A study by Deger et al. showed elevated levels of NE in plasma from 22 BD orally quiet patients compared to HC [14]. Further studies are required that measure NE, SLPI and α1AT levels in matched serum and saliva of BD patients to explore if there is a more robust correlation with clinical activity in the mouth, eyes, skin, joints and central nervous system (CNS).
Lakschevitz et al. have reported that oral and peripheral blood neutrophils from HCs behave differently. Based on microarray data, they found that HC salivary neutrophil RNA changed their transcriptome when they migrated from the blood to the oral environment. This alteration was especially evident during increased oral infection or inflammation [45, 46]. It has also been suggested that neutrophils are already primed by chemotactic factors such as IL‐8 or TNF‐α by the time they reach the oral cavity [47]. There is therefore a need to keep in mind that oral neutrophils are exposed to different microbial, antigen and toxin conditions in saliva when compared to blood, which can influence the release of potent anti‐microbial proteases such as NE into the saliva and oral cavity [48].
Importantly, during quiet oral episodes, increased NE degranulation in saliva would probably rely upon local anti‐protease activity such as SLPI to curtail epithelial damage. Our data suggest that SLPIs depletion in BDq saliva may be a key factor in the manifestation of oral ulcers. It may benefit BD patients to use a local, topical NE‐inhibitor, such as a compound containing rSLPI, to help control recurring oral ulceration [49].
Salivary proteases and NE activity
NE was confirmed to be enzymatically active in saliva and responsible for the majority of protease activity. It was anticipated that patients with ulcers would have increased NE activity, as the ulcerated wound would have initiated neutrophil recruitment to the site. However, BDq and RASq also had higher NE activity compared to HCs. Individuals with BD have been described as having hyperactive circulating neutrophils [16]. Our data add to this observation that proteolytic, active oral NE is also present. Under‐regulated, enzymatically active NE in BD saliva is likely to contribute to recurrent mucosal damage and ulceration characteristic of BD.
α1AT is not the main NE inhibitor in saliva
NE and α1AT showed a direct relationship – if NE increased, α1AT also increased. In HCs, α1AT was five times the NE concentration and approximately 10 times that of SLPI. SLPI and α1AT are competitors, in that they both inhibit NE. Both also reside inside neutrophils [33], presumably protecting neutrophils from unintentional self‐induced proteolytic damage. The majority of α1AT is produced in liver cells. Approximately 1–2 g/l is transferred to plasma and up to 10% diffuses into biological fluids such as saliva [50]. As a serpin, α1AT is thought to irreversibly bind and inactivate NE [51], yet in our enzyme assay salivary NE was proteolytically active. This leads us to deduce that despite the abundance of α1AT in saliva, it is not the main inhibitor of NE in the oral cavity. Also, various proteases can inactivate α1AT such as collagenase [52], therefore it is also possible that α1AT’s inhibitory site, which is susceptible to oxidation, is inactivated by reactive oxygen species generated by phagocytic cells and bacteria in saliva [53].
COLC–AZA may lower NE during BD ulceration
The combination of COLC and AZA medications is associated with decreased NE in BDa saliva. COLC is one of the recommended medications for treating mucocutaneous lesions by reducing the migration of neutrophils [18], and has been shown to modulate oxidative stress in BD neutrophils by regulating Ca2+ release [54]. In combination with AZA, we sought to determine whether this combination of systemic medication influences oral NE levels. We found that BDa patients taking both medications had significantly lower NE, which was surprising during ulceration. Despite the small sample size, the narrow range of their NE measurements indicates low variability. AZA is an immunosuppressive drug which lowers the proliferation of lymphocytes [55]. COLC accumulates in white blood cells (WBCs) (namely neutrophils) disrupting various inflammatory activation, degranulation and migration processes [56]. Coupled together, the drugs’ synergistic effects of reducing WBCs and proinflammatory activities, thus decreasing NE degranulation, may help to control inflammation. Further investigation is necessary to validate this data.
