

Abstract
Worrisome growth can be a sign of underlying pathology but usually reflects healthy variation. It is often recognized through short stature, which is defined by arbitrarily separating height, a physical trait on a continuum, into “normal” and “abnormal.” In some cases of worrisome growth, recombinant human growth hormone (rhGH) treatment is indicated to hasten growth/increase height. This review addresses the two most frequently treated indications for rhGH, growth hormone deficiency (GHD) and idiopathic short stature (ISS). A review of worrisome growth itself, of the history of GH treatment, of the blurry line between partial GHD and ISS, of the GH stakeholders, and of the outside pressures involved in these cases demonstrates the ambiguous platform upon which treatment decisions are made. The rhGH treatment decision process can be examined further by considering the three most impactful factors on parental height-related medical decision-making: treatment characteristics, child health, and psychosocial function. While it is important to note that treatment for classical GHD is uncontroversial and supported, treatment decisions for partial GHD and ISS are more complicated and require careful evaluation of both patient needs and the supporting evidence. As the rhGH community grows, physicians, parents, and patients are encouraged to engage in a shared decision-making process to navigate the many challenges facing the GH field. Although this review addresses GHD and ISS specifically, the issues discussed are often applicable to pediatrics as a whole.
Introduction
In 1997, pediatric endocrinologist Ron Rosenfeld, MD captured the crux of the growth hormone (GH) world: “[GH] deficiency is the easiest endocrine disorder to treat and the hardest to diagnose.”1 The steps of diagnosis, treatment decision-making, and actual treatment of children with rhGH introduce various issues found throughout medicine. Combining issues in defining “normal,” the reliability of data and testing, discrepancies in diagnostic cutoffs, demographic skew, patient/parent/pediatrician interaction, societal and commercial pressures, treatment safety, treatment efficacy and expectations, impact on psychosocial function, and considerations of cost and coverage, a discussion of pediatric rhGH treatment is a prismatic study of the problems that affect medicine as a whole. In addition, a focus on rhGH treatment of children involves the larger issue of pediatric medical decision-making, perhaps amplified by a chronic injectable medication borne by the child that in many cases may be considered elective: How do the patient, parent, and medical professionals each take part in treatment decisions?
Growth as a vital sign of pediatric health: what is “normal”?
Although short stature in and of itself is not a disease, growth failure can be a very sensitive sign of underlying health issues in children and adolescents.2 Nonetheless, most short children are healthy. How does one separate worrisome growth from healthy human variation? With height as a physical trait on a continuum, how do we define what is “normal” versus “abnormal”? Various aspects of growth and height - current height, genetic potential, height velocity, and body proportions - are considered by clinicians in the pursuit to separate suspicious from “normal” growth to ascertain the need for further investigation.2,3
How the child grows: Screening for growth failure and short stature
Growth begins prenatally and continues postnatally through puberty in a typical pattern, acting as an important indicator of overall health in childhood.2 For this reason, organizations such as the American Academy of Pediatrics emphasize the need to measure length/height and weight of a child routinely to generate growth curves as an integral part of pediatric health surveillance.2,4 Although fundamental and seemingly simple, such growth data often are not collected or incorrectly measured, leading to inaccuracies that can confound proper analysis.2,5
Once measured, length (recumbent) or height (standing) measurements are plotted on the appropriate growth charts to ascertain the child’s longitudinal growth pattern in comparison to age- and sex-matched children of the reference population.6 Current standards for practice in the USA are to plot recumbent length up to age 2 years on the World Health Organization (WHO) growth charts7 and afterwards, standing height on the growth charts created by the Centers for Disease Control and Prevention (CDC)8 (similar charts have been created for other countries from their own reference populations). It is important to match the child’s measurement position to that used to generate the growth chart on which it is plotted, because all humans lose height once they rise from a lying position. Inappropriate referrals for subspecialist care commonly occur from the artifactual drop in centiles that results from plotting a standing height measurement on a recumbent length chart. Weight-for-age, weight-for-length, and body mass index (BMI) growth charts complement length/height charts by showing how weight gain has proceeded, both itself and relative to height gain. Comparison of body segment length (arm span or sitting height) to standing height, or the ratio of upper-to-lower body segment lengths, allows for screening for disproportionate growth, which can suggest the presence of a skeletal dysplasia.2,9 Syndrome-specific growth charts have been created and should be used for children with certain genetic syndromes that include a distinct growth pattern.
Worrisome signs: Criteria for growth failure
The first two criteria for worrisome growth fall into the category of short stature.2 The simplest is that the child is “short” relative to the reference population (i.e. which centile do they plot on the growth chart). Because this compares individuals to population means for a highly variable trait, a more specific comparison (and second, more targeted criterion for growth failure) is that the child is “short” relative to expected based on his or her genetic potential, i.e. gender-adjusted mid-parental height (MPH). Calculating MPH involves averaging the heights of the patient’s parents after adjusting the height of the parent whose gender is opposite that of the child (to account for the average man’s height being 5 inches or 13 cm taller than the average woman’s).2,10 The clinician can compare the MPH to the child’s projected height by extrapolating current growth chart data to the point of growth cessation (around 20 years of age). Significant differences between the child’s projected height and potential (MPH) raise concerns regarding the child’s current growth.2
While height is a snapshot in time, height velocity provides a longitudinal trajectory that is highly informative as well; thus, the last two criteria for worrisome growth address cases of growth failure. Annualized height velocity (cm/yr) is calculated by taking the difference in height at two time points and dividing that difference by the number of months between the two measurements to obtain a velocity (cm/mo), which is then multiplied by 12 (mo/yr).2 This value can be compared to normal growth velocities based on age and pubertal maturity.6 Left unchecked, growth failure will eventually translate into downwardly crossing major centile lines on the growth chart, a characteristic indicator of prolonged and severe growth failure.2 In any case of growth failure - slow height velocity or downwardly crossing centile lines –timeliness determines if the growth pattern can be restored to its physiologic channel. Many diseases present with slow height velocity. Recognizing and investigating growth failure early allows for both catch-up growth and discovery and treatment of potential underlying pathology. However, in cases of unchecked growth failure, the underlying disease can go undiagnosed, and the ability to rechannel will disappear as growth plates fuse.2
A child must meet only one of the above four criteria (but may meet multiple) to be on a worrisome height/growth path. However, short stature does not automatically imply growth failure (and vice versa), although short stature is the most recognizable presentation of growth failure.2 A child may exhibit slow growth with normal stature if their baseline height (i.e. genetic potential) was very tall on the growth charts, or may exhibit normal height velocity with short stature if they have healthy variant, familial short stature.
Considerations of adult height: Will growth failure be sustained?
Beyond a child’s current height, patient-families and clinicians often are concerned about what adult height that child will achieve. Normally-growing children typically follow the same height centile, so tracing that centile to adulthood on the growth chart can estimate the expected adult height.2 This technique does not take into account variations in rates of growth; it will over-estimate adult height for early or fast maturers and under-estimate adult height for late maturers (i.e. children with constitutional delay of growth and puberty [CDGP]).
A second method to evaluate growth involves comparison of bone age and chronological age.2 Bone age is measured by an AP radiograph of the hand and wrist (unilateral suffices, and conventionally is taken on the left) that indicates the apparent stage of skeletal maturation. To interpret the bone age, clinicians compare a patient’s radiograph to sex-specific standards. One of the most commonly used standards is the Atlas of Greulich and Pyle, created from a collection of x-rays obtained in the 1930s from white children.11 While their current accuracy in the face of secular trends in growth and their generalizability to other racial/ethnic groups may be questionable, a discrepancy between bone and chronological ages is still useful clinically. A delayed bone age indicates more time remaining for growth than suggested by the chronological age and thus portends a taller adult height prognosis, while the converse is true for an advanced bone age.12 However, bone age does not provide information as to the cause of that discrepancy (i.e. the underlying diagnosis).
It may be more correct to think of bone age not as an actual age, but as a measure of the proportion of skeletal maturation achieved in the normal progression to an adult skeleton. Thus, combining a child’s bone age and current height data can predict the child’s expected adult height.13 There are multiple techniques for doing this, including the Bayley-Pinneau tables provided in the Greulich and Pyle Atlas.11 While seemingly scientific, adult height predictions carry considerable margins of error. A study found that across three commonly used adult height prediction algorithms, the same case input yielded different predicted adult height values.13
Growth failure and short stature may warrant less concern if height velocity is expected to increase or if predicted adult height is within the normal range. Thus, predicted adult height is traditionally considered in cases of worrisome growth to decide whether treatment is indicated. Caution should be used in basing decisions solely on adult height predictions, given the limitations in its accuracy.13
What do we call “short”?
The “normal” range in medicine is generally accepted as between −2 and +2 SD for any continuous variable. Following this statistical convention, “abnormal” short stature is defined as height more than 2 SD below the mean for age and sex, or more than 2 SD below the MPH.6,14 A more conservative cutoff of −2.25 SD (shortest 1.2%) was selected by the U.S. Food and Drug Administration (FDA) as the height threshold for the idiopathic short stature (ISS) indication for rhGH treatment.15 In the UK, an even more limited −2.65 SD is recommended for referral to a PCP.16 For a normally distributed variable like height, −2 SD statistically corresponds to the shortest 2.3% of the population. 17 Because growth is followed pictorially on the growth charts, “below the curves” is often used to demarcate “normal” from “abnormal,” making the cut-off 2% (WHO) or 5% (CDC),17 depending on the growth chart used. In addition, some definitions of short stature extend parameters to include not only age and sex, but also population group.18 Thus, for a continuous physical trait like height, where we draw the line between “normal” and “abnormal” is rather arbitrary. 2
The impact of growth concerns in a pediatric setting
Given that short height is easily recognized in the community, it often acts as a red flag for parents, prompting them to bring their child to a clinician to address the issue.2 In some cases, the PCP will refer the child to a pediatric endocrinologist, and sometimes the parents by-pass the PCP to seek subspecialist care directly. In fact, short stature is one of the most prevalent concerns of patients referred to pediatric endocrinologists.19
Although short stature is most commonly (about 90% of cases) the result of healthy genetic variation,19 growth failure should not be overlooked. Indeed, growth failure or short stature can reflect a larger, undiagnosed issue, such as various diseases (eg. celiac disease, inflammatory bowel disease, cystic fibrosis, HIV, renal disease), malnutrition, endocrine disorders, and psychosocial deprivation.2,19 A more detailed discussion of the causes of short stature is beyond the scope of this paper and reviewed elsewhere.2 Of note, many of these conditions are treated by interventions other than rhGH.
The various issues plaguing analysis of growth data (i.e. incorrect or missing data, subjective bone age interpretations, incongruent adult height predictions, and differing cutoffs for “abnormal” short stature) generate uncertainty in separating between “normal” and “abnormal.” This issue is discussed in more detail in context of GH deficiency (GHD) and ISS.
