
Fig. 10. Determination of stabilization of FBXL2 and IP3R3 using pulse-chase assay in neonatal cardiomyocytes from WT and FUNDC1−/− mice transfected with FUNDC1 or FBXL2 viral vector.
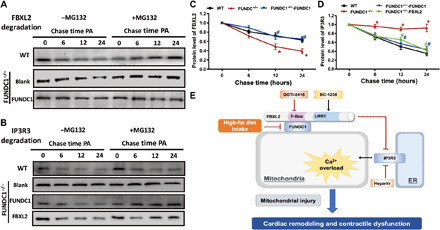
(A) Western blots analysis of palmitic acid (0.5 mM)–induced time-dependent FBXL2 degradation (0 to 24 hours) in neonatal cardiomyocytes from WT or FUNDC1−/− mice with or without FUNDC1 transfection. A cohort of cardiomyocytes was incubated with the proteasomal inhibitor MG132 throughout the course of study. (B) Western blots analysis of palmitic acid (0.5 mM)–induced time-dependent IP3R3 degradation (0 to 24 hours) in neonatal cardiomyocytes from WT or FUNDC1−/− mice with or without FUNDC1 or FBXL2 transfection. A cohort of cardiomyocytes was incubated with MG132 throughout the study. (C) Pooled data of FBXL2 degradation in the absence of MG132. (D) Pooled data of IP3R3 degradation in the absence of MG132. (E) Diagram depicting proposed mechanism for the role of FUNDC1 in HF diet–induced changes in cardiac remodeling and contractile defects. HF intake suppressed FUNDC1–mediated mitophagy, leading to disturbed interaction between FUNDC1 and FBXL2, thus prompting destabilization of FBXL2. FBXL2 loss facilitates IP3R3-mediated Ca2+ mobilization into mitochondria (mitochondrial Ca2+ overload) and RIP-mediated cell death, resulting in mitochondrial injury and cardiac geometric and functional anomalies.