Study limitations
There are some limitations to this study. First, RAS subjects were sought as a non‐systemic disease controls from general oral medicine clinics and from local advertisements, but recruitment was lower than expected compared to our BD cohort, which was recruited from dedicated specialist BD clinics. Individuals with RAS are thought to endure a unique immune dysregulation which, for unknown reasons, manifests solely in the oral cavity [57]. Furthermore, Ueta et al. have suggested that individuals with RAS may harbour an immune cell dysfunction based on the evidence that their neutrophils had suppressed superoxide production compared to HCs [58]. Overall, it was rationalized that the RAS group, although having small patient numbers, would still add further important insights into oral ulceration mechanisms for which very little is known.
Another limitation presented itself when directly swabbing mucosal ulcers too gently with cytology swabs, as this did not generate enough RNA for all analyses. In future collections, a softer swab that would be both gentle to the patient’s ulcerated mucosa and compatible with PCR assays would be used. Another consideration for future research would be to collect data on the subject’s smoking status, race, duration of treatment and alterations in treatment regimens. Also, relatively large volumes of saliva were necessary to run NE, SLPI, α1AT ELISAs and enzyme activity assays in duplicate. Therefore, it was not possible to run all the analyses on the same set of saliva samples, which resulted in unequally sized groups for the different assays. Although BD is a rare disorder, this research study benefited from a substantial number of participants, thanks to their attendance and recruitment at the Royal London Hospital’s specialized clinic.
In conclusion, this study was able to describe the depletion of SLPI in BD saliva during orally asymptomatic episodes and highlights its potential in balancing the damage caused by enzymatically active, excessive NE. As biomarkers, the collective measurements of NE and SLPI in saliva alongside oral epithelial SLPI mRNA could differentiate between RASq and BDq to assist clinical evaluation. Early intervention could be key to controlling the progression of inflammation associated with BD. During oral ulceration, the current combined treatment with COLC and AZA seems to lower NE in saliva, which could help to control inflammation. As a local, topical treatment, recombinant SLPI is commercially available and has been trialled in other inflammatory disorders [49, 59, 60]. This study also found that α1AT, the most well‐known NE‐inhibitor in the vasculature, did not seem to translate its anti‐protease activity in saliva – again highlighting the importance of SLPI as the local NE inhibitor.
Disclosures
All authors confirm there are no financial conflicts of interest.
Author contributions
T. N. and E. H‐P. contributed to conception, design, data acquisition, analysis and interpretation, drafted and critically revised the manuscript. F. F. and L. B. helped with the analysis and interpretation as well as a critical review the manuscript. I. K. assisted with data acquisition and critically revised the manuscript. All authors gave their final approval and agree to be accountable for all aspects of the work. Financial support for this study was provided by the James Paget PhD Studentship to T. N. T. N. is currently supported by the National Institute of Health (5T32GM103702‐05).
Supporting information
Table S1. BD subject’s clinical presentation at time of saliva collection for each ELISA NE ELISA.
Table S2. All Behçet’s patients’ medication regime at the time of saliva collection.
Fig. S1. The correlation of SLPI and NE in saliva was assessed for a subset of HC and BD patients. Spearman’s rho results revealed the inverse relationship of SLPI/NE for HC and BDa was as expected (when SLPI increased, NE decreased), however for BDq saliva samples, the strength of the inverse relationship was diminished providing evidence of protease‐inhibitor disruption.
Fig. S2. NE, SLPI, and α1AT levels in saliva from BDq (a‐c) and BDa (d‐f) with active disease manifestations at their most common systemic sites. The most common active symptoms in BD patients were their joints and skin ‐ particularly folliculitis. a, b. NE and SLPI in saliva were not significantly increased when BDq patients also had active joints or folliculitis. c. α1AT in BDq saliva was significantly higher in those with active joints. d‐f. BDa patients did not show any significant difference in NE, SLPI, or α1AT saliva levels during active joints or folliculitis. NS, no significance.
Fig. S3. The most stable four reference genes in oral buccal swabs among three patient groups: HC, RAS, and BD; the latter groups include samples directly obtained from oral ulcers. Each gene’s stability was determined by the mean Cq from all patient groups. Error bars represent the standard deviation.
