

Abstract

The combination of the predicted polymer market growth and the emergence of renewable feedstocks creates a fantastic opportunity for sustainable polymers. To replace fossil-based feedstock, there are only three alternative sustainable carbon sources: biomass, CO2, and existing plastics (via mechanical and/or chemical recycling). The ultimate circular feedstock would be CO2: it can be electrochemically reduced to formic acid derivatives that subsequently can be converted into useful monomers such as glycolic acid. This work is part of the European Horizon 2020 project “Ocean” in which the steps from CO2 to glycolic acid are developed. Polyglycolic acid (PGA) and poly(lactide-co-glycolide) (PLGA) copolyesters with high lactic acid (LA) content are well-known. PGA is very difficult to handle due to its high crystallinity. On the other hand, PLGAs with high LA content lack good oxygen and moisture barriers. The aim of this work is to understand the structure–property relationships for the mostly unexplored glycolic acid rich PLGA copolymer series and to assess their suitability as barrier materials. Thus, PLGA copolymers with between 50 and 91 mol % glycolic acid were synthesized and their properties were evaluated. Increased thermal stability was observed with increasing glycolic acid content. Only those containing 87 and 91 mol % glycolic acid were semicrystalline. A crystallization study under non-isothermal conditions revealed that copolymerization reduces the crystallization rate for PLGA compared to polylactic acid (PLA) and PGA. While PGA homopolymer crystallizes completely when cooled at 10 °C·min–1, the copolymers with 9 and 13% lactic acid show almost 10 times slower crystallization, which is a huge advantage vis-à-vis PGA for processing. The kinetics of this process, modeled with the Jeziorny-modified Avrami method, confirmed those observations. Barrier property assessment revealed great potential for these copolymers for application in barrier films. Increasing glycolic acid content in PLGA copolymers enhances the barrier to both oxygen and water vapor. At room temperature and a relative humidity below 70% the PLGA copolymers with high glycolic acid content outperform the barrier properties of polyethylene terephthalate.
Keywords: circular polyester, poly(lactide-co-glycolide), CO2 as feedstock, glycolide, oxygen and water barrier
Introduction
Poly(lactic-co-glycolic acid) (PLGA) are aliphatic polyesters of increasing interest for the biomedical field due to their biocompatibility and degradability in vivo.1 High molecular weight PLGA can only be produced via ring opening polymerization (ROP) of lactide (LAC) and glycolide (GL) and not via the monomers LA and GA.2 ROP has been shown to be a very useful route to produce polymers of industrial importance at commercial scale. These polyester syntheses can be carried out via different mechanisms such as anionic, cationic, enzymatic ,and coordination–insertion polymerization.3,4 Because of its solubility in various lactones, high catalytic activity, low toxicity, and ability to give high molecular weight polymers, tin(II) 2-ethylhexanoate Sn(Oct)2 has been the most widely used and studied catalyst for lactide and glycolide polymerization.5 According to Ryner et al.,6 the most accepted ROP mechanism involves a coordination–insertion mechanism initiated by the hydroxyl group of an added alcohol which coordinates to SnOct2, forming an initiating tin alkoxide complex. The rate-determining step of the ROP is then the nucleophilic attack of the alkoxide on the carbonyl carbon of the monomer.
The mechanical and physicochemical properties of PLGA are strongly determined by the ratio of the monomers. Most commercial PLGA grades currently available contain mainly lactide (between 50 and 95 mol %)7,8 with the exception of the absorbable suture material brand Vicryl (90% glycolide) from Ethicon.9 Copolymers with between 50 and 85 mol % lactide exhibit a tensile strength (σu between 41.4 and 55.2 MPa and a tensile modulus (E) between 1 and 4.3 GPa.10 These values tend to be close to those reported for PLA (σu = 60.4 MPa and E = 3.54 GPa11) and considerably lower than those for PGA, especially the tensile strength (σu = 117 MPa and E = 7.6 GPa12). Furthermore, materials with high contents of lactide exhibit an amorphous microstructure with glass transition temperatures (Tg) between 31 and 51 °C and thermal stability up to 240 °C.13,14 They have also shown longer degradation time with increasing lactide content and improved hydrophobicity.15 Studies for copolymers with higher glycolic acid content are scarce,16,17 and important properties (e.g., barrier properties, thermal stability and crystallization behavior) useful for determining their potential for a wider range of applications have not been reported yet. In 1979, Gilding and Reed published work on PLGA copolymers with high glycolide content.16 To the best of our knowledge, this is the only published research thus far that has focused on these types of materials. Although valuable observations concerning the reaction mechanism and their characterization have been provided, many important aspects of these copolymers are still unknown. The PLA and PLGA data reported in that work and references used for comparison all deal with l-lactic acid or l-lactide-based polymers.
Research on polymer crystallization is very relevant since it can influence properties such as permeability, transparency, toughness, and elasticity. For semicrystalline materials, these properties are affected by the size, orientation, shape, and regularity of the crystallites, as well as by the degree of crystallinity.18 Crystallization behavior of pure PLA has been reported frequently.18−20 For PGA, however, studies on crystallization behavior are scarce. Yu et al. investigated the spherulitic morphology,21 crystalline structure, and structural evolution of PGA in the crystallization and melting processes by time-resolved wide-angle X-ray diffraction (WAXD) and Fourier transform infrared (FTIR) spectroscopy. According to the authors, PGA exhibits very fast crystallization from the melt and it completely crystallizes under fast cooling conditions (10 °C·min1). A unique morphology during crystallization, consisting of hedrites, was observed. This differs from the normal characteristic spherulites found for PLA and other aliphatic polyesters with similar chemical structures. Furthermore, Chen et al. studied different composites with a PLA matrix and PGA fibers as fillers.22 The results showed that PGA fiber addition accelerates the crystallization of PLA under isothermal conditions. This trend, however, was not strongly affected by the fiber content. For semicrystalline PLGA copolymers, the influence of glycolide content on the crystallization process has not been reported. Understanding such behavior is very relevant since it can provide valuable insight for material processing and application assessment.
Another very relevant property for PLGA copolymers, which is affected by the glycolide content, is the barrier for water vapor and oxygen. Common oxygen barrier polymers include PET, ethylene vinyl alcohol (EVOH), poly(vinylidene chloride) (PVDC), poly(vinyl alcohol) (PVOH), polyacrylonitrile (PAN), nylons, and nylon-MXD6.23 In some cases, they are used in multilayer systems with good water barrier polymers such as polypropylene (PP) and polyethylene (PE). These materials, consumed massively in different applications, are fossil-based and not degradable, which means that when leaked into ecosystems, irreversible accumulation of polymers will occur. For this reason, the use of single-use packaging is under increasing pressure.
The barrier properties of PLA have been frequently studied and reviewed.24−30 PLA alone does not exhibit a good enough water vapor barrier to compete with commonly used packaging materials such as PET and polystyrene (PS). In terms of oxygen barrier, PLA shows a lower permeability coefficient than PS and a higher one than PET.31 Because of the aforementioned limitations regarding barrier, PLA use in packaging has been restricted to short shelf life products. Nevertheless, the growing environmental awareness of consumers has contributed to increasing and ongoing interest for this polymer in the packaging industry.32,33
PGA, on the other hand, possesses very good barrier properties. The company Kureha has used it for the production of containers, pouches, and mono-34 and multilayer35,36 sheets. According to Kureha, a PGA film of 20 μm thickness possesses an oxygen transmission rate (OTR) of about 1 cm3·m–2·day–1 (measured at 30 °C and 80% relative humidity (RH)) and a water vapor transmission rate (WVTR) of about 10 g·m–2·day–1 at the same conditions.37 This performance is superior to that of most commercially available barrier polymers.
Nevertheless, PGA is also a very brittle material (linked to its high degree of crystallinity), with very limited solubility and difficult processability. The latter is related to its thermal properties. PGA exhibits a relatively small difference between its melt Tm (222 °C) and crystallization temperatures (Tc = 192–198 °C).38 For extrusion of items such as films and sheets this fast crystallization upon cooling from the melt complicates obtaining amorphous preforms. Consequently, transparent products are difficult to achieve. PGA also displays a small difference between its Tg (40–45 °C) and cold crystallization temperatures (Tcc = 75 °C).39 This is problematic for stretching (of films, sheets, and fibers, etc.) or stretch blow molding, since this is limited to a narrow temperature range. Furthermore, PGA has been reported to generate gases upon melt processing, which have been attributed to low molecular weight products produced in the molten state. Lastly, PGA is limited in its application due to its very fast degradation rate12 (2 weeks by hydrolysis at 99 °C). Taking into account the aforementioned limitations, copolymerization with low amounts of lactic acid/lactide is an attractive strategy for enhancing processability and solubility properties while also tuning the material properties (e.g., degradation rate).