GH: History and uses
The history of human GH treatment began in 1958, when Dr. Maurice Raben reported the first successful treatment of an abnormally short patient with human growth hormone (hGH) extracted from pituitary glands of cadavers.20 The need for cadaveric pituitary glands and the purification process to isolate hGH limited the amount available for treatment; only the most severe cases of GHD were treated, with doses determined by availability.21 In 1985, after 30 years of hGH use for GHD, cases of Creutzfeld-Jacob Disease (CJD) began to be reported in treated patients, totaling 194 cases around the world.21 The U.S. prohibited use of hGH that same year, and many other countries followed suit.22 Serendipitously, also in 1985, the FDA and European Medicines Agency (EMA) approved recombinant hGH (rhGH) for treating GHD.22
The introduction of rhGH allowed for the rapid expansion of rhGH treatment, both in dosage and indications.23 Criteria for GHD were relaxed, and rhGH received approval for treating many other pediatric conditions (eg. chronic renal insufficiency, Turner syndrome, Prader-Willi syndrome, and small-for-gestational age without catch-up growth [SGA]).23,24 In 2003, the U.S. FDA approved rhGH treatment for ISS.15 A handful of other countries, such as Australia and New Zealand, also approved rhGH for ISS, although their criteria for ISS remain more conservative.19 EMA has yet to approve ISS as an indication for rhGH treatment.25 However, globally, rhGH has been used to treat non-GHD indications well before approval (if ever) by the countries’ governments, as off-label prescribing has accounted for a large portion of overall rhGH use.23,26 rhGH enabled expansion of treatment to short but otherwise healthy children. Socially, its use for ISS made it clear that rhGH could change as well as treat, moving pediatric endocrinology into the shaky realm of medical cosmetology and medicalizing short stature as variation warranting pharmacologic treatment.27
rhGH treatment for classical GHD is universally accepted and strongly evidence based. However, treatment for partial GHD and ISS is controversial, and treatment decisions may differ based on indication. Indeed, distinction between GHD and ISS is necessary to determine whether the issue involves both metabolic health and height (GHD) or height only (ISS), and whether screening for other pituitary dysfunction is indicated. In addition, the determination of GHD or ISS in a patient can help predict responsiveness to rhGH therapy, which may factor in the decision to treat. Unfortunately, a clear distinction between GHD and ISS remains evasive.
GHD and ISS: Definitions and distinction
GHD
Pediatric GHD most often is recognized through short stature and/or growth failure, though some patients can present with hypoglycemia and in boys, microphallus. It can be categorized by cause, severity, extent, and duration (Figure 1). However, subcategories of and criteria for GHD differ depending on the source. In this review, names of different classes of GHD are consistent with those in the GH guidelines set forth by the Drug and Therapeutics, and Ethics Committees of the Pediatric Endocrine Society (PES).15 In some cases, the cause of GHD is known. For example, GHD can be genetic, can be associated with pituitary malformation or disorder, or can result from insult to the pituitary gland and its neuronal control (through infection, brain surgery or irradiation, tumors, autoimmune or infiltrative diseases).22,28 In such cases, GHD is referred to as organic. However, more commonly, the cause of GHD remains unknown, termed idiopathic GHD.19 Regardless of its cause, GHD can be classical or partial. Classical GHD refers to very low secretion of GH; it is commonly organic in nature.15 Partial GHD is present when GH is secreted, but at levels slightly lower than “normal” (“normal” is defined below). GHD can be either isolated, where GH is the only pituitary hormone deficient, or part of Multiple Pituitary Hormone Deficiency (MPHD). MPHD often presents as classical GHD, while isolated GHD is more likely to be partial. Finally, GHD can be classified based on its timeline; persistent GHD continues into adulthood, while in non-persistent GHD, GH levels reach the lower adult standard for “normal” secretion (GH production physiologically declines with age such that normal production in adults is much lower than that in youth).15,29 MPHD usually is also persistent GHD.
Figure 1: Classifications of Growth Hormone Deficiency (GHD).
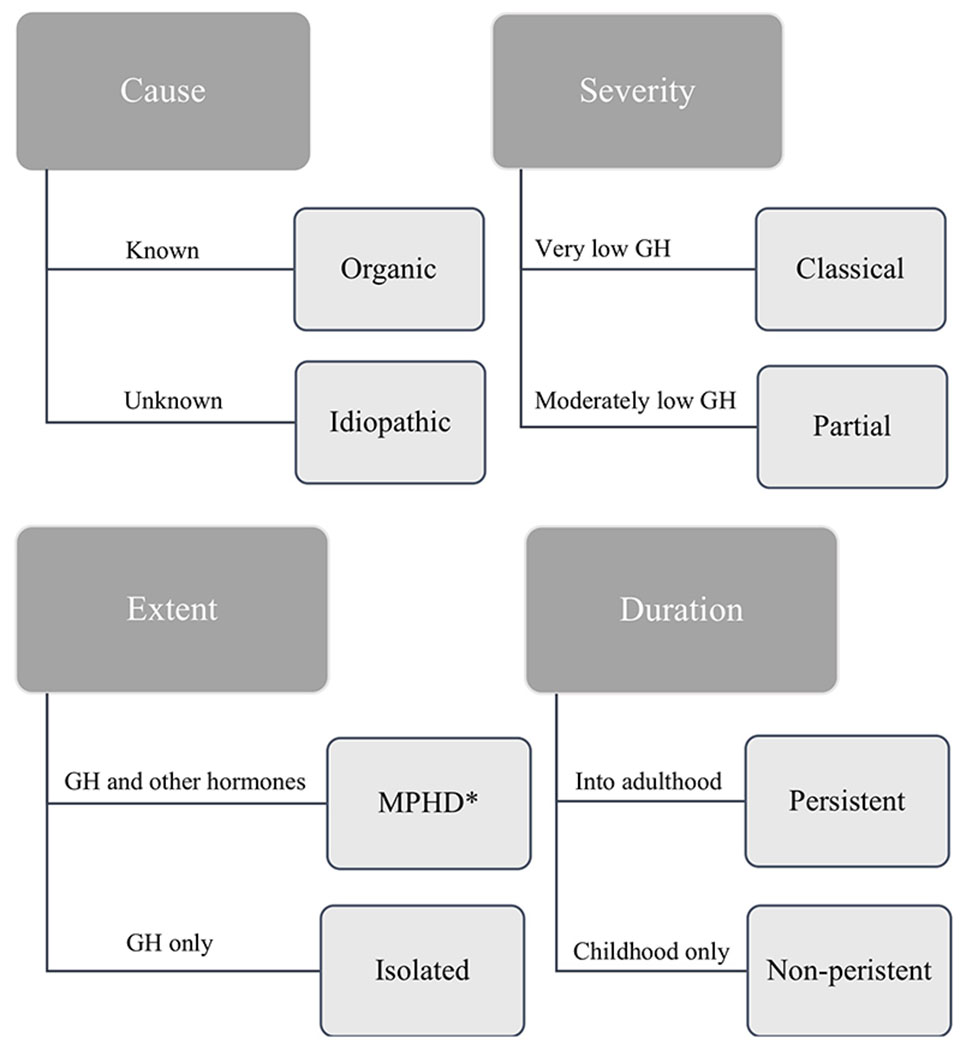
*MPHD refers to Multiple Pituitary Hormone Deficiency.
GHD is relatively rare in the population, with an estimated prevalence of 1:3500 children.30 The most common case of GHD is idiopathic, partial, isolated GHD, which controversially exists on the arbitrarily-defined continuum of “sufficient” and “insufficient” GH secretion.31 There is a philosophical debate whether partial GHD even exists;23,32 some endocrinologists feel it is merely an artifactual concept created by the limitations of provocative testing, while others believe a partial deficiency is a biologically plausible condition.
GHD in any form can affect body composition through increased adiposity and reduced lean muscle mass, decreased bone mineral density, and unfavorable lipid metabolism, with increased LDL and lowered HDL levels.15 Although rhGH treatment following growth plate fusion provides no additional height gain, rhGH treatment is continued through adulthood in cases of persistent GHD for metabolic, bone, and cardiovascular health, and in some studies, improved quality of life (QoL).15 Thus, for patients with GHD, rhGH treatment both improves health and increases height.15
ISS
ISS refers to the condition in which a short child presents with normal birth weight, normal levels of GH secretion, and no identifiable abnormalities to explain their growth pattern.14 It is not a diagnosis but a justification for rhGH treatment. ISS was created as an acknowledgement of limitations in our diagnostic capabilities that would exclude children who could potentially benefit from rhGH treatment from receiving it (see below). The ISS indication for rhGH treatment was defined by the U.S. FDA in 2003 as fulfilling 3 criteria: height below −2.25 SD (≤ 1.2nd percentile), growth rates unlikely to permit attainment of adult height in the normal range, and exclusion (via diagnostic evaluation) of other causes of short stature that should be observed or treated by other means.15 The term, “ISS,” has been redefined through the years, with the broadest being a child whose short stature (wherever you draw the line) is unexplained by current testing. By grouping it within diagnostic classifications of short stature/growth failure, ISS has been transformed conceptually into a diagnostic category.
Using the same term for an indication for rhGH treatment and for a diagnostic category can lead to misalignment. For example, familial short stature (FSS) and CDGP have traditionally represented healthy variants that do not require rhGH treatment2 (indeed, the second stipulation set forth by the FDA was included intentionally to underscore the inappropriateness of treating CDGP with GH; those children will reach normal heights on their own). FSS refers to children with height channeling at a normal velocity and trajectory for their MPH range but who are short compared to the reference population (i.e. growth chart).2,19 CDGP refers to late bloomers, whose current height is short, but whose delayed puberty and bone age portend an extended time to grow, with an adult height normal for their MPH range.2,19 Both FSS and CDGP present with short stature as a result of genetic influences, but the specific gene(s) involved in these conditions remain(s) unidentified.19 Conceptually, since the specific genes are unknown, the argument can be made that the two fall in the idiopathic category of short stature (i.e. ISS). By calling them “ISS,” does that now change expectations for whether they should be treated with GH? Further, as genomics advance and are used to screen more children, additional gene(s) controlling height and growth will be discovered, likely including those that lead to the polygenic traits of FSS and CDGP. By identifying a genetic cause, will that reframe FSS and CDGP from polygenic, healthy variant growth patterns to genetically induced “conditions,” thereby further medicalizing them?
Distinguishing between GHD and ISS
Both GHD and ISS present with short stature and/or growth failure. In addition to the heterogeneous definitions of “short,” the process of diagnosis introduces further ambiguity, especially between partial GHD and ISS. As a result, differentiating between the two often becomes a subjective endeavor. The diagnostic process in a case of worrisome growth involves evaluating growth, laboratory data, provocative GH testing (when indicated), and radiological data; multiple issues at each level generate uncertainty.