Acknowledgements
We would like to thank all study participants, as well as the Behçet’s clinic team at Barts and The London School of Medicine and Dentistry for consenting patients and collecting samples.
References
- 1. Direskeneli H. Autoimmunity vs autoinflammation in Behcet’s disease: do we oversimplify a complex disorder? Rheumatology 2006; 45:1461–5. [DOI] [PubMed] [Google Scholar]
- 2. Hedayatfar A. Behcet’s disease: autoimmune or autoinflammatory? J Ophthalmic Vis Res 2013; 8:291–3. [PMC free article] [PubMed] [Google Scholar]
- 3. Direskeneli H, Keser G, D’Cruz D et al Anti‐endothelial cell antibodies, endothelial proliferation and von Willebrand factor antigen in Behcet’s disease. Clin Rheumatol 1995; 14:55–61. [DOI] [PubMed] [Google Scholar]
- 4. Petrushkin H, Norman PJ, Lougee E et al KIR3DL1/S1 allotypes contribute differentially to the development of Behcet disease. J Immunol 2019; 203:1629–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wallace GR. HLA‐B*51 the primary risk in Behcet disease. Proc Natl Acad Sci USA 2014; 111:8706–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fortune F. Can you catch Behcet’s disease? J Lab Clin Med 2003; 141:5–6. [DOI] [PubMed] [Google Scholar]
- 7. Freysdottir J, Lau S, Fortune F. Gammadelta T cells in Behcet’s disease (BD) and recurrent aphthous stomatitis (RAS). Clin Exp Immunol 1999; 118:451–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kokturk A. Clinical and pathological manifestations with differential diagnosis in Behcet’s disease. Patholog Res Int 2012; 2012:690390 10.1155/2012/690390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lehner T. Immunological aspects of recurrent oral ulceration and Behcet’s syndrome. J Oral Pathol 1978; 7:424–30. [DOI] [PubMed] [Google Scholar]
- 10. Natah SS, Hayrinen‐Immonen R, Hietanen J, Malmstrom M, Konttinen YT. Immunolocalization of tumor necrosis factor‐alpha expressing cells in recurrent aphthous ulcer lesions (RAU). J Oral Pathol Med 2000; 29:19–25. [DOI] [PubMed] [Google Scholar]
- 11. Dalghous AM, Freysdottir J, Fortune F. Expression of cytokines, chemokines, and chemokine receptors in oral ulcers of patients with Behcet’s disease (BD) and recurrent aphthous stomatitis is Th1‐associated, although Th2‐association is also observed in patients with BD. Scand J Rheumatol 2006; 35:472–5. [DOI] [PubMed] [Google Scholar]
- 12. Impellizzieri D, Ridder F, Raeber ME et al IL‐4 receptor engagement in human neutrophils impairs their migration and extracellular trap formation. J Allergy Clin Immunol 2019; 144:267–79.e4. [DOI] [PubMed] [Google Scholar]
- 13. Girard D, Paquin R, Beaulieu AD. Responsiveness of human neutrophils to interleukin‐4: induction of cytoskeletal rearrangements, de novo protein synthesis and delay of apoptosis. Biochem J 1997; 325(Pt 1):147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deger O, Orem A, Akyol N, Bahadir S, Yildirmis S. Polymorphonuclear leukocyte elastase levels in patients with Behcet’s disease. Clin Chim Acta 1995; 236:129–34. [DOI] [PubMed] [Google Scholar]
- 15. Eksioglu‐Demiralp E, Direskeneli H, Kibaroglu A, Yavuz S, Ergun T, Akoglu T. Neutrophil activation in Behcet’s disease. Clin Exp Rheumatol 2001; 19(5 Suppl 24):S19–24. [PubMed] [Google Scholar]
- 16. Verity DH, Wallace GR, Vaughan RW, Stanford MR. Behcet’s disease: from Hippocrates to the third millennium. Br J Ophthalmol 2003; 87:1175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sensi A, Gavioli R, Spisani S et al HLA B51 antigen associated with neutrophil hyper‐reactivity. Dis Markers 1991; 9:327–31. [PubMed] [Google Scholar]