Here, we report on our studies regarding the effects of glycolic acid content on the properties of copolymers derived from glycolide and lactide. Copolymers containing from 50 to 91 mol % glycolide were synthesized via ROP and characterized in terms of structure, density, thermal stability, and molecular weight distribution. For semicrystalline copolymers, the crystallization behavior was studied under non-isothermal crystallization conditions. Furthermore, the potential of PLGA copolymers as barrier materials was assessed by measuring the oxygen and water permeability at 30 °C and 70% RH, using hot pressed films. In addition, the effect of relative humidity and temperature on the water permeability was assessed for the two copolymers with higher glycolic acid content.
Experimental Section
Materials
Lactide and glycolide were acquired from Corbion (The Netherlands). Sn(Oct)2 was purchased from Alfa Aesar (96% purity), and 1-dodecanol was obtained from Merck (98%). Toluene was purchased from Acros (99.8% purity). Deuterated chloroform was obtained from Eurisotop, and hexafluoroisopropanol-d2 was acquired from Sigma-Aldrich.
Copolymer Synthesis
A series of lactide and glycolide copolymers was prepared on a 20 g scale by ring opening polymerization using the following general procedure.
Prior to polymerization, the glassware was cleaned and dried at 120 °C in an oven overnight. Lactide and glycolide were weighed in a 250 mL three neck round-bottom flask, equipped with a mechanical stirrer. Subsequently, the system was evacuated under vacuum and flushed three times with nitrogen for 5 min, each time at room temperature. Then, 0.60 g (0.03 wt % relative to the monomer feed) of catalyst Sn(Oct)2 and 0.20 g (0.01 wt % relative to the monomer feed) of initiator 1-dodecanol were added to the flask (prepared separately as a toluene solution). The toluene was removed under reduced pressure (10 mbar). The flask was heated in a silicone oil bath initially at 180 °C under nitrogen atmosphere. The temperature was gradually increased to 200 °C and then stirred at 100 rpm for 4 h. The product was recovered, ground, and dried for about 12 h at 40 °C in a vacuum oven (10 mbar). Copolymers with increasing glycolide content (50, 60, 70, 80, 85, and 90 mol %) were prepared using this procedure.
While the concentration of catalyst and initiator were kept constant for all syntheses, the reaction temperature was increased gradually to 220 °C for the copolymers with 85 and 90 mol % glycolide. Furthermore, the reaction time was reduced to 3 h for 80 and 85% glycolide and to 2 h for 90 mol % glycolide. Under these conditions, both monomers reached high conversion with little discoloration of the final product. As was observed in prior experiments, longer reaction times for these three samples led to additional transesterification side reactions, broader molecular weight distributions, and strong polymer discoloration.
PLA and PGA were synthesized as reference materials according to the protocol described by Kaihara et al.40
Structure and Thermal Stability of PLGA Copolymers
Nuclear Magnetic Resonance
1H nuclear magnetic resonance (NMR) spectroscopy was used to determine the built-in ratio of LA and GA in the copolymer for each sample. The measurements were performed on a Bruker AMX 400 (1H, 400.13 MHz). Samples with less than 70 mol % glycolide content were dissolved in deuterated chloroform. The remaining samples were dissolved in deuterated hexafluoroisopropanol.
Gel Permeation Chromatography
Gel permeation chromatography (GPC) was used to determine the molecular weight, relative to poly(methyl methacrylate) (PMMA) calibration standards, for all samples. A Merck-Hitachi LaChrom HPLC system, equipped with two PL gel 5 μm MIXED-C (300 × 7.5 mm2) columns using hexafluoroisopropanol as mobile phase, was used. Calculation of the molecular weight was done with the CirrusPL DataStream software package.
Thermogravimetric Analysis
Thermogravimetric analyses (TGAs) were performed to determine the thermal stability and mass loss as a function of temperature increase. The measurements were carried out on a TGA/DSC 3+ STARe system from Mettler Toledo. Approximatively 7 mg of each sample was introduced in a sealed aluminum sample vessel (40 μm). The test was subsequently conducted at a heating rate of 10 °C·min–1 from room temperature to 400 °C under nitrogen atmosphere with a flow rate of 50 mL·min–1.
Differential Scanning Calorimetry
The thermal transitions were determined using a differential scanning calorimeter DSC 3+ STARe system from Mettler Toledo. Samples with a mass of approximatively 5 mg were introduced in sealed aluminum pans (40 μm). Two scans were carried out. First, each sample was heated from room temperature to 230 °C (10 °C·min–1). Subsequently they were cooled to room temperature at the same rate and finally heated again to 230 °C at 10 °C·min–1. The Tg, Tcc, and melting temperature were recorded from the second scan. The percentage of crystallinity for the semicrystalline samples was calculated according to eq 1.28
| 1 |
Here ΔHm represents the enthalpy of fusion, ΔHcc is the enthalpy of cold crystallization, and ΔHmC is the heat of melting of the purely crystalline sample.
Crystallization Behavior of Semicrystalline Copolymers
To study the effect of glycolide content on the crystallization process, a non-isothermal crystallization test of all PLGA copolymers containing 80 mol % or more glycolic acid was carried out using DSC. Each copolymer sample was first heated from 0 to 230 °C (10 °C·min–1) and maintained at that temperature for 3 min to remove the thermal history. Subsequently, the samples were cooled at different cooling rates (10, 7, 5, 3, 1, and 0.5 °C·min–1) to 0 °C. The Tc and enthalpy of crystallization (ΔHc) were recorded. Finally, samples were heated for a second time from 0 to 230 °C (10 °C·min–1). For comparison, a similar method was carried out for PLA and PGA synthesized in the laboratory. PLA and PGA were first heated from 0 to 190 and 240 °C, respectively, at 10 °C·min–1. PLA and PGA were respectively kept at these temperatures for 3 min and then cooled at the different cooling rates mentioned before. In the second heating scan, PLA and PGA were heated to 190 and 240 °C, respectively, at 10 °C·min–1. All measurements were performed under nitrogen atmosphere, using a flow of 50 mL·min–1.
The effect of glycolide content on the spherulitic growth and morphology of the semicrystalline samples was observed with a polarized optical microscope (Olympus BX53) mounted with an Olympus DP26 camera and equipped with a Linkam Hotstage HFSX350. The ground samples were first sandwiched between two glass slides and molten at 240 °C for 3 min. They were subsequently cooled to 40 °C at 10 °C·min–1. The crystalline morphology was recorded during this process.
Measurement of Barrier Properties from PLGA Films
PLGA films were prepared via compression molding using a thermal press (Carver Auto Four/3015-NE,H). The polymer powder was first dried (12 h at 40 °C) and kept under vacuum until processing. The polymer was subsequently sandwiched between two poly(tetrafluoroethylene) (PTFE) films (0.14 mm thickness) and 2 aluminum plates (3 mm thickness each). For copolymers with 50, 60, 70, and 80 mol % glycolide, the sandwich was held for 1 min at 180 °C with a force of 0.5 ton to guarantee complete melting of the material. Subsequently, a pressure of 1 ton was applied for 30 s and finally the pressure was increased to 5 tons for another 30 s. For copolymers with 85 and 90 mol % glycolic acid the same procedure was used, except with a pressing temperature between 210 and 220 °C. The films were cooled by putting the PTFE sandwich in contact with two cold aluminum plates. Sample thickness was measured with an electronic micrometer at 12 different points, and an average of these values was taken as the film thickness. Films with a thickness between 170 μm (for copolymers with up to 70 mol % glycolide) and 100 μm (for copolymers with between 80 and 90 mol % glycolide) were tested for oxygen and moisture barrier.
Because PGA has a very high barrier to both oxygen and water, it would be necessary to prepare film samples below 50 μm thickness to be able to detect permeant. However, sufficiently thin PGA films could not be made using this technique as compression molded films prepared with such a highly crystalline material exhibit high brittleness and tend to crack very easily during demolding. This is accompanied by other microdefects (e.g., bubbles and pinholes) that are prone to appear in thinner films. For this reason, the oxygen permeability (OP) and the water permeability (WP) values for commercial PGA were used as a comparison.