Growth data
Since both GHD and ISS present with worrisome but proportionate growth, concerning growth data function only to prompt further investigation, not to distinguish between the two. Pediatricians often then screen a child exhibiting worrisome growth for common non-GHD conditions that manifest through short stature and/or slow height velocity, such as hypothyroidism, Turner syndrome, SGA, systemic diseases, and malnutrition.2 This is especially true if the child exhibits disproportionate growth, which would exclude GHD or ISS. However, milder cases of skeletal dyplasia are often missed, leading to a misdiagnosis of ISS.19 In these situations, the underlying pathological cause for the disproportionate growth is consequently overlooked.19
The initial analysis of growth as worrisome depends on growth data being collected and properly measured.2 Unfortunately, PCPs often fail in this regard; a 2000 U.S. study found that in 35% of well-child visits, PCPs (in pediatric and family practices) did not collect growth data and/or document a present abnormality.33 Further, a survey by the WHO found that inaccurate plotting and interpretation of growth charts was universal, with only 20% of health care workers in various regions responding that they experienced “no problems” using growth charts.34
Accurate auxological data also include a complete, accurate family history, especially when FSS is suspected. Evaluation of an extended pedigree is the only way to distinguish FSS from a heritable, as of yet undiagnosed growth problem.2 If the whole family has heights below population norms, then FSS seems likely; however, the repeated presence of short stature in an otherwise normally statured family suggests underlying pathology.2 Other issues in interpretation of growth data involve the bone age and predicted adult height limitations discussed above.
Laboratory data
Circulating concentrations of the GH mediator, insulin-like growth factor (IGF)-I, and its binding protein, IGFBP-3, are often measured via immunoassay or mass spectroscopy as a screen for GHD.12,22,35 Because transcription of both genes is GH-dependent, normal levels of IGF-I and IGFBP-3 reflect normal GH activity (i.e. exclude GHD), while low levels of one or both suggest a possible diagnosis of GHD.36 Since serum levels of these molecules do not experience as much diurnal fluctuation as GH, they serve as convenient indicators of GH secretion that are measurable on a random blood sample.22,35 However, like all diagnostic data for worrisome growth, the tests are limited. Principally, accurate measurements of IGF-I levels are difficult to obtain, because the molecule must first be dissociated from IGFBP-3 and the other IGFBPs to be recognized by an immunoassay.35,37 Additionally, the results of the test may not accurately reflect GH secretion because IGF-I and IGFBP-3 are continuously broken down and their circulating concentrations depend on confounding factors such as sex, ethnicity, age, nutritional status, sex hormones, and hepatic and renal function.22,35,38–39 Furthermore, the separation methods and assays used to measure these levels are not standardized, precluding meaningful comparisons across assays.35,37 Nonetheless, these tests are used to screen for GHD, as normal results allow many children to avoid unnecessary provocative GH testing, which is much more involved and invasive.
Provocative GH testing
Provocative GH testing (a.k.a. GH stimulation testing) involves administration of a pharmacologic secretagogue followed by serial blood sampling to measure the GH response. Serum GH concentration is then measured by an immunoassay.40
A diagnosis of classical GHD through significantly low levels of serum GH is uncontroversial and well supported.15 However, there are four glaring issues in the testing process that render distinction between partial GHD and ISS almost impossible: the variable nature of GH secretion, the questionable reliability of the test itself, the lack of standardization or harmonization in GH assays, and the blurred quantitative line between partial GHD and ISS.
GH is secreted in pulses principally during deep sleep, and its circulating concentration is affected by a multitude of other factors, such as BMI, the amount of GH-binding protein (GHBP) in the blood, the expression of GH receptors, the chemical form of the specific GH molecule measured (two circulating isoforms – 20 and 22 kDa), and the secretagogue used (e.g. clonidine, arginine, glucagon, insulin, or L-DOPA).12,37,40 Healthy children with no signs of worrisome growth often fail to meet the “normal” threshold of GH concentration, suggesting inaccuracy in the test, in the accepted cutoff, or in both.15 In an attempt to reduce false failures on these grounds, a child’s GH levels must fall below accepted values on two separate tests to be considered GHD.12,15 Although this may improve test specificity in detecting adequate GH secretion, it fails to address the fact that healthily growing children are failing to meet the supposed threshold of “normal” GH secretion. Other factors such as obesity or delayed puberty can skew the results of the test, also generating false failures.12,15 Sex hormone-priming (i.e. administering estrogen or testosterone in preparation of the test) may mitigate the effect of delayed puberty on decreased GH levels, although its use varies among countries.12,15 In medicine and science as a whole, reproducibility suggests credibility; results of the provocative GH test usually fail to meet this standard, as the same individual may show different results on repeat testing on different days.15
Even in the unrealistic case of consistent GH secretion and precise provocative testing, the reported results of the test are still problematic. The assays used to analyze the blood samples may use monoclonal or polyclonal antibodies (or both) and are not standardized or harmonized, which leads to differing values measured based on the assay used.37,40 A study comparing results of three reference assays found differences in classification (GHD or non-GHD) in 36 of 132 patients (27%).41 In 2011, an international conference of the Growth Hormone Research Society (GRS), the IGF Society, and the International Federation of Clinical Chemistry produced a consensus statement on standardizing GH assays, urging the widespread adoption of the GH reference standard produced by the WHO;37 the 2016 PES guidelines and 2019 GRS workshop report strongly endorsed this recommendation.12,15 Several countries, such as Finland, Germany, and Japan, have pursued standardization efforts.15,40 Although these measures are moving the field in the right direction, further work is needed to achieve uniform analysis of blood samples.
Even if the preceding impediments were surmounted, the pediatric endocrinologist must interpret the numbers. This proves especially difficult in cases that seem to walk the poorly-defined line between partial GHD and ISS. Very low peak serum GH levels (i.e. less than 5 μg/L) point to classical GHD.15 However, the transition from “normal” GH secretion to partial deficiency remains unclear.23 The traditional criterion for GHD, established in 1990, is arbitrarily defined as two provocative tests (i.e. two different secretagogues not administered simultaneously) eliciting peak GH levels below the “normal” 10 μg/L.22 The not uncommon failure of normally-growing children to meet this threshold and changes in GH assays since 1990 have prompted some countries, such as Japan, to lower the cutoff for GHD to 6 or 7 μg/L.42,43 All of these issues have led leaders in the GH field to recommend, by the 2016 PES guidelines15 and the 2019 GRS workshop report,12 the avoidance of relying on provocative GH test results as the sole criterion for diagnosing GHD. Without a clear marker for adequate GH secretion, the distinction of partial GHD versus ISS often falls to the judgement of the pediatric endocrinologist, who must integrate all the clinical clues to reach a diagnosis.
In practical terms, this translates into a failure of provocative GH test results to reliably predict responsiveness to rhGH treatment. The ultimate goal of such diagnostic testing is to direct treatment (involving years of daily injections of an expensive medication with potential side effects) to only those patients who will respond with a significant increase in their height velocity and adult height attainment. The ability to predict responsiveness to rhGH of the patient based on indication (GHD or ISS) will be discussed along with treatment efficacy.
Radiological data
Magnetic resonance imaging (MRI) of the pituitary is obtained as standard of care before initiating rhGH treatment for patients with GHD, because rhGH treatment is contraindicated in cases of active malignancy.15 Furthermore, pituitary MRI findings can support the interpretation of GH test results. For example, a diagnosis of classical GHD may be supported by the presence of pituitary malformation (especially in the case of MPHD or commonly associated conditions like septo-optic dysplasia or holoprosencephaly).15,22 However, most patients with isolated GHD have normal pituitary glands on MRI, which neither supports nor excludes a diagnosis of GHD.
Overall, the diagnostic process for worrisome growth is fraught with challenges that can hinder proper diagnosis; but who are the patients being evaluated, and what driving forces brought them to the endocrinologist in the first place?
The GH stakeholders
Patient
Patients can present for rhGH treatment of GHD and ISS from the neonatal period through puberty, with the median age of rhGH initiation (across the 4 U.S. rhGH registries) of 11 years for females and 12 years for males.44 The demographics of patients at both the subspecialist referral and rhGH treatment stages are consistently and concerningly skewed.
One of the most glaring demographic skews in the GH world concerns the evaluation and treatment of males. Review of new patient evaluations for short stature/growth failure in a U.S. endocrine subspecialist clinic found that males outnumbered females almost 2:1.45 Not only were males seen more often, they presented with less severe height deficits and lower prevalence of underlying disease.45 Males also outnumbered females 2:1 for all indications across the 4 U.S. registries of pediatric rhGH recipients, and 3:1 for ISS.44 Review of the Pfizer International Growth Study (KIGS) database also showed that the majority of pediatric patients receiving rhGH treatment were male.26 The disparity was greatest for indications such as ISS and idiopathic GHD, where the reason for treatment is most subjective.26 Among the top 10 countries with over 1000 patients enrolled in KIGS, the percentage of male rhGH recipients ranged from 53% (UK) to 65% (Asia); U.S.A. was 64% and Italy 59%.26 Although the values across Asia, U.S.A., and Europe/Australia/New Zealand all exceeded 50%, the over 10% difference between Asia or U.S.A. and the UK suggests that factors other than the patient’s sex impact the gender bias in rhGH treatment.26
Racial disparities also are prevalent in subspecialist referral and rhGH treatment frequencies. In a U.S. study, 91% of families pursuing evaluation by a pediatric endocrinologist in a diverse, urban environment were white (compared to 72% of the local population).46 In a U.S. pediatric rhGH registry, white children comprised around 80% of those treated for GHD and 85% of those treated for ISS, much higher than the expected value (based on census data and different growth rates among racial and ethnic groups) of 63%.47 Interestingly, the gender bias cut across racial lines, with similar male predominance seen among white and African American subjects.47
Another consideration is socioeconomic status. Low socioeconomic status is a known risk factor for short stature.48 However, a 1999 study found that 49% of children seen by pediatric endocrinologists for short stature (compared to 23% in the population) came from families with high annual income (≥ $50,000).46
Altogether, the skewed statistics connote overinterest in treating short, primarily white boys from higher socioeconomic groups, with the potential for delayed or missed diagnosis of underlying pathology in short girls and in children from racial-ethnic minorities and/or lower socioeconomic strata.45,47,49
Parent and clinician
Although differences between the referred/treated population and the general population are readily apparent, the cause of these biases cannot be pinpointed to a singular source, instead the complicated product of personal, societal, and commercial pressures.
Increased parental concern increases rates of both subspecialist referral by PCPs50 and rhGH prescription,51 independent of the degree of short stature. Parents of children referred for evaluation by a pediatric endocrinologist were found to be mostly white and to have a higher income and education level than the general population.46 When asked “How short is too short?” for an adult, responses of 1,820 parents of pediatric primary care patients seemed to match these observed biases; respondents who were female, white, taller, from nonurban primary practices, and wealthier generally provided a taller acceptable height threshold.52 Further, the median reported acceptable height threshold for males was 13 cm (5 inches) taller than that for females.52 It was unclear whether this higher threshold for males was due to a belief that males should be taller or simply reflected the difference in mean heights found between men and women in society.52 Another study found that parents generally considered short stature as more of an issue for males than females.46 Taken together, the data suggest that parental attitudes toward height account for at least part of the gender, racial, and socioeconomic skew seen in the referred and rhGH-treated worrisome growth populations.