- 18. Alpsoy E. New evidence‐based treatment approach in Behcet’s disease. Patholog Res Int 2012; 2012:871019 10.1155/2012/871019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fouret P, du Bois RM, Bernaudin JF, Takahashi H, Ferrans VJ, Crystal RG. Expression of the neutrophil elastase gene during human bone marrow cell differentiation. J Exp Med 1989; 169:833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kessenbrock K, Dau T, Jenne DE. Tailor‐made inflammation: how neutrophil serine proteases modulate the inflammatory response. J Mol Med 2011; 89:23–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Le Joncour A, Martos R, Loyau S et al Critical role of neutrophil extracellular traps (NETs) in patients with Behcet’s disease. Ann Rheum Dis 2019; 78:1274–82. [DOI] [PubMed] [Google Scholar]
- 22. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 2006; 6:173–82. [DOI] [PubMed] [Google Scholar]
- 23. Heutinck KM, ten Berge IJ, Hack CE, Hamann J, Rowshani AT. Serine proteases of the human immune system in health and disease. Mol Immunol 2010; 47:1943–55. [DOI] [PubMed] [Google Scholar]
- 24. Stockley RA. Neutrophils and the pathogenesis of COPD. Chest 2002; 121(5 Suppl):151S–5S. [DOI] [PubMed] [Google Scholar]
- 25. Reilly CF, Travis J. The degradation of human lung elastin by neutrophil proteinases. Biochim Biophys Acta 1980; 621:147–57. [DOI] [PubMed] [Google Scholar]
- 26. Piccioni PD, Kramps JA, Rudolphus A, Bulgheroni A, Luisetti M. Proteinase/proteinase inhibitor imbalance in sputum sol phases from patients with chronic obstructive pulmonary disease. Suggestions for a key role played by antileukoprotease. Chest 1992; 102:1470–6. [DOI] [PubMed] [Google Scholar]
- 27. Jacobsen LC, Sorensen OE, Cowland JB, Borregaard N, Theilgaard‐Monch K. The secretory leukocyte protease inhibitor (SLPI) and the secondary granule protein lactoferrin are synthesized in myelocytes, colocalize in subcellular fractions of neutrophils, and are coreleased by activated neutrophils. J Leukoc Biol 2008; 83:1155–64. 10.1189/jlb.0706442. [DOI] [PubMed] [Google Scholar]
- 28. Doumas S, Kolokotronis A, Stefanopoulos P. Anti‐inflammatory and antimicrobial roles of secretory leukocyte protease inhibitor. Infect Immun 2005; 73:1271–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sallenave JM. The role of secretory leukocyte proteinase inhibitor and elafin (elastase‐specific inhibitor/skin‐derived antileukoprotease) as alarm antiproteinases in inflammatory lung disease. Respir Res 2000; 1:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thompson RC, Ohlsson K. Isolation, properties, and complete amino‐acid‐sequence of human secretory leukocyte protease inhibitor, a potent inhibitor of leukocyte elastase. Proc Natl Acad Sci USA 1986; 83:6692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pederson ED, Stanke SR, Whitener SJ, Sebastiani PT, Lamberts BL, Turner DW. Salivary levels of alpha 2‐macroglobulin, alpha 1‐antitrypsin, C‐reactive protein, cathepsin G and elastase in humans with or without destructive periodontal disease. Arch Oral Biol 1995; 40:1151–5. [DOI] [PubMed] [Google Scholar]
- 32. Huynh C, Roch‐Arveiller M, Meyer J, Giroud JP. Gingival crevicular fluid of patients with gingivitis or periodontal disease: evaluation of elastase‐alpha 1 proteinase inhibitor complex. J Clin Periodontol 1992; 19:187–92. [DOI] [PubMed] [Google Scholar]
- 33. Clemmensen SN, Jacobsen LC, Rorvig S et al Alpha‐1‐antitrypsin is produced by human neutrophil granulocytes and their precursors and liberated during granule exocytosis. Eur J Haematol 2011; 86:517–30. [DOI] [PubMed] [Google Scholar]