The OP was determined using a Totalperm (Permtech s.r.l) instrument. Calibration of the system was carried out with a standard PET film provided by Permtech (Italy), according to the ASTM F1927-14 standard.
In this study, the series of glycolide and lactide copolymers was tested at 30 °C and 70% RH. The measurements were done in duplicates, and each one concluded when the collected data had reached a tolerance level of 0.5%. The system reports the oxygen transmission rate (OTR) at the established conditions; these values were normalized by the film thickness (x) to determine the oxygen permeability as follows:24
| 2 |
| 3 |
The water permeability (WP) was measured using the same Totalperm (Permtech s.r.l) instrument calibrated for water vapor according to the ASTM E96/E96M-15 standard. The test concludes once the variation between the recorded results reaches a tolerance level of 0.5%.
Initially, the samples were tested at 30 °C and 70% RH. Subsequently, the influence of temperature on the WP was studied at 30 and 40 °C for the films with the highest content of glycolic acid (80 and 90%). Similarly, the effect of RH was tested at 10, 50, 70 and 90% RH for these samples. The measured WVTR was normalized by the film thickness (x) to calculate the water permeability as follows:24
| 4 |
| 5 |
Density Determination of PLGA Films by a Density Gradient Column
The measurements were carried out in a density gradient column using a column filler and sweeper from H&D Fitzgerald Ltd., according to the ASTM D1501-10 standard. The column was filled by mixing two miscible liquids of known densities creating a density gradient. The “heavy liquid” is a calcium nitrate tetrahydrate/water solution (ρ = 1503 kg·m–3) and the “light liquid” is Milli-Q purified water (ρ = 997.5 kg·m–3). The density of the solution was measured with a density meter for liquids (Mettler Toledo DM40). Calibrated glass floats of known density were used as reference material for the column calibration. All of the measurements were taken at room temperature (21 ± 1 °C).
Two density columns with different ranges were used: the first one between 1477 and 1428 kg·m–3 and the second one between 1440 and 1349 kg·m–3. These ranges were chosen by testing various samples of each copolymer in calcium nitrate tetrahydrate/water solutions with different densities. Following the ASTM D1505-10 standard the columns were calibrated with correlation factors (R2) of 0.9929 and 0.9956, providing a density measurement with good accuracy. The polymer films were first cut in small triangles, immersed in water for about 3 min, and then carefully introduced in the density column. Measurements were taken after 1 h. The test for each copolymer was done in triplicate.
Results and Discussion
Effect of Glycolide Content on Structure and Thermal Stability of PLGA Copolymers
Each copolymer composition was measured by NMR spectroscopy. Samples with an initial glycolic acid content of 70 mol % and higher were not soluble in any of the deuterated organic solvents commonly used in the laboratory. For this reason hexafluoroisopropanol-d2 (HFIP) was used.41 Unfortunately, since (deuterated) HFIP is an expensive and harmful solvent, its general use in several analytical techniques is limited. Therefore, chloroform-d was used as solvent for the remaining samples.
The ratio of glycolide to l-lactide in the copolymer was determined by the integral of the peaks corresponding to the methylene group hydrogens (CH2) of glycolide and the area under the peaks of the methyl group hydrogens (CH3) or the methine group (CH) of the lactide according to eqs 6 and 7.
The determined ratio between lactic and glycolic acids in the copolymers is shown in Table 1. In a previous work, Gilding et al.16 reported the synthesis of polyglycolic acid using Sn(Oct)2 as a catalyst. According to their research, when glycolide is polymerized over 3–4 h (94–96%), the molecular weight distribution broadens significantly due to transesterification reactions between the existing polymer chains. This dual polymerizing and depolymerizing effect with Sn(Oct)2 when used at temperatures above 200 °C has been reported before.42 In line with the previous remarks, 4 h was selected as the maximum reaction time for the PLGA copolymers reported in this work. At longer reaction time, also dark brown coloration was observed.
| 6 |
| 7 |
As shown in Table 1, there is a common trend for higher incorporation of glycolide in the copolymer with respect to the designed amount in the feed. This is in line with previous reports and results from the much higher reactivity of glycolide compared to lactide in ROP. From literature14,43 it is known that the polymerization of these two monomers starts with the most reactive monomer polymerizing preferentially with itself: a glycolide ended growing chain will tend to add another glycolide unit, while a lactide ended chain will also tend to add a glycolide unit. As the reaction progresses, lactide is built into the polymer chains that are rich in glycolide. In the end some unreacted lactide remains. This means that the resulting copolymers are likely richer in glycolide than the starting monomer mixture would suggest, as can be seen in Table 1.
Table 1. GL/LAC Built-in Ratios, Molecular Weight Distribution, and Thermal Transitions for PLGA Copolymers, PLA, and PGA.
| GL in feed (mol %) | LAC in feed (mol %) | GL in copol (mol %) | LAC in copol (mol %) | Mn (kg/mol) | PDI | Tg (°C) | microstructure | Tcc (°C) | Tm (°C) | degree of crystallinity (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 50 | 50 | 52 | 48 | 23.6 | 2.2 | 44 | amorphous | |||
| 60 | 40 | 61 | 39 | 29.3 | 2.6 | 43 | amorphous | |||
| 70 | 30 | 72 | 28 | 27.2 | 2.7 | 43 | amorphous | |||
| 80 | 20 | 82 | 18 | 33.1 | 3.5 | 42 | amorphous | |||
| 85 | 15 | 87 | 13 | 28.9 | 2.9 | 41 | semicrystalline | 133 | 190 | 5 |
| 90 | 10 | 91 | 9 | 35.9 | 3.4 | 41 | semicrystalline | 113 | 199 | 29 |
| 0 | 100 | 0 | 100 | 35.8 | 2 | 58 | semicrystalline | 103 | 174 | 25 |
| 100 | 0 | 100 | 0 | 41 | semicrystalline | 222 | 48 |
In addition, broad composition ranges within the initially formed chains and those formed toward the end are more expected due to the significant reactivity difference between glycolide and lactide. For all cases, the glycolide conversion reached 99%. In the case of lactide, however, copolymers with initial feeds of 50, 60, and 70 mol % showed a maximum conversion of 96%. For the remaining copolymers, a small fraction of residual lactide was found (between 1 and 2%) together with some oligomer formation.
Under similar reaction conditions, higher addition of glycolide induces broadening of the molecular weight distribution. Although the reaction time was reduced for these high glycolic acid containing copolymers (above 70 mol % glycolide in the feed), the required temperatures for polymerization (up to 220 °C) seem to promote chain transfer reactions when using Sn(Oct)2 as a catalyst.
Although both PLA and PGA are semicrystalline, copolymerization induces a loss of crystallinity and as a consequence materials obtained from glycolide between 50 and 80 mol % in the feed are amorphous according to DSC, when submitted to 10 °C·min–1 heating and cooling cycles. The samples with 87 and 91 mol % of built-in glycolic acid showed a crystalline phase in the thermogram with a Tcc at 133 and 113 °C and a Tm of 190 and 199 °C, respectively.
The degree of crystallinity was calculated using eq 1, taking 191.344 and 93.7 J·g–145 as the heats of melting for purely crystalline samples (ΔHmC) of PGA and PLA, respectively. At these conditions the degree of crystallinity is higher for the copolymer with greater glycolide content. However, when compared to the degree of crystallinity of PGA, it is observed that a small incorporation of lactide units can reduce the ability for the polymer chains to adopt an ordered configuration and, therefore, delay the formation of crystalline regions. Furthermore, the broad molecular weight distribution determined for PLGA13/87 and PLGA9/91 implies high heterogeneity in chain length. This can also influence the crystallization process, since long and short chains tend to crystallize at different rates. The TGA curves of PLA, PGA, and the PLA/PGA copolymers, scanned under nitrogen atmosphere, are shown in Figure 1. For all of the samples, the thermal degradation occurred in a single-stage process.
Figure 1.