The same holds true for clinicians at both the primary care and subspecialty levels. When evaluating patients for worrisome growth, PCPs in a U.S. urban setting ordered GH/IGF testing twice as often for boys than girls.49 PCPs from another U.S. city were found to refer male patients to a pediatric endocrinologist more often than female patients.50 The same study found that family concern and physician characteristics (i.e. older, female, shorter) influenced referral, independent of actual height/growth data, implying that the physician, as well as family, mindset in part determines the potential rhGH candidate pool.50 Pediatric endocrinologists are thus already presented with predominantly male cases of worrisome growth, but studies have shown that they carry their own biases. A nation-wide U.S. survey with hypothetical cases found that pediatric endocrinologists recommended rhGH treatment for males 1.3 times more often than females despite otherwise identical case vignettes.51
Societal pressures
On a larger scale, the societal view that taller is better fuels concern regarding short stature and commoditizes centimeters of increased height. The term “heightism” refers to the prejudice that short stature hinders success and tall stature facilitates it.2,53 Multiple studies reviewed elsewhere have shown that taller stature is associated with markers of social and financial success, especially for males, while other studies have suggested that other factors, such as education level, income, and methodological flaws within the studies themselves, mitigate the association between stature and success.2,54 A more exhaustive list of studies and their findings regarding the outcomes of short stature has been compiled.54
The subsequent question is not whether height actually produces the difference in success outcomes, but whether people believe this effect exists. Regardless of the data, does the general public embrace heightism? Various studies reviewed elsewhere have found that people, beginning at a young age, perceive taller individuals as more capable and successful.54 The correlation seems to be stronger for males.46,54 One review suggests two possible reasons for the perception that taller stature leads to greater success: an evolutionary/biological instinct and/or a focusing illusion (i.e. overweighting the effect of height on success by virtue of concentrating on it).55 Irrespective of cause, heightism is undoubtedly engrained in our society, even in childhood. A child may positively associate “big” and negatively associate “little” based on common phrases celebrating the “big and strong” child and encouraging the child to act like a “big kid.”56
The media can fuel heightism. Shows such as The Little Couple and Little People, Big World emphasize the difference between short people and the general population and portray their navigation through the “big world” as entertainment. The media can also heighten anxiety surrounding short stature and misinform the concerned parent. In the U.S. in 2014, 59% of adults reported using the internet as a source for health information;57 55% reported this in Europe in 2019.58 Given the large number of people looking to the internet for information, it is common for incorrect information to mislead the reader. After parents in 38% of focus groups reported using the internet to research rhGH treatment, study investigators analyzed the content of websites providing information about rhGH; they found that the sites tended to deemphasize (or completely failed to mention) both risk and cost associated with rhGH and that 44% of the sites evaluated were commercially sponsored.59 Such content positions parents to form initial opinions about treatment without access to all necessary information. It is imperative that the clinician provides families with complete and correct information, as well as tips to recognize reputable websites if necessary.59 As the internet pervades everyday life, this standard of practice applies to all clinical situations, not just those of worrisome growth.
rhGH, like many other specialty drugs, remains extensively intertwined with financial forces. A 2004 study proposed that an inch increase in height was associated with a 1.8% increase in wages in the U.S. and a 2.2% increase in the U.K.;60 such results, when taken in the context of rhGH treatment, promote increasing height as an investment for future financial success. The danger when taking cohort-level data and applying them at an individual level lies in making the incorrect assumption that each inch directly translates into monetary gain.55 Further, studies investigating rhGH are often industry-sponsored. An analysis of previous studies (1958-2014) of psychological outcomes following short stature treatment (of various indications) with GH (pituitary GH and/or rhGH) found that 82.1% of randomized and 33.3% of nonrandomized studies were industry-sponsored.61 Physicians rely on such studies to make educated decisions regarding treatment, introducing industry bias and publication bias (the greater likelihood of publishing a study with positive results than one with negative results) into clinical practice.61 Such sweeping industry involvement in the research of rhGH carries the potential to entangle education, promotion, and persuasion.23 Furthermore, rhGH manufacturers can become involved in the screening process, by sponsoring school-based screening programs32 or financially supporting screening programs through patient advocacy foundations62 in an effort to increase the ascertainment and referral of short children for evaluation and possible rhGH treatment. In the U.S., the primary responsibility for growth screening rests with PCPs.4
With an understanding of normal vs worrisome growth, GHD, ISS, and the various stakeholders in rhGH, the following question can now be explored: How do all these complicated, often ambiguous aspects translate in a realized medical setting to result in a decision regarding rhGH treatment?
Treatment decision
It is important to recall that rhGH treatment is supported by strong evidence and accepted for classical GHD. Issues in treatment decisions arise in the gray areas of partial GHD and ISS. Of note, the rhGH decision-making process involves the patient, parent and physician triad. However, although the child reaps the benefits and bears the burdens of a decision to treat or not treat, research investigating the role of the child in the decision-making process is lacking. Thus, while the term “triad” will be used through the remainder of this review, the gap in research regarding the child’s role in the triad is acknowledged. The following consideration of the rhGH treatment decision-making process is separated into the three main concern categories (in decreasing order) reported by parents of primary care patients as greatly impacting their decision to seek medical care for a child’s short stature: treatment characteristics, child health, and psychosocial function.63
rhGH characteristics and child health
rhGH is administered as a daily subcutaneous injection. The frequency and method of administration may prove challenging for some families. Specific dosing varies based on indication, weight, and individual response. As such, response to dosing must be continually monitored, which involves regular visits with an endocrinologist, repeated blood tests and x-rays, and doses must be adjusted if necessary.15
The distinct treatment factors that most impact parental height-related medical decision-making are side effects and proven efficacy.63 Thus, all possible side effects and realistic treatment outcome expectations should be explained and discussed with families deciding whether to initiate rhGH.
Treatment safety
Potential adverse effects associated with rhGH therapy, for both GHD and ISS, include intracranial hypertension, slipped capital femoral epiphysis (SCFE), and progression of scoliosis.15 The physician must screen for their development via appropriate history and physical examination at monitoring clinic visits.15 If potential MPHD is suspected, physicians must re-evaluate the adrenal and thyroid axes; GHD is often the first and sometimes only pituitary hormone deficiency to present, and rhGH therapy may uncover or exacerbate other hormone insufficiencies.15 rhGH therapy reduces insulin sensitivity; as a result, for patients already at risk for diabetes mellitus, possible development of the disease should be monitored.15 An early increase in development of second neoplasms has been reported with rhGH treatment of patients with GHD due to malignancy and radiation treatment;64,65 however, these patients have higher risk for second neoplasms at baseline, and a subsequent study suggested that radiation treatment (rather than rhGH) was primarily responsible for the increased risk.66 Reports have suggested that rhGH treatment of children without increased cancer risk at baseline does not increase the incidence of malignancy, but data are insufficient to know whether rhGH treatment affects incidence in children with cancer-predisposition syndromes.64,67
Post-marketing surveillance studies (i.e. rhGH registries) were initially required by the FDA of all approved rhGH manufacturers. With over 600,000 patient-years experience recorded worldwide, such data have been helpful in understanding rhGH treatment and its safety. However, the data provided are less clear-cut given the discrepancies in tests, assays, and cutoffs used across the many clinical practices from which the data originated, as well as the voluntary ascertainment nature of such studies. Other factors limiting the accuracy of the data include dosage changes and differences among recipients over the decades, lack of control population, and potential manifestation of adverse events only post-treatment.23 Thus, the SAGhE (Safety and Appropriateness of Growth Hormone Treatments in Europe) consortium was created to conduct the largest and longest, industry-independent cohort study of adults who had been treated with rhGH for various indications in childhood since 1984 (n= 24,232 patients from 8 countries).68 Preliminary analyses of national subgroups yielded contradictory and at times alarming findings.69–71 Subsequently, cancer-focused analyses of the entire cohort found that increased incidence and mortality risks of some site-specific cancers were largely predicated on a prior cancer history, except for a higher incidence (based on small numbers of cases) of bone and bladder cancers in those without previous malignancy.72 Incidence of Hodgkin lymphoma increased with longer follow-up (after 20 years) for both overall cohort and the non-previous cancer group, and a GH dose-dependent increase in cancer mortality was found only in the group treated with GH after previous cancer.72 Continued vigilance and study are required, as the results were neither completely negative nor internally consistent to support a causal role for GH treatment in cancer risk. A 2016 consensus statement by the European Society of Paediatric Endocrinology, the GRS, and PES generally supported the long-term safety of rhGH treatment but cautioned that the area requires more research.64 Regardless, it is important to note that rhGH treatment in the case of ISS, unlike GHD, yields no health benefits. Thus, the threshold of risk tolerable for height gain alone remains more unclear.
Treatment efficacy
Treatment efficacy depends on the observed response to treatment and the chosen definition of success, and for rhGH, can be partially gauged by indication. On one extreme, patients with classical GHD and very low provocative test results respond dramatically to even low doses of rhGH, achieving normal adult heights (compared to adult heights of −4 to −6 SD in historical patients with untreated idiopathic isolated GHD).73 On the other extreme, response to therapy in children with ISS is highly variable and difficult to predict.15,19 In a randomized, controlled trial of children with ISS, mean height gain achieved through rhGH treatment (approximately 6 year treatment duration) was 8 cm (1.3 SDS) with a large (no gain to 3SDS) and dose-dependent range in response.74 Models by Ranke and Lindberg using the KIGS database suggest that factors including younger age at rhGH initiation, taller parents, taller predicted height, and greater first-year response to rhGH may correlate to greater total response to rhGH treatment, though research regarding predictors of rhGH responsiveness are limited by disuniformity in registry data.75 Partial GHD falls in between. Some patients with peak GH levels >10 μg/L (i.e. non-GHD, so ISS) respond well to rhGH treatment as if they had GHD, while some patients with peak GH levels <10 μg/L (i.e. GHD) behave more like patients with ISS and are poor responders to rhGH treatment. This underscores the imperative for quality, controlled studies and better diagnostics.