- 34. Lee WL, Downey GP. Leukocyte elastase: physiological functions and role in acute lung injury. Am J Respir Crit Care Med 2001; 164:896–904. [DOI] [PubMed] [Google Scholar]
- 35. Ginzberg HH, Cherapanov V, Dong Q et al Neutrophil‐mediated epithelial injury during transmigration: role of elastase. Am J Physiol Gastrointest Liver Physiol 2001; 281:G705–717. [DOI] [PubMed] [Google Scholar]
- 36. International Study Group for Behcet’s Disease . Criteria for diagnosis of Behcet’s disease. Lancet 1990; 335:1078–80. [PubMed] [Google Scholar]
- 37. Angelov N, Moutsopoulos N, Jeong MJ, Nares S, Ashcroft G, Wahl SM. Aberrant mucosal wound repair in the absence of secretory leukocyte protease inhibitor. Thromb Haemost 2004; 92:288–97. [DOI] [PubMed] [Google Scholar]
- 38. Cox SW, Rodriguez‐Gonzalez EM, Booth V, Eley BM. Secretory leukocyte protease inhibitor and its potential interactions with elastase and cathepsin B in gingival crevicular fluid and saliva from patients with chronic periodontitis. J Periodont Res 2006; 41:477–85. [DOI] [PubMed] [Google Scholar]
- 39. Pateel D, Seema H, Kale A. Role of salivary leukocyte protease inhibitor in periodontal disease progression. J Indian Soc Periodont 2010; 14:109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weldon S, McNally P, McElvaney NG et al Decreased levels of secretory leucoprotease inhibitor in the Pseudomonas‐infected cystic fibrosis lung are due to neutrophil elastase degradation. J Immunol 2009; 183:8148–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seoudi N, Bergmeier LA, Drobniewski F, Paster B, Fortune F. The oral mucosal and salivary microbial community of Behcet’s syndrome and recurrent aphthous stomatitis. J Oral Microbiol 2015; 7:27150 10.3402/jom.v7.27150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Galeone M, Colucci R, D’Erme AM, Moretti S, Lotti T. Potential infectious etiology of Behcet’s disease. Patholog Res Int 2012; 2012:595380 10.1155/2012/595380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sallenave JM, Shulmann J, Crossley J, Jordana M, Gauldie J. Regulation of secretory leukocyte proteinase inhibitor (SLPI) and elastase‐specific inhibitor (ESI/elafin) in human airway epithelial cells by cytokines and neutrophilic enzymes. Am J Respir Cell Mol Biol 1994; 11:733–41. [DOI] [PubMed] [Google Scholar]
- 44. Saitoh H, Masuda T, Shimura S, Fushimi T, Shirato K. Secretion and gene expression of secretory leukocyte protease inhibitor by human airway submucosal glands. Am J Physiol Lung Cell Mol Physiol 2001; 280:L79–87. [DOI] [PubMed] [Google Scholar]
- 45. Lakschevitz FS, Aboodi GM, Glogauer M. Oral neutrophil transcriptome changes result in a pro‐survival phenotype in periodontal diseases. PLOS ONE 2013; 8:e68983 10.1371/journal.pone.0068983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lakschevitz FS, Aboodi GM, Glogauer M. Oral neutrophils display a site‐specific phenotype characterized by expression of T‐cell receptors. J Periodontol 2013; 84:1493–503. [DOI] [PubMed] [Google Scholar]
- 47. Nakahara H, Sato EF, Ishisaka R et al Biochemical properties of human oral polymorphonuclear leukocytes. Free Radic Res 1998; 28:485–95. [DOI] [PubMed] [Google Scholar]
- 48. Hirschfeld J. Neutrophil subsets in periodontal health and disease: a mini review. Front Immunol 2019; 10:3001 10.3389/fimmu.2019.03001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Camper N, Glasgow AM, Osbourn M et al A secretory leukocyte protease inhibitor variant with improved activity against lung infection. Mucosal Immunol 2016; 9:669–76. [DOI] [PubMed] [Google Scholar]