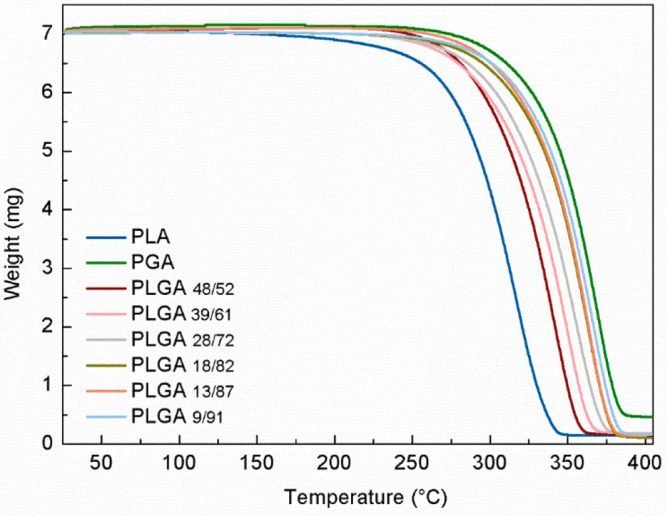
TGA thermogram for PLGA copolymers, PLA, and PGA using a heating rate of 10 °C·min–1.
All PLGA copolymers were thermally stable up to 250 °C, and they show increasing resistance to thermal degradation with increasing content of glycolide. A mass loss of 5% occurs between 271 and 292 °C, and the maximum degradation rate occurs between 364 and 385 °C. Of all the copolymers, the highest temperature values correspond to PLGA9/91. This is not unexpected since semicrystalline materials tend to be more thermally stable than their amorphous counterparts. In a highly ordered state, the mobility of the polymer chains is more restricted. PGA, for example, possesses the most ordered molecular packaging (relative to the degree of crystallinity) and thus requires higher temperatures to transform to an unordered fluid state and consequently to decompose.
Crystallization Behavior of Semicrystalline PLGA Copolymers
The exothermic effects recorded for PLGA13/87 and PLGA9/91 during the first cooling cycle are shown in Table 2.
Table 2. Crystallization Temperatures of PLGA, PLA, and PGA Samples at Different Cooling Rates.
| sample | ϕ (°C·min–1) | To (°C) | Tc (°C) | sample | ϕ (°C·min–1) | To (°C) | Tc (°C) |
|---|---|---|---|---|---|---|---|
| PLGA13/87 | 10 | 145 | 117 | PGA | 10 | 198 | 196 |
| 7 | 147 | 119 | 7 | 198 | 196 | ||
| 5 | 148 | 124 | 5 | 200 | 198 | ||
| 3 | 148 | 130 | 3 | 201 | 199 | ||
| 1 | 152 | 143 | 1 | 205 | 203 | ||
| 0.5 | 164 | 154 | 0.5 | 206 | 205 | ||
| PLGA9/91 | 10 | 153 | 129 | PLA | 10 | 116 | 101 |
| 7 | 156 | 137 | 7 | 119 | 103 | ||
| 5 | 159 | 142 | 5 | 122 | 108 | ||
| 3 | 162 | 148 | 3 | 129 | 120 | ||
| 1 | 168 | 158 | 1 | 136 | 130 | ||
| 0.5 | 177 | 167 | 0.5 | 139 | 135 |
The crystallization range for PLGA13/87 narrows as the cooling rate decreases. This is accompanied by a shift of the Tc toward higher values. Also, an increase in exothermic enthalpy change is observed with slower cooling of the samples. For this copolymer, the degree of crystallinity increases from 5 to 25% when cooled at between 10 and 0.5 °C·min–1. Although a similar trend is observed for PLGA9/91, the exothermic range is narrower and at 10 °C·min–1 the sample already exhibits 29% crystallinity. With decreasing cooling rate (ϕ), the crystallinity reaches 34% at 0.5 °C·min–1.
Other parameters, such as Tc and onset temperature of crystallization (To), are also shown in Table 2. As expected, under all cooling conditions, the copolymers crystallize at temperatures within the crystallization range of PGA and PLA individually.
PGA showed complete crystallization at a cooling rate of 10 °C·min–1, with a ΔHc that remained fairly constant with different cooling rates. These observations are in agreement with data previously reported for PGA.12,21 When comparing the crystallization temperatures of semicrystalline PLGA samples with PGA, it becomes apparent that with a small inclusion of lactide, the resulting copolymers not only crystallize at lower temperatures but also in a broader temperature range. These thermal variations will have an impact on material processing. The difference between Tm and the onset crystallization temperature for PLGA13/87 and PLGA9/91 cooled at 10 °C·min–1 (45–46 °C) is higher than that of PGA (26.3 °C). Due to this broader range, obtaining transparent items (films, sheets, and so on) upon extrusion of PLGA13/87 and PLGA9/91 would be easier. As mentioned before, this is still very challenging for PGA.
For PLA, crystallization occurs at lower temperatures, but in a narrower range than both PLGA13/87 and PLGA9/91 copolymers.
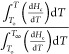 |
8 |
To better understand the non-isothermal crystallization mechanism, the relative crystallinity (X) as a function of temperature was calculated. Using eq 8, the exothermic events during cooling can be integrated along the crystallization range. Here, T∞ corresponds to the end temperature of crystallization and Hc is the enthalpy of crystallization.46
Figure 2 shows the relative crystallinity evolution as a function of temperature at different cooling rates for PLGA13/87, PLGA9/91, PGA, and PLA. Both copolymers PLGA13/87 and PLGA9/91 clearly exhibit broader crystallization ranges with increasing cooling rate. The glycolide content in the copolymers has a significant influence in the crystallization mechanism. As shown, with this small difference in glycolide content, the crystallization temperatures for PLGA13/87 and PLGA9/91 are increased from 117 to 154 °C at 10 °C·min–1 and from 129 to 177 °C at the slowest cooling rate of 0.5 °C·min–1. For PGA, the crystallization range broadness remains constant despite the cooling rate, while for PLA it narrows with slower cooling. Nevertheless, this effect for PLA is less significant than for the copolymers.
Figure 2.

Evolution of relative degree of crystallinity as a function of temperature for non-isothermal crystallization of PLGA13/87, PLGA9/91, PGA, and PLA at different cooling rates.
As mentioned before, crystal formation for PGA was similar under all tested cooling conditions. This differs from PLA, which shows a behavior closer to that of the copolymers: the degree of crystallization is enhanced with the decreasing cooling rate.
The progression of the relative degree of crystallinity as a function of time can be determined using eq 9.18
| 9 |
Here, T represents the crystallization temperature at time t and ϕ is the cooling rate in °C·min–1. Figure 3 illustrates the resulting plots for the copolymers and PLA and PGA homopolymers.
Figure 3.

Evolution of relative degree of crystallinity as a function of time for non-isothermal crystallization of PLGA13/87, PLGA9/91, PLA, and PGA at different cooling rates.
PLA and PGA homopolymers exhibit a higher crystallization rate than both PLGA13/87 and PLGA9/91 copolymers. This difference is more significant for PGA, which crystallizes very rapidly even at the highest cooling rates. Overall, higher crystallization temperatures, induced by slower cooling rates, are a consequence of slow crystallization. For each sample individually, the crystallization rate does not seem to be highly impacted by the cooling rate between 10 and 3 °C·min–1. However, there was a substantial delay in crystallization when samples were cooled at 1 and 0.5 °C·min–1. This trend was common for all samples. Importantly, the much longer crystallization times for PLGA13/87 and PLGA9/91 in comparison to those for PLA and PGA confirm that small inclusions of lactic acid in glycolic acid rich samples have a detrimental effect on their ability to crystallize. This effect becomes more significant with increasing lactic acid content (from 20 mol %) leading to amorphous copolymers.
Spherulitic behavior and morphology were observed during non-isothermal crystallization from the melt at 10 °C·min–1 for both PLGA samples (Figure 4).
Figure 4.

Optical micrograms taken for PLGA13/87 cooled down from the melt at 10 °C·min–1 at (a) 145, (b) 115, and (c) 85 °C and for PLGA9/91 at (d) 155, (e) 130, and (f) 100 °C.
According to the polarized optical microscopy images, PLGA13/87 starts crystallizing at lower temperatures than PLGA9/91 and both temperature ranges remain below the crystallization temperature of PGA. These results support the crystallization data obtained by DSC. Yu et al. reported the formation of hedrites with a high degree of complexity during isothermal crystallization of PGA.21 This unique morphology differs from other polyesters with comparable chemical structure, such as PLA, which shows well-defined spherulites during crystallization.20 As shown in the images of Figure 4, both copolymers exhibit a spherulitic morphology that resembles that of PLA. This is an unexpected observation, since both samples contain a very high amount of glycolide yet no hedrite-like crystals are observed. While PLGA9/91 exhibits higher spherulite size than PLGA13/87, the latter shows denser growth. In both cases, amorphous areas with no spherulite formation remained.