Defining potential rhGH success in treating GHD is two-fold, involving both improvements in health and increase in height. In terms of health benefits, rhGH was shown effective for patients with GHD, increasing bone mineral density and lean body mass, and lowering LDL and overall cholesterol levels.15 According to the 2016 PES guidelines, the primary goal of rhGH treatment for pediatric GHD is to accelerate height velocity and normalize adult height in context of the genetic potential of the patient (most often measured by adult height SDS – MPH SDS).15 Treatment generally has proven successful in doing so; for example, for a registry-drawn sample of treated/untreated children with idiopathic, isolated GHD, the mean difference between adult height SDS and MPH SDS was found to be −0.4 SD for the treated group and −4.7 SD for the untreated group.73 Treatment for pediatric GHD should be stopped before growth plate fusion, usually indicated by height velocity below 2-2.5 cm/yr and confirmed by bone age, in order to avoid inducing acromegalic changes.15 At this point, it is essential to evaluate for possible persistent GHD and to continue treatment at adult (i.e. lower) doses in these cases to maintain the health benefits of replacing deficient GH secretion.15 Pediatric rhGH treatment may be considered “successful” and stopped before attainment of a terminal height velocity or fused bone age if an auxological goal is met, or if the triad decides that the benefits of treatment no longer outweigh the risks and burdens.15
For ISS, “successful” rhGH treatment does not include health benefits and therefore is measured solely through changes in height and growth. Defining a “successful” gain in height or increase in velocity reintroduces a common theme in the GH world: the need to set a goal/cutoff arbitrarily on the continuum of a physical trait. As such, the requirements for adequate increase in height and/or velocity are not well-defined. First-year response to rhGH treatment is often used as a surrogate indicator of likely overall treatment success,15 because first-year response is generally correlated to total height gain.75 In a consensus statement produced by PES, the GRS, and the European Society for Pediatric Endocrinology, adequate response was set to an increase in height SDS at least 0.3-0.5 SDs within the first year of rhGH initiation.14 If this height gain is not achieved, the triad should consider stopping therapy, as the patient will continue incurring the burdens of daily injections and exposure to potential side effects with low likelihood of any benefit.15
Overall “success,” with subsequent termination, of rhGH therapy for ISS can be defined in multiple ways, based on the various possible goals of therapy. The first consideration in defining the treatment goal involves when the increase in height is desired. The treatment goal may focus on immediate increases in height or long-term height gain (Figure 2). Therapy may be considered “successful” when short-term growth hits a set accelerated rate, allowing for an increase in current height.3,14 On the other hand, “success” may be determined long-term, by considering adult height attained.3,14 In that case, therapeutic “success” can be defined by adult height within the population “normal” range or by adult height matching the child’s genetic potential.3, 14 If height within the “normal” range is considered the measure of “successful” treatment, the treatment goal must be further defined by where it lies within the “normal” range (i.e. just above −2 SD, a certain centile, or the maximal height achievable by that patient). Regardless of desired outcome, once growth plates are fused, rhGH treatment must be stopped, similar to non-persistent GHD.
Figure 2: Potential Treatment Goals for Idiopathic Short Stature (ISS).
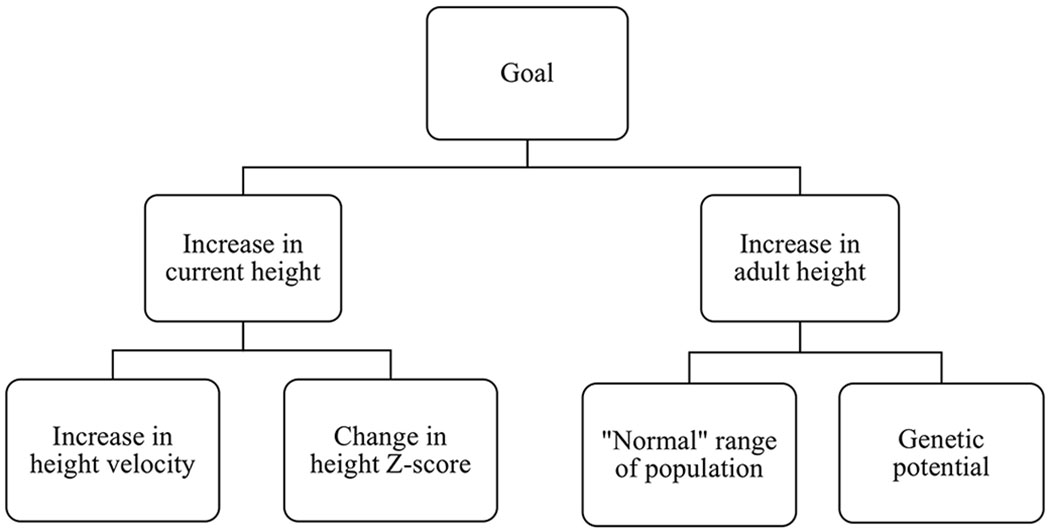
The overarching goals of treating ISS can focus on short-term or long-term outcomes. Each one can have different metrics of success.
Treatment success in any case will be compromised if the patient does not adhere to the proper use of rhGH, decreasing potential height gain.76 Given the burden of administering a daily, self-injected drug, non-adherence is common among both ISS and GHD patients; in fact, a study found that 66% of patients (of various treatment indications) receiving rhGH miss more than 1 dose each week.77 Factors shown to reduce adherence include lack of understanding regarding treatment/the consequences of missed doses, inadequate contact with health-care providers, and inadequate training.76 Efforts to boost adherence have led to the development of long-acting rhGH (LAGH) products, allowing for less frequent injections (reviewed by GRS in 2016).78 Several LAGH products are in testing phases; some have been approved in China, South Korea, and Europe for use for GHD, and others have been abandoned in testing phases or removed from the market.79 Further studies are required to ensure the safety and efficacy of this future technology.79
In light of the variable response to therapy, the clinician must calibrate patient and parent expectations of rhGH treatment for GHD and especially ISS to realistic targets. Treatment goals may not be met, and even if treatment is carried out to the quantitatively-defined “success,” patients and parents may still be unhappy with the result. A study found that children treated for GHD and their parents viewed quantitative treatment success as failure, because the results of therapy did not match their expectations.80
Psychosocial function
In its constitution, the WHO defines health as “a state of complete physical, mental and social well-being and not merely the absence of disease or infirmity.”81 When deciding whether to treat both GHD and ISS, one of the main considerations is the amount of height that can be gained through rhGH therapy. By the WHO definition, potential physical benefits and improvements in psychosocial function via increased height are considered improvements in health.
Short stature as a physical impairment
A child with short stature may have difficulty in everyday life as a result of height. For example, activities such as reaching elevated objects, going on amusement park rides, qualifying for sports teams, or driving without modifications to a car may become an issue in the current and/or future life of a child with short stature. These physical limitations are often directly correlated with the severity of the height deficit and may be ameliorated through rhGH therapy.15 Further, some patient-families seek rhGH treatment to prevent perceived disadvantage in sports performance due to the child’s height, although other factors like skill, practice, and talent also play a role.
Short stature as a determinant of psychosocial function
The idea that height gain is psychosocially beneficial to a patient rests on the validity of two statements: that short stature impedes psychosocial function and that height gain can improve the patient’s psychosocial well-being. Both are supported by scant data. When deciding whether to initiate rhGH treatment, the psychosocial functioning of the child in context of current and future stature is considered. The psychosocial component of the treatment decision is especially pertinent for children with ISS, where height gain is the only physical benefit to treatment; thus, a psychosocial justification of treatment relies on the translation of height increase to psychosocial improvement. The prevalence of patients being treated with rhGH for ISS (both approved indication where available and off-label elsewhere) manifests the widespread belief that short stature indeed hinders the child psychosocially and that increased height leads to improvements in psychosocial functioning.
Traditionally, it has been assumed that short stature causes psychological, behavioral and educational issues. While clinic-based studies have associated short stature with increased psychosocial stress (e.g. teasing, bullying and juvenilization),82 it is also reported that this increase is unrelated to the severity of the patient’s short stature82,83 and is marginal in the overall population.84 Furthermore, researchers have cautioned that increased psychosocial stress does not automatically imply impaired psychosocial functioning.54 Indeed, several studies and reviews have concluded that there is no direct link between short stature and overall functioning in psychosocial domains in referred82,85 and overall84,85 populations. Other studies, such as the Wessex growth study, reported slight association between short stature and psychological development in the general population but were unable to directly relate measured deficits to short stature itself, finding social class a better predictor of psychosocial development and likely confounder.86
One reported trend is that patients referred for short stature more often present with psychosocial issues compared to the general population,85 although some studies have altogether contradicted this, finding referred populations comparable to the general population.82 A potential explanation for the belief that patient psychosocial function differs from population norms is that psychosocial issues may increase patient-parent concern and subsequent referral, creating an ascertainment bias.85 Additionally, the psychosocial impairment measured in the study was reported by parents of the referred children,85 who may be inaccurate judges of the true functioning of the child. Indeed, a similar study found that boys referred to a pediatric endocrinologist for short stature were reported by parents to be less socially competent and to exhibit more problems with behavior and emotion than a normative sample, but this observed psychosocial impairment was not matched in the boys’ self-reports.83 Together, these findings suggest that parent report alone may not be a reliable measurement of psychosocial function, and that male patients, already over-represented in the referred and treated populations, may be at greater risk for incongruencies between self and parent perceptions of psychosocial issues arising from short stature. Similar issues may be present when physician perception is used as a measure of patient psychosocial functioning. A survey of U.S. pediatric endocrinologists found that 83.5% of respondents believed that short stature (3rd to 5th percentile) sometimes (69.7%), often (13.5%), or always (0.3%) impairs emotional well-being in a child.51 It seems that whether patients and parents believe that the child will be psychosocially impaired may influence parent/PCP report just as strongly as whether the child actually exhibits such impairment (Figure 3). Subsequently, study data based on parent and pediatrician perceptions may blur preexisting beliefs with patient reality.
Figure 3: Hall of Mirrors.