- 50. de Serres F, Blanco I. Role of alpha‐1 antitrypsin in human health and disease. J Intern Med 2014; 276:311–35. [DOI] [PubMed] [Google Scholar]
- 51. Korkmaz B, Moreau T, Gauthier F. Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie 2008; 90:227–42. [DOI] [PubMed] [Google Scholar]
- 52. Michaelis J, Vissers MC, Winterbourn CC. Human neutrophil collagenase cleaves alpha 1‐antitrypsin. Biochem J 1990; 270:809–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rosenberg S, Barr PJ, Najarian RC, Hallewell RA. Synthesis in yeast of a functional oxidation‐resistant mutant of human alpha‐antitrypsin. Nature 1984; 312:77–80. [DOI] [PubMed] [Google Scholar]
- 54. Korkmaz S, Erturan I, Naziroglu M, Uguz AC, Cig B, Ovey IS. Colchicine modulates oxidative stress in serum and neutrophil of patients with Behcet disease through regulation of Ca(2)(+) release and antioxidant system. J Membr Biol 2011; 244:113–20. [DOI] [PubMed] [Google Scholar]
- 55. Nair JR, Moots RJ. Behcet;s disease. Clin Med 2017; 17:71–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Leung YY, Yao Hui LL, Kraus VB. Colchicine – update on mechanisms of action and therapeutic uses. Semin Arthritis Rheum 2015; 45:341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Natah SS, Konttinen YT, Enattah NS, Ashammakhi N, Sharkey KA, Hayrinen‐Immonen R. Recurrent aphthous ulcers today: a review of the growing knowledge. Int J Oral Maxillofac Surg 2004; 33:221–34. [DOI] [PubMed] [Google Scholar]
- 58. Ueta E, Umazume M, Yamamoto T, Osaki T. Leukocyte dysfunction in oral mucous membrane diseases. J Oral Pathol Med 1993; 22:120–5. [DOI] [PubMed] [Google Scholar]
- 59. Maffia PC, Guerrieri D, Villalonga X et al Cementoin‐SLPI fusion protein binds to human monocytes and epithelial cells and shows higher biological activity than SLPI. Sci Rep 2018; 8:5332 10.1038/s41598-018-23680-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McElvaney NG, Doujaiji B, Moan MJ, Burnham MR, Wu MC, Crystal RG. Pharmacokinetics of recombinant secretory leukoprotease inhibitor aerosolized to normals and individuals with cystic fibrosis. Am Rev Respir Dis 1993; 148(4 Pt 1):1056–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. BD subject’s clinical presentation at time of saliva collection for each ELISA NE ELISA.
Table S2. All Behçet’s patients’ medication regime at the time of saliva collection.
Fig. S1. The correlation of SLPI and NE in saliva was assessed for a subset of HC and BD patients. Spearman’s rho results revealed the inverse relationship of SLPI/NE for HC and BDa was as expected (when SLPI increased, NE decreased), however for BDq saliva samples, the strength of the inverse relationship was diminished providing evidence of protease‐inhibitor disruption.
Fig. S2. NE, SLPI, and α1AT levels in saliva from BDq (a‐c) and BDa (d‐f) with active disease manifestations at their most common systemic sites. The most common active symptoms in BD patients were their joints and skin ‐ particularly folliculitis. a, b. NE and SLPI in saliva were not significantly increased when BDq patients also had active joints or folliculitis. c. α1AT in BDq saliva was significantly higher in those with active joints. d‐f. BDa patients did not show any significant difference in NE, SLPI, or α1AT saliva levels during active joints or folliculitis. NS, no significance.
Fig. S3. The most stable four reference genes in oral buccal swabs among three patient groups: HC, RAS, and BD; the latter groups include samples directly obtained from oral ulcers. Each gene’s stability was determined by the mean Cq from all patient groups. Error bars represent the standard deviation.