Non-isothermal Crystallization Kinetics
The crystallization kinetics were studied with the Jeziorny-modified Avrami model.47 This method has been previously applied and reported for studying polymer crystallization under non-isothermal conditions.48−51
Typically, the Avrami model alone is used to analyze crystallization kinetics under isothermal conditions according to eq 10, with Xt being the relative crystallinity as a function of time, K the Avrami (crystallization) rate constant, n the Avrami exponent, and t the crystallization time.
| 10 |
Equation 10 can be modified for non-isothermal conditions, leading to the Jeziorny-modified Avrami model, which includes a correction for the cooling rate:
| 11 |
| 12 |
Here n is related to the type of nucleation and growth mechanism and K is a rate constant that takes into account nucleation and growth rate parameters. In addition, K can be corrected (Kc) for variable cooling rates. Equation 11 implies that n and K can be determined from the slope and intercept of the resulting (linearized) plot of log[− ln(1 – Xt)] vs log t (Figure 5).18,52
Figure 5.

Avrami plots for log[− ln(1 – Xt)] as a function of log t at different heating rates for PLGA13/87, PLGA9/91, PLA, and PGA.
For all curves some nonlinearity is observed. This was typically observed below 1% and above 90% relative crystallinity. Because of this, the area between both values was selected as the range to fit the Avrami linear model. Table 3 presents the calculated n, K, Kc, and the R2 correlation coefficient using the Jeziorny-modified Avrami method. As shown, for both copolymers the n values stay within a similar range (3.3–3.6). This could suggest a similar crystallization mechanism. However, a consistent trend was not identified for all evaluated samples, which makes it difficult to draw conclusions on the nature of a general crystallization process.
Table 3. Parameters for Non-isothermal Crystallization Kinetics of PLGA13/87, PLGA9/91, PLA, and PGA Obtained from Jeziorny-Modified Avrami Model.
| sample | ϕ (°C·min–1) | n | K (min–1) | Kc (min–1) | R2 |
|---|---|---|---|---|---|
| PLGA13/87 | 10 | 3.42 | 0.13 | 0.82 | 0.99 |
| 7 | 3.35 | 8.8.10–3 | 0.71 | 0.99 | |
| 5 | 3.39 | 63 × 10–3 | 0.58 | 1.00 | |
| 3 | 3.16 | 55 × 10–3 | 0.38 | 0.99 | |
| 1 | 3.32 | 16 × 10–3 | 16 × 10–3 | 0.99 | |
| 0.5 | 3.62 | 5.6 × 10–3 | 3.16 × 10–5 | 0.99 | |
| PLGA9/91 | 10 | 3.34 | 0.18 | 0.84 | 1.00 |
| 7 | 3.41 | 0.15 | 0.77 | 1.00 | |
| 5 | 3.48 | 0.11 | 0.64 | 0.99 | |
| 3 | 3.53 | 0.07 | 0.40 | 0.99 | |
| 1 | 3.49 | 0.02 | 20 × 10–3 | 0.99 | |
| 0.5 | 3.59 | 6.7 × 10–3 | 4.52 × 10–5 | 0.99 | |
| PLA | 10 | 3.67 | 0.30 | 0.88 | 0.99 |
| 7 | 3.45 | 0.20 | 0.80 | 0.99 | |
| 5 | 3.11 | 0.18 | 0.71 | 0.99 | |
| 3 | 3.41 | 0.12 | 0.49 | 0.99 | |
| 1 | 3.55 | 0.05 | 51 × 10–3 | 0.99 | |
| 0.5 | 3.56 | 0.02 | 7 × 10–4 | 0.99 | |
| PGA | 10 | 2.97 | 2.73 | 1.10 | 0.99 |
| 7 | 3.19 | 2.92 | 1.16 | 0.99 | |
| 5 | 3.41 | 2.01 | 1.15 | 0.99 | |
| 3 | 3.41 | 1.04 | 1.01 | 0.99 | |
| 1 | 4.09 | 0.20 | 0.20 | 0.99 | |
| 0.5 | 3.52 | 0.09 | 9.1 × 10–3 | 0.99 |
For Kc, a steady trend was identified for all of the samples. Kc decreases with decreasing cooling rate, which implies that crystallization takes longer when the cooling rate is lowered. Furthermore, Kc values for PLGA13/87 are lower than those for PLGA9/91 and both are below the values determined for PLA and PGA. This is also in line with the DSC analysis where PGA and PLA showed faster crystallization compared with the copolymers.
Effect of Glycolide Content on Density and Barrier Properties of PLGA Films
Figure 6a shows the densities for PLGA films as a function of glycolide percentage in the copolymer. The reported values are averages of measurements performed in triplicate. The measured density increases with glycolide content: from 1.38 g.cm–3 for PLA to 1.50 g.cm–3 for PLGA9/91.
Figure 6.

Density (a), oxygen permeability (b), and water permeability (c) (measured at 70% RH and 30 °C) as a function of glycolide content in PLGA copolymers.
Nevertheless, the results remain within the expected density range between PLA and PGA (1.2–1.7 g.cm–3). All copolymers have a density higher than 1, which implies that they would sink when/if leaked into marine ecosystems. Because PLGA plastics are expected to degrade through hydrolysis as a first step, it is important to understand their degradation mechanism and the effect of their degradation products on aquatic media/ecosystems. For this group of copolymers a tailored degradation can be achieved by tuning the lactide content.
Specific barrier properties are required for polyesters, depending on the intended application. In the packaging industry, for example, oxygen and water vapor permeability are key properties, as water and oxygen often have a large impact on product shelf life. For PLA and PGA these properties have been studied.31,37 Although these barrier properties are important performance characteristics of PLGA copolymers, no reports on this topic were found so far.
The compression molded films were analyzed by DSC using the same method described before. Although the films were cooled rapidly using cold aluminum plates, a degree of crystallinity between 2 and 10% was measured for PLGA13/87 and PLGA9/91, respectively. Obtaining this information is very relevant, since crystallinity is well-known to influence barrier properties of polymers. Furthermore, Tm of the same semicrystalline samples remained unaffected by the processing, but Tg decreased with respect to the unprocessed polymers (34.5 and 35.7 °C, respectively). High temperature and pressure during compression molding can induce thermal degradation of the polymer. For the amorphous copolymers, films showed a decrease in Tg during the first heating scan (Tg between 43 and 37 °C) compared to that of the unprocessed material.
Panels b and c of Figure 6 show the oxygen and water permeability, respectively, for copolymers with between 52 and 91 mol % glycolic acid content, at 30 °C and 70% RH. For comparison, a PLA film prepared with the same compression molding protocol was also tested. The results obtained for WP and OP for PLA match with those reported by Siró et al. for commercial PLA (Biophan 111) measured under similar conditions.53
PLA has a much higher OP than the other tested samples, and copolymerization with glycolide significantly improves the barrier to both oxygen and water vapor. PLGA films with between 52 and 72 mol % glycolic acid exhibit similar OP and WP values, with a slight decrease for those with increased glycolic acid content. This trend becomes more evident at the highest glycolic acid contents, with both OP and WP below 1.0. When compared to values for PGA reported by the Kureha Co. (OP = 0.013 mm·cm3·m–2·day–1·bar–1 at 20 °C and 80% RH; WP = 0.165 g·mm·m2·day–1·bar–1 at 40 °C and 90% RH), PLGA copolymers exhibit a higher transmission for both permeants. The negative effect of lactic acid introduction on the barrier properties was expected, since PLA alone has much higher permeability coefficients for water and oxygen than PGA.
The transport of a gas or vapor through a polymer film depends on both its solubility within the polymer matrix and the ability of the gas or vapor to diffuse though the matrix.54 It has been demonstrated before that crystallinity influences both the solubility and the diffusion coefficient. Crystalline regions tend to be much more dense and well-arranged than amorphous regions, which helps to prevent penetrant sorption and reduces penetrant solubility.55 This is in line with the much better WP of PLGA9/91 compared to the amorphous samples. The crystalline regions act as barriers to diffusion and force the water molecules to go through a more tortuous path compared to the more permeable amorphous regions. In the case of commercial PGA, the much higher crystallinity in comparison to the tested copolymers (45–55%) contributes to this trend, which leads to superior performance.