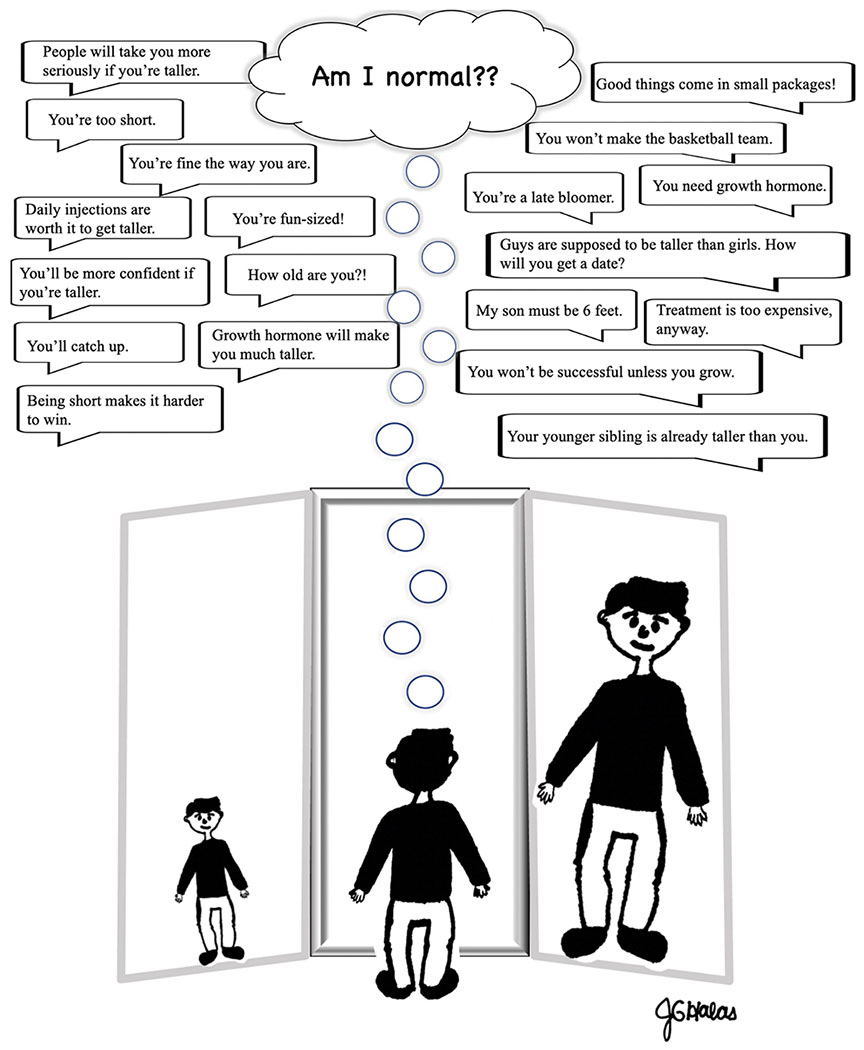
Identity formation including, “Am I normal?” is an important aspect of personal development in adolescence, and youth are affected by social influences from many sources in their environment.
A phenomenon complicating the interaction between height and psychosocial function is perceived height. One study found that both patients and their parents tended to overestimate patient height, and that larger overestimations translated to better psychosocial functioning and greater satisfaction with current height; this study suggests that perceived height is a stronger indicator of psychosocial function and patient/parent satisfaction compared to actual height.87
rhGH therapy as a treatment to improve psychosocial function
To justify rhGH treatment on psychosocial grounds, not only must an impairment be present but also it must be supported that rhGH treatment can ameliorate such issues. The weak connection of short stature to psychosocial functioning has been discussed. Although psychosocial issues cannot be directly attributed to short stature, rhGH candidates may still exhibit them due to separate, or compounding, factors. Thus, a discussion of the effect of rhGH treatment on psychosocial functioning remains relevant.
Generally, studies using parent and patient ratings have been unable to support psychosocial88 or quality of life (QoL)89 improvements in children treated for ISS compared to placebo88 or non-treated89 control groups, with slight improvements in behavior reported by parents88 or in QoL reported by the pediatrician.89 Nonetheless, some argue that rhGH therapy is indeed beneficial psychosocially in cases of short stature. In addition, pediatricians tend to report improvements in QoL for treated groups in the absence of matching patient or parent reports.89 These findings illustrate the widespread belief among pediatricians that rhGH therapy will benefit the patient; in a U.S. survey, 31.5% of pediatric endocrinologists responded that rhGH was likely or very likely “to have a positive impact on emotional well-being in short non-GHD children, even if rhGH therapy does not have a major impact on their adult height.”51 In a study of patients with GHD, improvements in QoL were reported by mothers and patients receiving rhGH treatment for GHD developed post-malignancy, but not for those with idiopathic GHD.90
QoL is analyzed in studies through various measures, such as the Pediatric Quality of Life Inventory,90 the TNO-AZL Children’s Quality of Life Questionnaire,89 and the Idiopathic Short Stature Quality of Life Questionnare.89 Additional studies use the Quality of Life in Short Stature Youth (QoLISSY) questionnaire, the only validated (in Europe and subsequently the U.S.) measure of QoL in children with short stature.91 Many of the available measures are described and evaluated elsewhere,91,92 and the PES guidelines acknowledge the need to develop more appropriate, accurate QoL measures for children with short stature.15
Overall, the GH field suffers a paucity of well-designed studies supporting psychosocial benefits of rhGH therapy for both ISS and GHD. In light of this ambiguity, the 2016 PES guidelines advise that the physician evaluate and monitor psychosocial development in an individualized manner, and psychological counseling should be offered, in lieu of or in addition to rhGH treatment, if necessary.15 These recommendations are especially true for cases of ISS, where psychosocial functioning is the main justification for treatment.
Additional considerations: Costs and coverage
rhGH therapy is expensive, with median annual cost (in 2016) of $28,805 USD [interquartile range, 75th-25th percentiles, of $31,383] according to U.S. national insurance claims93 or calculated, for cases of ISS, as $52,634 USD (2015) for every inch of height gained.94 Its high cost factors into treatment decisions, and necessitates the involvement of medical insurance and policy decisions. Given bureaucratic, economic, and cultural differences, the way in which insurance affects rhGH cost and coverage varies across countries. Among the 10 main users of a major rhGH brand, U.S.A. is the only country with a commercial third-party payor health system.26 The nine other countries (Japan, Italy, Turkey, Sweden, Australia, Germany, Spain, France, UK) have a system in which the government functions as the primary payor, although supplements to the national plan from private insurance companies are available in some countries (eg. Spain).26 Different approaches to cost-containing efforts have been taken. For example, Japan adopted a tiered governmental cost support for rhGH therapy, with 70% coverage for cases of height −2.0 SD and 90% coverage for height −2.5 SD or shorter.95 On the other hand, in the U.S., commercial insurance companies have adopted formulary brand preference strategies; defined their own, more conservative criteria for GHD than those outlined by PES; and started denying overage for ISS in toto.93 A U.S. study using commercial claims data of rhGH from 2004 to 2016 reported that, comparing per capita costs in 2004 constant dollars, although mean total expenditures of rhGH decreased by 26%, mean total patient financial burden increased by 163% while mean estimated insurance-paid amounts decreased by 28%.93 This translates to an increase in the patient’s share of spending from 1.4% in 2004 to 3.9% in 2016.93 The increasing financial burden on patients occurs across U.S. healthcare and is especially evident for specialty drugs, as insurance companies shield themselves from the drugs’ high costs.93
Shared decision-making
In summary, the GH dilemma includes issues at every step in the path to potential treatment for GHD and ISS. In detecting worrisome growth, the field faces the ultimate challenge of defining “normal.” This already arbitrary endeavor is further complicated due to missing or incorrectly measured growth data, to the subjective interpretation of bone ages and the uncertainty in predicted adult heights, and to inconsistent cutoffs of “short.” In distinguishing between GHD and ISS, growth data, laboratory data and provocative testing are all fraught with limitations and potential pitfalls. Within provocative testing especially, the pulsatile and highly variable nature of GH secretion, accuracy of the test itself, lack of standardized assays, and unclear threshold demarcating partial GHD from ISS further compounds the ambiguity.
All this being said, rhGH treatment for classical GHD is uncontroversial and well supported. For non-GHD short stature, it behooves the clinician to inform the patient-family that not treating is an option. When deciding whether to initiate rhGH treatment, commercial and societal pressures influence the triad of patient, parent and pediatrician. In addition, questions remain regarding the long-term safety of rhGH, the limit of tolerable risk for treatment of ISS, reliable predictors of rhGH response in ISS, and what construes an adequate response to therapy. Especially relevant in the case of ISS, quality designed, well controlled, unbiased studies are still needed to clarify the impact of both short stature and rhGH treatment on psychosocial functioning. The high cost of rhGH influences treatment decision-making on the personal level, and at the societal level, leads to considerations of worthy resource allocation and who should be the responsible payor. Reviewing rhGH treatment for GHD and ISS also introduces the broader issues of demographic skews in patient populations, the interplay between the decision-making triad (especially the lack of research regarding the child’s role), and the difference between quantitative success and treatment expectations. Consequently, while all issues have been reviewed in the context of rhGH therapy for GHD and ISS, many of them can be applied to other conditions and to the broader scope of pediatric care in general.
Value in healthcare has been defined as quality over cost.96 Thus, improving quality and/or reducing cost leads to an increase in patient-care value. As part of its Quality of Health Care in America project, the Institue of Medicine (IOM) outlined six primary goals of and ten recommendations to improve the quality of U.S. national health care.96 Among them are patient-centeredness and evidence-based decision making.96 In the rhGH process, the triad must navigate numerous diagnostic and decision-making challenges in the pursuit of quality healthcare. In line with the IOM, the 2016 PES guidelines suggested that the key to this is an integration of patient needs and gathered evidence, recommending: “In the USA, for children who meet FDA criteria, we suggest a shared decision-making approach to pursuing rhGH treatment for a child with ISS. The decision can be made on a case-by-case basis after assessment of physical and psychological burdens, and discussion of risks and benefits.”15 In practice, this approach calls the physician to identify and appropriately treat any underlying pathology, to have frank conversations about potential benefits and side effects, to remain receptive to patient and parent needs and concerns, and to set realistic expectations so patient-families can make well-informed decisions.
Acknowledgements
This work was supported by NIH grant R01 HD097129 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (A.G.).
AG served as a member of the Steering Committee, Pfizer International Growth Study Database and as a consultant for the Pediatric Endocrine Society GH Deficiency Knowledge Center, sponsored by Sandoz.
Footnotes
Conflicts of Interest:
JGH has no potential conflicts to disclose.