PLGA copolymers perform really well as barrier to oxygen and water as compared to PET, a common barrier polymer. PET films (10% degree of crystallinity) were prepared with compression molding and tested with the same protocol described here for PLGA films. They showed an OP of 4.5 mm·cm3·m–2·day–1·bar–1 and a WP of 0.83 g·mm·m–2·day–1·bar–1 at 30 °C and 70% RH. These values are higher than those reported for commercial PET, for which typically chain orientation is induced during the stretching process. This processing induces lower permeability coefficients. The non-oriented PET shows higher WP than the entire range of PLGA copolymers studied in this work. The OP, however, is improved in comparison to PLLA but only superior to PET for the samples with richer glycolide content. Since PET is a very high demand polymer in the packaging industry, the fact that PLGA copolymers, depending on the composition, have a comparable or improved barrier to oxygen and water suggests good potential for this application field. It is, however, important to consider that PET exhibits higher Tg (60–80 °C) and Tm (253–255 °C) than PLGA, which allows it to retain its properties over a wider temperature range. Furthermore, PET is known for its high mechanical strength and good flexibility. A commercial PET film (23 μm), for example, displays a tensile strength (σu) of 200 MPa, a tensile modulus (E) of 3.9 GPa and elongation of 30%.56 Less flexibility is displayed by PGA films (20 μm) with elongation of 40%, σu = 380 MPa and E = 7 GPa.12 When comparing both PET and PGA with a commercial PLA film (100 μm), higher elongation at break is reported (180%), but both σu (103 MPa) and E (3.44 GPa) are inferior.57 Although mechanical properties for the series of copolymers here studied have not been determined, behavior between that of PGA and PLA, depending on the lactide to glycolide ratio, should be expected.
Furthermore, PET, even in its partly bio-based form (Coca-Cola’s “plant-bottle” PET), is neither degradable nor compostable.2 Therefore, using PLGA copolymers (bio- and CO2-based and (bio)degradable) for applications such as packaging can avoid the infinite accumulation of plastics such as PET that leak into the environment (an estimated 8 million tons per year). In addition, composting of food wastes contaminated with (bio)degradable plastic packaging is a much better end-of-life option than (open air) burning, which is often the only solution for food contaminated nondegradable plastics in areas without a waste collection infrastructure.
Effect of Temperature and Relative Humidity on WP of Glycolic Acid Rich Copolymers
Barrier properties of plastics are affected by different factors, which include the following: fractional free volume, chemical structure, chain orientation, crystallinity, temperature, and relative humidity. This section of the study focuses on water permeability, because PLA and PGA are known to degrade preferentially through hydrolysis. PGA, particularly, has shown a faster degradation rate than PLA.58,59 Therefore, it is important to evaluate how the water barrier performance of glycolic acid rich copolymers is affected by different moisture conditions and temperature changes (close to the Tg). Figure 7 shows the water permeability for PLGA13/87 and PLGA9/91 vs the relative humidity (between 10 and 90%) at 30 and 40 °C.
Figure 7.

Water permeability vs relative humidity at 30 and 40 °C for (a) PLGA13/87 and (b) PLGA9/91.
As observed in Figure 7, the rate of moisture sorption is very dependent on temperature and relative humidity. As a general trend, more water is permeated through the film with increasing RH and temperature. For 30 °C this growth is gradual, and at the highest RH, the barrier to water is still very good. Clearly, an increase in temperature (closer to the Tg) is detrimental for barrier performance, and the increase in WP with increasing relative humidity is much more important. For PLGA13/87, for example, more than double the amount of water is permeated at 40 °C under equal RH conditions (above 10% RH) compared to 30 °C. PLGA9/91 also takes up more moisture at higher temperature.
For comparison, the thermo-compressed commercial PET was tested under the same temperature and RH conditions. At 40 °C and 90% RH it showed a WP of 1.8 g·mm·m–2·day–1·bar–1. When compared to the WP of PLGA13/87 (2.5 g·mm·m–2·day–1·bar–1) it is clear that, from a practical point of view, PET performs better at high temperatures. Evidently, PLGA13/87 does not perform well at a service life temperature close to or above 40 °C. In contrast, PLGA9/91 exhibits lower WP (0.87 g·mm·m–2·day–1·bar–1) than PET at the same conditions (1.8 g·mm·m2·day–1·bar–1), demonstrating that even at temperatures close to the Tg and at high moisture content, it performs better as a barrier to water.
Conclusions
While previous publications have focused on lactic acid rich PLGA copolymers, this study focused on mapping the structure–property relationships for glycolic acid rich PLGA copolymers. For this purpose, PLGA copolymers with between 50 and 91 mol % glycolide were synthesized by ROP.
The higher reactivity of glycolide compared to lactide resulted in slightly higher glycolide content than the feed ratio would suggest. An increased thermal stability was observed with increasing glycolic acid content, although the differences were small. The density increased with increasing glycolic acid content. In all cases, the densities were significantly higher than that of water (1.0).
The PLGA copolymers show significantly different crystallization behavior than both PLA and PGA homopolymers, which exhibit a semicrystalline structure as observed in DSC when heated and cooled at 10 °C·min–1. The samples with 87 and 91 mol % glycolic acid were semicrystalline, whereas those with 50–80% glycolide content were amorphous. A crystallization study under non-isothermal conditions revealed that copolymerization reduces the crystallization rate for PLGA copolymers almost 10 times compared to PLA and PGA homopolymers at 10 °C·min–1 cooling rate. This is an advantage vis-à-vis PGA for processing into, among others, films. The kinetics of this process, modeled with the Jeziorny-modified Avrami method, were in line with the more difficult crystallization for these copolymers. The crystallization rate decreased with decreasing cooling rates. Furthermore, the crystal morphology observed by polarization optical microscopy revealed spherulite-like crystals, the structure of which is more closely related to that of PLA.
Finally, the barrier property assessment revealed that increasing glycolic acid content has a beneficial effect on the barrier to both oxygen and water vapor. At room temperature and a relative humidity below 70%, these copolymers outperform non-oriented PET. At 40 °C, only PLGA9/91 remains less permeable than PET. In all cases, the water permeability was highly dependent on the temperature and relative humidity conditions.
Their great potential as barrier polymers and their possible future production from CO2, as well as the fact that these materials are (chemically) recyclable and biodegradable, make these materials very interesting for future applications in areas such as films for packaging. Currently, however, glycolide, the main monomer for the production of high GA content PLGA materials, is expensive and is produced at limited scale. It is for this reason that we are developing a cheaper route from CO2 in this European Ocean Program.
Acknowledgments
We thank the following people for their contribution to this research: Nils Leoné and Prof. Jules Harings of the Biobased Materials group at Maastricht University for their help carrying out the POM measurements; Julia Flaceliere of AVANTIUM Renewable Polymers for her guidance and help with carrying out the polymer density measurements. This project has received funding from the European Union’s Horizon 2020 research and innovation program under Grant Agreement No. 767798.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsapm.0c00315.
Detailed reaction conditions, NMR, TGA, DSC, and Totalperm (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Silva A.; Cardoso B.; Silva M.; Freitas R.; Sousa R. Synthesis, Characterization, and Study of PLGA Copolymer in Vitro Degradation. J. Biomater. Nanobiotechnol. 2015, 06, 8–19. 10.4236/jbnb.2015.61002. [DOI] [Google Scholar]
- Murcia Valderrama M. A.; van Putten R.-J.; Gruter G.-J. M. The Potential of Oxalic - and Glycolic Acid Based Polyesters (Review). Towards CO2 as a Feedstock (Carbon Capture and Utilization - CCU). Eur. Polym. J. 2019, 119, 445–468. 10.1016/j.eurpolymj.2019.07.036. [DOI] [Google Scholar]
- Kricheldorf H. Syntheses and Application of Polylactides. Chemosphere 2001, 43, 49–54. 10.1016/S0045-6535(00)00323-4. [DOI] [PubMed] [Google Scholar]
- Storey R.; Mullen B.; Desai G.; Sherman J.; Tang C. Soluble Tin(II) Macroinitiator Adducts for the Controlled Ring-Opening Polymerization of Lactones and Cyclic Carbonates. J. Polym. Sci., Part A: Polym. Chem. 2002, 40, 3434–3442. 10.1002/pola.10448. [DOI] [Google Scholar]
- Kaplan D. L.Biopolymers from Renewable Resources; Springer Science & Business Media, 2013. [Google Scholar]
- Ryner M.; Stridsberg K.; Albertsson A.-C.; von Schenck H.; Svensson M. Mechanism of Ring-Opening Polymerization of 1,5-Dioxepan-2-One and L-Lactide with Stannous 2-Ethylhexanoate. A Theoretical Study. Macromolecules 2001, 34 (12), 3877–3881. 10.1021/ma002096n. [DOI] [Google Scholar]
- Chu C. C. 11 - Materials for Absorbable and Nonabsorbable Surgical Sutures. In Biotextiles as Medical Implants 2013, 275–334. 10.1533/9780857095602.2.275. [DOI] [Google Scholar]
- Singh G.; Tanurajvir K.; Ravinder K.; Kaur A. Recent Biomedical Applications and Patents on Biodegradable Polymer-PLGA. Int. J. Pharmacol. Pharm. Sci. 2014, 1, 30–42. [Google Scholar]
- Plus Antibacterial Sutures Evidence Summary. Technical, Clinical, and Economic Data Supporting Triclosan-coated Sutures. https://www.jnjmedicaldevices.com/sites/default/files/user_uploaded_assets/pdf_assets/2019-06/103240-181127_Plus_EvidenceSummary_192_CA.pdf (accessed 2020-06-20).