References
- 1.Rosenfeld RG. Ten axioms in the evaluation of growth failure. Endocrinologist. 1997;7:148–52. [Google Scholar]
- 2.Grimberg A, Lifshitz F. Worrisome Growth. In: Lifshitz F, editor. Pediatric Endocrinology 5th ed. Vol. 2, Growth, adrenal, sexual, thyroid, calcium, and fluid balance disorders. New York: Informa Healthcare USA, Inc.; 2007. p. 1–50. [Google Scholar]
- 3.Allen DB, Cuttler L. Clinical practice. Short stature in childhood — challenges and choices. N Engl J Med. 2013;368(13):1220–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bright Futures/AAP Recommendations for Preventive Pediatric Health Care (Periodicity Schedule) [Internet]. American Academy of Pediatrics; ©2019. [cited 2019 Dec 16]. Available from: https://www.aap.org/en-us/documents/periodicity_schedule.pdf. [Google Scholar]
- 5.Lipman TH, Hench KD, Benyi T, Delaune J, Gilluly KA, Johnson L, et al. A multicenter randomised controlled trial of an intervention to improve the accuracy of linear growth measurement. Arch Dis Child. 2004. April;89(4):342–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanner JM, Whitehouse RH. Clinical longitudinal standards for height, weight, height velocity, weight velocity, and stages of puberty. Arch Dis Child. 1976. March;51(3):170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.National Center for Health Statistics: WHO growth charts [Internet]. Centers for Disease Control and Prevention; ©2009. [cited 2019 Dec 16]. Available from: https://www.cdc.gov/growthcharts/who_charts.htm#The%20WHO%20Growth%20Charts. [Google Scholar]
- 8.National Center for Health Statistics: clinical growth charts [Internet]. Centers for Disease Control and Prevention; ©2000. [cited 2019 Dec 16]. Available from: https://www.cdc.gov/growthcharts/clinical_charts.htm [Google Scholar]
- 9.Wit JM, Oostdijk W, Losekoot M, van Duyvenvoorde HA, Ruivenkamp CA, Kant SG. Mechanisms in endocrinology: Novel genetic causes of short stature. Eur J Endocrinol. 2016. April;174(4):R145–73. [DOI] [PubMed] [Google Scholar]
- 10.Tanner JM, Goldstein H, Whitehouse RH. Standards for children’s height at ages 2–9 years allowing for heights of parents. Arch Dis Child. 1970. December;45(244):755–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greulich WW, Pyle SI. Radiographic atlas of skeletal development of the hand and wrist. Stanford (CA): Stanford University Press; 1950. [Google Scholar]
- 12.Collett-Solberg PF, Ambler G, Backeljauw PF, Bidlingmaier M, Biller BMK, Boguszewski MCS, et al. Diagnosis, genetics, and therapy of short stature in children: A Growth Hormone Research Society international perspective. Horm Res Paediatr. 2019;92(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Topor LS, Feldman HA, Bauchner H, Cohen LE. Variation in methods of predicting adult height for children with idiopathic short stature. Pediatrics. 2010. November;126(5):938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen P, Rogol AD, Deal CL, Saenger P, Reiter EO, Ross JL, et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: A summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab. 2008. November; 93(11):4210–7. [DOI] [PubMed] [Google Scholar]
- 15.Grimberg A, Divall SA, Polychronakos C, Allen DB, Cohen LE, Quintos JB, et al. Guidelines for growth hormone and insulin-like growth factor-I treatment in children and adolescents: Growth hormone deficiency, idiopathic short stature, and primary insulin-like growth factor-I deficiency. Horm Res Paediatr. 2016;86(6):361–97. [DOI] [PubMed] [Google Scholar]
- 16.Hall DM. Growth monitoring. Arch Dis Child. 2000;82:10–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Growth chart training: using the WHO growth charts [Internet]. Centers for Disease Control and Prevention; [reviewed 15 Apr 2015; cited 2019 Dec 16]. Available from: https://www.cdc.gov/nccdphp/dnpao/growthcharts/who/using/assessing_growth.htm. [Google Scholar]
- 18.Ranke MB. Towards a consensus on the definition of idiopathic short stature. Horm Res. 1996;45 Suppl 2:64–6. [DOI] [PubMed] [Google Scholar]
- 19.Cutfield WS, Albert BB. Growth hormone treatment for idiopathic short stature. Pediatr Endocrinol Rev. 2018. September;16(Suppl 1):113–22. [DOI] [PubMed] [Google Scholar]
- 20.Raben MS. Treatment of a pituitary dwarf with human growth hormone. J Clin Endocrinol Metab. 1958. August;18(8):901–3. [DOI] [PubMed] [Google Scholar]
- 21.Laron Z The era of cadaveric pituitary extracted human growth hormone (1958 – 1985): Biological and clinical aspects. Pediatr Endocrinol Rev. 2018. September;16 Suppl 1:11–6. [DOI] [PubMed] [Google Scholar]
- 22.Ranke MB. Pediatric growth hormone deficiency (GHD) in the recombinant human GH (rhGH) era. Pediatr Endocrinol Rev. 2018. September;16 Suppl 1:63–9. [DOI] [PubMed] [Google Scholar]
- 23.Grimberg A, Allen DB. Growth hormone treatment for growth hormone deficiency and idiopathic short stature: New guidelines shaped by the presence and absence of evidence. Curr Opin Pediatr. 2017. August;29(4):466–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen DB. Lessons learned from the hGH era. J Clin Endocrinol Metab. 2011. October; 96(10):3042–7. [DOI] [PubMed] [Google Scholar]
- 25.Watson SE, Rogol AD. Recent updates on recombinant human growth hormone outcomes and adverse events. Curr Opin Edocrinol Diabetes Obes. 2013. February; 20(1):39–43. [DOI] [PubMed] [Google Scholar]
- 26.Grimberg A, Stewart E, Wajnrajch MP. Gender of pediatric recombinant human growth hormone recipients in the United States and globally. J Clin Endocrinol Metab. 2008. June;93(6):2050–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen DB. Growth promotion ethics and the challenge to resist cosmetic endocrinology. Horm Res Paediatr 2017;87:145–52. [DOI] [PubMed] [Google Scholar]
- 28.Domené S, Domené HM. Genetic mutations in the GH/IGF axis. Pediatr Endocrinol Rev. 2018;16 Suppl 1:39–62. [DOI] [PubMed] [Google Scholar]
- 29.Jørgensen JO, Hermansen K, Stochholm K, Juul A. Adult growth hormone deficiency: from transition to senescence. Pediatr Endocrinol Rev. 2018. September;16(Suppl 1):70–9. [DOI] [PubMed] [Google Scholar]
- 30.Lindsay R, Feldkamp M, Harris D, Roberston J, Rallison M. Utah Growth Study: Growth standards and the prevalence of growth hormone deficiency. J Pediatr. 1994. July;125(1):29–35. [DOI] [PubMed] [Google Scholar]
- 31.Hindmarsh P, Smith PJ, Brook CGD, Matthews DR. The relationship between height and growth hormone secretion in short prepubertal children. Clin Endocrinol (Oxf). 1987. November;27(5):581–91. [DOI] [PubMed] [Google Scholar]
- 32.Committee on Drugs and Committee on Bioethics. Considerations related to the use of recombinant human growth hormone in children. Pediatrics. 1997. January; 99(1):122–9. [DOI] [PubMed] [Google Scholar]
- 33.Chen RS, Shiffman RN. Assessing growth patterns-routine but sometimes overlooked. Clin Pediatr (Phila). 2000. February;39(2):97–102. [DOI] [PubMed] [Google Scholar]
- 34.de Onis M, Wijnhoven TM, Onyango AW. Worldwide practices in child growth monitoring. J Pediatr. 2004. April;144(4):461–5. [DOI] [PubMed] [Google Scholar]
- 35.Ketha H, Singh RJ. Clinical assays for quantitation of insulin-like-growth-factor-1 (IGF1). Methods. 2015. June 15;81:93–8. [DOI] [PubMed] [Google Scholar]
- 36.Blum WF, Albertsson-Wikland K, Rosberg S, Ranke MB. Serum levels of insulin-like growth factor (IGF-I) and IGF binding protein 3 reflect spontaneous growth hormone secretion. J Clin Endocrinol Metab. 1993. June;76(6):1610–6. [DOI] [PubMed] [Google Scholar]
- 37.Clemmons DR. Consensus statement on the standardization and evaluation of growth hormone and insulin-like growth factor assays. Clin Chem. 2011. April;57(4):555–9. [DOI] [PubMed] [Google Scholar]
- 38.Hawkes CP, Grimberg A. Insulin-like growth factor-I is a marker for the nutritional state. Pediatr Endocrinol Rev. 2015. December;13(2):499–511. [PMC free article] [PubMed] [Google Scholar]
- 39.Cruickshank JK, Heald AH, Anderson S, Cade JE, Sampayo J, Riste LK, et al. Epidemiology of the insulin-like growth factor system in three ethnic groups. Am J Epidemiol. 2001. September 15;154(6):504–13. [DOI] [PubMed] [Google Scholar]
- 40.Katsumata N Standardization of growth hormone and insulin-like growth factor-I measurement. Pediatr Endocrinol Rev. 2018. September;16 Suppl 1:28–32. [DOI] [PubMed] [Google Scholar]
- 41.Hauffa BP, Lehmann N, Bettendorf M, Mehls O, Dörr HG, Partsch CJ, Schwarz HP, et al. Central reassessment of GH concentrations measured at local treatment centers in children with impaired growth: consequences for patient management. Eur J Endocrinol. 2004. March;150(3):291–7. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka T, Tachibana K, Shimatsu A, Katsumata N, Tsushima T, Hizuka N, Fujieda K, Yokoya S, Irie M. A nationwide attempt to standardize growth hormone assays. Horm Res. 2005;64(Suppl 2):6–11. [DOI] [PubMed] [Google Scholar]
- 43.Binder G, Huller E, Blumenstock G, Schweizer R. Auxology-based cut-off values for biochemical testing of GH secretion in childhood. Growth Horm IGF Res 2011;21:212–8. [DOI] [PubMed] [Google Scholar]
- 44.Grimberg A, Heurta-Saenz L, Grundmeier R, Ramos MJ, Pati S, Cucchiara AJ, et al. Gender bias in U.S. pediatric growth hormone treatment. Sci Rep. 2015. June 9;5:11099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grimberg A, Kutikov JK, Cucchiara AJ. Sex differences in patients referred for evaluation of poor growth. J Pediatr. 2005. February;146:212–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finkelstein BS, Singh J, Silvers JB, Marrero U, Neuhauser D, Cuttler L. Patient attitudes and preferences regarding treatment: GH therapy for childhood short stature. Horm Res. 1999;51 Suppl 1:67–72. [DOI] [PubMed] [Google Scholar]
- 47.Grimberg A, Lindberg A, Wajnrajch M, Cucchiara AJ, Caamcho-Hübner C. Racial/ethnic disparities in US pediatric growth hormone treatment. Horm Res Paediatr. 2018;90(2):102–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finch BK, Beck AN. Socio-economic status and z-score standardized height-for-age of U.S.-born children (ages 2–6). Econ Hum Biol. 2011. July;9(3):272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grimberg A, Feemster KA, Pati S, Ramos M, Grundmeier R, Cucchiara AJ, et al. Medically underserved girls receive less evaluation for short stature. Pediatrics. 2011. April;127(4):696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cuttler L, Marinova D, Mercer MB, Connors A, Meehan R, Silvers JB. Patient, physician, and consumer drivers: Referrals for short stature and access to specialty drugs. Med Care. 2009. August;47(8):858–65. [DOI] [PubMed] [Google Scholar]
- 51.Cuttler L, Silvers JB, Singh J, Marrero U, Finkelstein B, Tannin G, et al. Short stature and growth hormone therapy: A national study of physician recommendation patterns. JAMA. 1996. August 21;276(7):531–7. [PubMed] [Google Scholar]
- 52.Cousounis PA, Lipman TH, Ginsburg K, Cucchiara AJ, Grimberg A. How short is too short according to parents of primary care patients. Endocr Pract. 2014. November;20(11):1113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schumacher A On the significance of stature in human society. J Hum Evol. 1982;11:697–701. [Google Scholar]
- 54.Sandberg DE, Colsman M. Growth hormone treatment of short stature: Status of the quality of life rationale. Horm Res. 2005;63(6):275–83. [DOI] [PubMed] [Google Scholar]
- 55.Sandberg DE, Gardner M. Short stature: Is it a psychosocial problem and does changing height matter? Pediatr Clin N Am. 2015;62:963–82. [DOI] [PubMed] [Google Scholar]
- 56.Underwood LE, Rieser PA. Is it ethical to treat short healthy children with growth hormone? Acta Paediatr Scand Suppl. 1989;362:18–23. [DOI] [PubMed] [Google Scholar]
- 57.Fox S Health topics: 80% of internet users look for health information online. Washington (D.C.): Pew Research Center’s Internet & American Life Project; 2011. February 33 p. [Google Scholar]
- 58.Individuals using the internet for seeking health-related information [Internet]. [place unknown]: Eurostat. 2006-2019 [cited 2019 Dec 16]. Available from: https://ec.europa.eu/eurostat/databrowser/view/tin00101/default/table?lang=en.