- Van de Velde K.; Kiekens P. Biopolymers: Overview of Several Properties and Consequences on Their Applications. Polym. Test. 2002, 21 (4), 433–442. 10.1016/S0142-9418(01)00107-6. [DOI] [Google Scholar]
- Phuong V. T.; Coltelli M.-B.; Cinelli P.; Cifelli M.; Verstichel S.; Lazzeri A. Compatibilization and Property Enhancement of Poly(Lactic Acid)/Polycarbonate Blends through Triacetin-Mediated Interchange Reactions in the Melt. Polymer 2014, 55 (17), 4498–4513. 10.1016/j.polymer.2014.06.070. [DOI] [Google Scholar]
- About Kuredux|Kuredux; Kureha Corp., Tokyo, Japan, http://www.kuredux.com/en/about/index.html (accessed 2020-06-20).
- Wang N.; Wu X. S.; Li C.; Feng M. F. Synthesis, Characterization, Biodegradation, and Drug Delivery Application of Biodegradable Lactic/Glycolic Acid Polymers: I. Synthesis and Characterization. J. Biomater. Sci., Polym. Ed. 2000, 11 (3), 301–318. 10.1163/156856200743715. [DOI] [PubMed] [Google Scholar]
- D’Avila Carvalho Erbetta C.; Alves R. J.; Resende J. M.; Fernando de Souza Freitas R.; Geraldo de Sousa R. Synthesis and Characterization of Poly(D, L-Lactide-co-Glycolide) Copolymer. J. Biomater. Nanobiotechnol. 2012, 03, 208–225. 10.4236/jbnb.2012.32027. [DOI] [Google Scholar]
- Vargha-Butler E. I.; Kiss E.; Lam C. N. C.; Keresztes Z.; Kálmán E.; Zhang L.; Neumann A. W. Wettability of biodegradable surfaces. Colloid Polym. Sci. 2001, 279, 1160–1168. 10.1007/s003960100549. [DOI] [Google Scholar]
- Gilding D. K.; Reed A. M. Biodegradable Polymers for Use in Surgery—Polyglycolic/Poly(Actic Acid) Homo- and Copolymers: 1. Polymer 1979, 20 (12), 1459–1464. 10.1016/0032-3861(79)90009-0. [DOI] [Google Scholar]
- Gorrasi G.; Meduri A.; Rizzarelli P.; Carroccio S.; Curcuruto G.; Pellecchia C.; Pappalardo D. Preparation of Poly(Glycolide-Co-Lactide)s through a Green Process: Analysis of Structural, Thermal, and Barrier Properties. React. Funct. Polym. 2016, 109, 70–78. 10.1016/j.reactfunctpolym.2016.10.002. [DOI] [Google Scholar]
- Chen Y.; Yao X.; Gu Q.; Pan Z. Non-Isothermal Crystallization Kinetics of Poly (Lactic Acid)/Graphene Nanocomposites. J. Polym. Eng. 2013, 33, 163–171. 10.1515/polyeng-2012-0124. [DOI] [Google Scholar]
- Kawai T.; Rahman N.; Matsuba G.; Nishida K.; Kanaya T.; Nakano M.; Okamoto H.; Kawada J.; Usuki A.; Honma N.; Nakajima K.; Matsuda M. Crystallization and Melting Behavior of Poly (l-Lactic Acid). Macromolecules 2007, 40 (26), 9463–9469. 10.1021/ma070082c. [DOI] [Google Scholar]
- Liu Y.; Wang L.; He L.; Fan Z.; Li S.-M. Non-Isothermal Crystallization Kinetics of Poly(L-Lactide). Polym. Int. 2010, 59, 1616–1621. 10.1002/pi.2894. [DOI] [Google Scholar]
- Yu C.; Bao J.; Xie Q.; Shan G.; Bao Y.; Pan P. Crystallization Behavior and Crystalline Structural Changes of Poly(Glycolic Acid) Investigated: Via Temperature-Variable WAXD and FTIR Analysis. CrystEngComm 2016, 18, 7894–7902. 10.1039/C6CE01623E. [DOI] [Google Scholar]
- Chen H.; Ma C.; Bai W.; Chen D.; Xiong C. Isothermal Crystallization and Melting Behavior of Composites Composed of Poly(L-Lactic Acid) and Poly(Glycolic Acid) Fibers. J. Macromol. Sci., Part B: Phys. 2014, 53, 1715–1725. 10.1080/00222348.2014.898998. [DOI] [Google Scholar]
- Finnigan B.Barrier Polymers. In The Wiley Encyclopedia of Packaging Technology, 3rd ed.; John Wiley & Sons: New York, 2009; pp 305–309, 10.1002/9780470541395. [DOI] [Google Scholar]
- Karkhanis S. S.; Stark N. M.; Sabo R. C.; Matuana L. M. Water Vapor and Oxygen Barrier Properties of Extrusion-Blown Poly(Lactic Acid)/Cellulose Nanocrystals Nanocomposite Films. Composites, Part A 2018, 114, 204–211. 10.1016/j.compositesa.2018.08.025. [DOI] [Google Scholar]
- Sonchaeng U.; Iñiguez-Franco F.; Auras R.; Selke S.; Rubino M.; Lim L.-T. Poly(Lactic Acid) Mass Transfer Properties. Prog. Polym. Sci. 2018, 86, 85–121. 10.1016/j.progpolymsci.2018.06.008. [DOI] [Google Scholar]
- Arrieta M. P.; Fortunati E.; Dominici F.; Rayón E.; López J.; Kenny J. M. PLA-PHB/Cellulose Based Films: Mechanical, Barrier and Disintegration Properties. Polym. Degrad. Stab. 2014, 107, 139–149. 10.1016/j.polymdegradstab.2014.05.010. [DOI] [Google Scholar]
- Courgneau C.; Domenek S.; Lebossé R.; Guinault A.; Avérous L.; Ducruet V. Effect of Crystallization on Barrier Properties of Formulated Polylactide. Polym. Int. 2012, 61 (2), 180–189. 10.1002/pi.3167. [DOI] [Google Scholar]
- Colomines G.; Ducruet V.; Courgneau C.; Guinault A.; Domenek S. Barrier Properties of Poly(Lactic Acid) and Its Morphological Changes Induced by Aroma Compound Sorption. Polym. Int. 2010, 59 (6), 818–826. 10.1002/pi.2793. [DOI] [Google Scholar]
- Holm V. K.; Ndoni S.; Risbo J. The Stability of Poly(Lactic Acid) Packaging Films as Influenced by Humidity and Temperature. J. Food Sci. 2006, 71, E40–E44. 10.1111/j.1365-2621.2006.tb08895.x. [DOI] [Google Scholar]
- Lehermeier H. J.; Dorgan J. R.; Way J. D. Gas Permeation Properties of Poly(Lactic Acid). J. Membr. Sci. 2001, 190 (2), 243–251. 10.1016/S0376-7388(01)00446-X. [DOI] [Google Scholar]
- Auras R. A.; Singh S. P.; Singh J. J. Evaluation of Oriented Poly(Lactide) Polymers vs. Existing PET and Oriented PS for Fresh Food Service Containers. Packag. Technol. Sci. 2005, 18 (4), 207–216. 10.1002/pts.692. [DOI] [Google Scholar]
- Pandey K.; Antil R.; Saha S.; Jacob J.; Balavairavan B. Poly(Lactic Acid)/Thermoplastic Polyurethane/Wood Flour Composites: Evaluation of Morphology, Thermal, Mechanical and Biodegradation Properties. Mater. Res. Express 2019, 6 (12), 125306. 10.1088/2053-1591/ab5398. [DOI] [Google Scholar]
- Mullapudi S. S.; Pandey K.; Maiti S. N.; Saha S. PLA/EVA/Teak Wood Flour Biocomposites for Packaging Application: Evaluation of Mechanical Performance and Biodegradation Properties. J. Package Technol. Res. 2018, 2 (3), 191–201. 10.1007/s41783-018-0037-2. [DOI] [Google Scholar]
- Kawakami Y.; Sato N.; Hoshino M.; Kouyama T.; Shiiki Z.. Oriented Polyglycolic Acid Film and Production Process Thereof. US 5853639 A, Dec. 29, 1998.