- 59.Cousounis P, Lipman TH, Ginsburg KR, and Grimberg A. Internet informs parents about growth hormone. Horm Res Paediatr. 2013;80(2):86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Persico N, Postlewaite A, Silverman D. The effect of adolescent experience on labor market outcomes: The case of height. J Polit Econ. 2004;112:1109–53. [Google Scholar]
- 61.Gardner M, Boshart ML, Yeguez CE, Desai KM, Sandberg DE. Coming up short: Risks of bias in assessing psychological outcomes in growth hormone therapy for short stature. J Clin Endocrinol Metab. 2016. January;101(1):23–30. [DOI] [PubMed] [Google Scholar]
- 62.Voss LD. Growth hormone therapy for the short normal child: Who needs it and who wants it? J Pediatr. 2000;136(1):103–6. [DOI] [PubMed] [Google Scholar]
- 63.Grimberg A, Cousounis P, Cucchiara AJ, Lipman TH, Ginsburg KR. Parental concerns influencing decisions to seek medical care for a child’s short stature. Horm Res Paediatr. 2015;84(5):338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Allen DB, Backeljauw P, Bidlingmaier M, Biller BM, Boguszewski M, Burman P, et al. GH safety workshop position paper: A critical appraisal of recombinant human GH therapy in children and adults. Eur J Endocrinol. 2016. February;174(2):P1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ergun-Longmire B, Mertens AC, Mitby P, Qin J, Heller G, Shi W, et al. Growth hormone treatment and risk of second neoplasms in the childhood cancer survivor. J Clin Endocrinol Metab. 2006. September;91(9):3494–8. [DOI] [PubMed] [Google Scholar]
- 66.Swerdlow AJ, Cooke R, Beckers D, Butler G, Carel JC, Cianfarani S, et al. Risk of meningioma in European patrients treated with growth hormone in childhood: Results from the SAGhE cohort. J Clin Endocrinol Metab. 2019. March 1;104(3):658–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raman S, Grimberg A, Waguespack SG, Miller BS, Sklar CA, Meacham LR, Patterson BC. Risk of neoplasia in pediatric patients receiving growth hormone therapy–a report from the Pediatric Endocrine Society Drugs and Therapeutics Committee. J Clin Endocrinol Metab. 2015. June;100(6):2192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Swerdlow AJ, Cooke R, Albertsson-Wikland K, Borgström B, Butler G, Cianfarani S, et al. Description of the SAGhE cohort: A large European study of mortality and cancer incidence risks after childhood treatment with recombinant growth hormone. Horm Res Paediatr. 2015;84(3):172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carel JC, Ecosse E, Landier F, Meguellati-Hakkas D, Kaguelidou F, Rey G, et al. Long-term mortality after recombinant growth hormone treatment for isolated growth hormone deficiency or childhood short stature: preliminary report of the French SAGhE study. J Clin Endocrinol Metab. 2012. February;97(2):416–25. [DOI] [PubMed] [Google Scholar]
- 70.Poidvin A, Touzé E, Ecosse E, Landier F, Béjot Y, Giroud M, et al. Growth hormone treatment for childhood short stature and risk of stroke in early adulthood. Neurology. 2014. August 26;83(9):780–6. [DOI] [PubMed] [Google Scholar]
- 71.Savendahl L, Maes M, Albertsson-Wikland K, Borgström B, Carel JC, Henrard S, et al. Long-term mortality and causes of death in isolated GHD, ISS, and SGA patients treated with recombinant growth hormone during childhood in Belgium, The Netherlands, and Sweden: Preliminary report of 3 countries participating in the EU SAGhE study. J Clin Endocrinol Metab. 2012. February; 97(2):E213–E217. [DOI] [PubMed] [Google Scholar]
- 72.Swerdlow AJ, Cooke R, Beckers D, Borgström B, Butler G, Carel JC, et al. Cancer risks in patients treated with growth hormone in childhood: The SAGhE European cohort study. J Clin Endocrinol Metab. 2017. May 1;102(5):1661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wit JM, Kamp GA, Rikken B. Spontaneous growth and response to growth hormone treatment in children with growth hormone deficiency and idiopathic short stature. Pediatr Res. 1996. February;39(2):295–302. [DOI] [PubMed] [Google Scholar]
- 74.Albertsson-Wikland K, Aronson AS, Gustafsson J, Hagenäs L, Ivarsson SA, Jonsson B, et al. Dose-dependent effect of growth hormone on final height in children with short stature without growth hormone deficiency. J Clin Endocrinol Metab 2008;93:4342–50. [DOI] [PubMed] [Google Scholar]
- 75.Ranke MB, Lindberg A, Price DA, Darendeliler F, Albertsson-Wikland K, Wilton P, et al. Age at growth hormone therapy start and first-year responsiveness to growth hormone are major determinants of height outcome in idiopathic short stature. Horm Res. 2007;68(2):53–62. [DOI] [PubMed] [Google Scholar]
- 76.Fisher BG, Acerini CL. Understanding the growth hormone therapy adherence paradigm: A systematic review. Horm Res Paediatr. 2013;79(4):189–96. [DOI] [PubMed] [Google Scholar]
- 77.Cutfield WS, Derraik JGB, Gunn AJ, Reid K, Delany T, Robinson E. Non-compliance with growth hormone treatment in children is common and impairs linear growth. PLoS One. 2011. January 31;6(1):e16223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Christiansen JS, Backeljauw PF, Bidlingmaier M, Biller BM, Boguszewski MC, Casanueva FF, et al. Growth Hormone Research Society perspective on the development of long-acting growth hormone preparations. Eur J Endocrinol. 2016. June;174(6):C1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lal RA, Hoffman AR. Long-acting growth hormone preparations in the treatment of children. Pediatr Endocrinol Rev. 2018. September;16 Suppl 1:162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rotnem D, Cohen DJ, Hintz R, Gene M. Psychological sequelae of relative “treatment failure” for children receiving human growth hormone replacement. J Am Acad Child Psychiatry. 1979. Summer;18(3):505–20. [DOI] [PubMed] [Google Scholar]
- 81.Constitution of the World Health Organization. Am J Public Health Nations Health. 1946. November;36(11):1315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zimet GD, Cutler M, Litvene M, Dahms W, Owens R, Cuttler L. Psychological adjustment of children evaluated for short stature: A preliminary report. J Dev Behav Pediatr. 1995. August;16(4):264–70. [PubMed] [Google Scholar]
- 83.Sandberg DE, Brook AE, Campos SP. Short stature: A psychosocial burden requiring growth hormone therapy? Pediatrics.1994. December;94(6 Pt 1):832–40. [PubMed] [Google Scholar]
- 84.Lee JM, Appugliese D, Coleman SM, Kaciroti N, Corwyn RF, Bradley RH, et al. Short stature in a population-based cohort: Social, emotional, and behavioral functioning. Pediatrics. 2009. September;124(3):903–10. [DOI] [PubMed] [Google Scholar]
- 85.Kranzler JH, Rosenbloom AL, Proctor B, Diamond FB Jr, Watson M. Is short stature a handicap? A comparison of the psychosocial functioning of referred and nonreferred children with normal short stature and children with normal stature. J Pediatr. 2000. January;136(1):96–102. [DOI] [PubMed] [Google Scholar]
- 86.Downie AB, Mulligan J, Stratford RJ, Betts PR, Voss LD. Are short normal children at a disadvantage? The Wessex growth study. BMJ. 1997. January 11;314(7074):97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hunt L, Hazen RA, Sandberg DE. Perceived versus measured height. Which is the stronger predictor of psychosocial functioning? Horm Res. 2000;53(3):129–38. [DOI] [PubMed] [Google Scholar]
- 88.Ross JL, Sandberg DE, Rose SR, Leschek EW, Baron J, Chipman JJ, et al. Psychological adaptation in children with idiopathic short stature treated with growth hormone or placebo. J Clin Endocrinol Metab. 2004. October;89(10):4873–8. [DOI] [PubMed] [Google Scholar]
- 89.Theunissen NC, Kamp GA, Koopman HM, Zwinderman KA, Vogels T, Wit JM. Quality of life and self-esteem in children treated for idiopathic short stature. J Pediatr. 2002. May;140(5);507–15. [DOI] [PubMed] [Google Scholar]
- 90.Sheppard L, Eiser C, Davies HA, Carney S, Clarke SA, Urquhart T, et al. The effects of growth hormone treatment on health-related quality of life in children. Horm Res. 2006;65(5):243–9. Epub 2006 Mar 30. [DOI] [PubMed] [Google Scholar]
- 91.Bullinger M, Sommer R, Pleil A, Mauras N, Ross J, Newfield R, Silverman L, et al. Evaluation of the American-English Quality of Life in Short Stature Youth (QoLISSY) questionnaire in the United States. Health Qual Life Outcomes. 2015. April 2;13: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brütt AL, Sandberg DE, Chaplin J, Hartmut W, Noeker M, Kołtowska-Häggström. Assessment of health-related quality of life and patient satisfaction in children and adolescents with growth hormone deficiency or idiopathic short stature – part 1: A critical evaluation of available tools. Horm Res. 2009;72(2):65–73. [DOI] [PubMed] [Google Scholar]
- 93.Grimberg A, Kanter GP. U.S. growth hormone use in the idiopathic short stature era: Trends in insurer payments and patient financial burden. J Endocr Soc. 2019. August 28;3(11):2023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee JM, Davis MM, Clark SJ, Hofer TP, Kemper AR. Estimated cost-effectiveness of growth hormone therapy for idiopathic short stature. Arch Pediatr Adolesc Med. 2006. March;160(3):263–9. [DOI] [PubMed] [Google Scholar]
- 95.Chihara K Diagnosis guideline for pediatric growth hormone deficiency In: Study Report for Pituitary Dysfunction. Tokyo, Japan: Japan Ministry of Health, Labour and Welfare; 2005. p. 118–20. [Google Scholar]
- 96.Institute of Medicine (US) Committee on Quality of Health Care in America Crossing the quality chasm: A new health system for the 21st century. Washington (DC): National Academics Press (US); 2001. [PubMed] [Google Scholar]