- Hokari Y.; Yamane K.; Wakabayashi J.; Suzuki T.. Polyglycolic Acid Resin-Based Layered Sheet and Method of Producing the Same. US 20090081396 A1, Mar. 26, 2009.
- Yamane K.; Kato R.; Tobita H.. Method for Producing Multilayer Stretch-Molded Article. EP 1674240 B1, Dec. 17, 2008.
- Yamane K.; Sato H.; Ichikawa Y.; Sunagawa K.; Shigaki Y. Development of an Industrial Production Technology for High-Molecular-Weight Polyglycolic Acid. Polym. J. 2014, 46 (11), 769–775. 10.1038/pj.2014.69. [DOI] [Google Scholar]
- Kawakami Y.; Sato N.; Hoshino M.; Kouyama T.; Shiiki Z.. Polyglycolic Acid Sheet and Production Process Thereof. EP 0805176 A1, Nov. 5, 1997.
- Yamane K.; Miura H.; Ono T.; Nakajima J.; Itoh D.. Crystalline Polyglycolic Acid, Polyglycolic Acid Composition and Production Process Thereof. US 20030125508 A1, Jul. 3, 2003.
- Kaihara S.; Matsumura S.; Mikos A. G.; Fisher J. P. Synthesis of Poly(L-Lactide) and Polyglycolide by Ring-Opening Polymerization. Nat. Protoc. 2007, 2 (11), 2767–2771. 10.1038/nprot.2007.391. [DOI] [PubMed] [Google Scholar]
- Ayyoob M.; Lee D. H.; Kim J. H.; Nam S. W.; Kim Y. J. Synthesis of Poly(Glycolic Acids) via Solution Polycondensation and Investigation of Their Thermal Degradation Behaviors. Fibers Polym. 2017, 18 (3), 407–415. 10.1007/s12221-017-6889-1. [DOI] [Google Scholar]
- Avgoustakis K.; Nixon J. R. Biodegradable Controlled Release Tablets 1: Preparative Variables Affecting the Properties of Poly(Lactide-Co-Glycolide) Copolymers as Matrix Forming Material. Int. J. Pharm. 1991, 70 (1), 77–85. 10.1016/0378-5173(91)90166-L. [DOI] [Google Scholar]
- Grijpma D. W.; Nijenhuis A. J.; Pennings A. J. Synthesis and Hydrolytic Degradation Behaviour of High-Molecular-Weight l-Lactide and Glycolide Copolymers. Polymer 1990, 31 (11), 2201–2206. 10.1016/0032-3861(90)90096-H. [DOI] [Google Scholar]
- Chu C. C. Differential Scanning Calorimetric Study of the Crystallization Kinetics of Polyglycolic Acid at High Undercooling. Polymer 1980, 21 (12), 1480–1482. 10.1016/0032-3861(80)90153-6. [DOI] [Google Scholar]
- Fischer E. W.; Sterzel H. J.; Wegner G. Investigation of the Structure of Solution Grown Crystals of Lactide Copolymers by Means of Chemical Reactions. Kolloid Z. Z. Polym. 1973, 251 (11), 980–990. 10.1007/BF01498927. [DOI] [Google Scholar]
- Hay J. N.; Sabir M. Crystallization Kinetics of High Polymers. Polyethylene Oxide—Part II. Polymer 1969, 10, 203–211. 10.1016/0032-3861(69)90031-7. [DOI] [Google Scholar]
- Jeziorny A. Parameters Characterizing the Kinetics of the Non-Isothermal Crystallization of Poly(Ethylene Terephthalate) Determined by d.s.c. Polymer 1978, 19 (10), 1142–1144. 10.1016/0032-3861(78)90060-5. [DOI] [Google Scholar]
- Sun Z.; Wang X.; Guo F.; Jiang C.; Pan Q. Isothermal and Nonisothermal Crystallization Kinetics of Bio-Sourced Nylon 69. Chin. J. Chem. Eng. 2016, 24 (5), 638–645. 10.1016/j.cjche.2015.12.021. [DOI] [Google Scholar]
- Coburn N.; Douglas P.; Kaya D.; Gupta J.; McNally T. Isothermal and Non-Isothermal Crystallization Kinetics of Composites of Poly(Propylene) and MWCNTs. Advanced Industrial and Engineering Polymer Research 2018, 1 (1), 99–110. 10.1016/j.aiepr.2018.06.001. [DOI] [Google Scholar]
- Zeng Y.; Liu Y.; Wang L.; Huang H.; Zhang X.; Liu Y.; Min M.; Li Y. Effect of Silver Nanoparticles on the Microstructure, Non-Isothermal Crystallization Behavior and Antibacterial Activity of Polyoxymethylene. Polymers 2020, 12 (2), 424. 10.3390/polym12020424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courgneau C.; Ducruet V.; Avérous L.; Grenet J.; Domenek S. Nonisothermal Crystallization Kinetics of Poly(Lactide)—Effect of Plasticizers and Nucleating Agent. Polym. Eng. Sci. 2013, 53 (5), 1085–1098. 10.1002/pen.23357. [DOI] [Google Scholar]
- Li J.; Wang Y.; Wang X.; Wu D. Development of Polyoxymethylene/Polylactide Blends for a Potentially Biodegradable Material: Crystallization Kinetics, Lifespan Prediction, and Enzymatic Degradation Behavior. Polymers (Basel, Switz.) 2019, 11 (9), 1516. 10.3390/polym11091516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siró I.; Plackett D.; Sommer-Larsen P. A Comparative Study of Oxygen Transmission Rates through Polymer Films Based on Fluorescence Quenching. Packag. Technol. Sci. 2010, 23 (6), 301–315. 10.1002/pts.895. [DOI] [Google Scholar]
- Miller K. S.; Krochta J. M. Oxygen and Aroma Barrier Properties of Edible Films: A Review. Trends Food Sci. Technol. 1997, 8 (7), 228–237. 10.1016/S0924-2244(97)01051-0. [DOI] [Google Scholar]
- Dhoot S. N.; Freeman B. D.; Stewart M. E.. Barrier Polymers. Encyclopedia of Polymer Science and Technology; American Cancer Society, 2002; 10.1002/0471440264. [DOI] [Google Scholar]
- NIIR Board of Consultants & Engineers. Handbook on Pet Film and Sheets, Urethane Foams, Flexible Foams, Rigid Foams, Speciality Plastics, Stretch Blow Moulding, Injection Blow Moulding, Injection and Co-Injection Preform Technologies; Asia Pacific Business Press, 2018. [Google Scholar]
- Ford L.Ingeo Biopolymer 4032D Technical Data Sheet https://www.natureworksllc.com/~/media/Technical_Resources/Technical_Data_Sheets/TechnicalDataSheet_4032D_films_pdf.pdf (accessed 2020-5-10).
- Elsawy M. A.; Kim K.-H.; Park J.-W.; Deep A. Hydrolytic Degradation of Polylactic Acid (PLA) and Its Composites. Renewable Sustainable Energy Rev. 2017, 79, 1346–1352. 10.1016/j.rser.2017.05.143. [DOI] [Google Scholar]
- Williams D. F.; Mort E. Enzyme-Accelerated Hydrolysis of Polyglycolic Acid. J. Bioeng. 1977, 1 (3), 231–238. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.