
Figure 1.
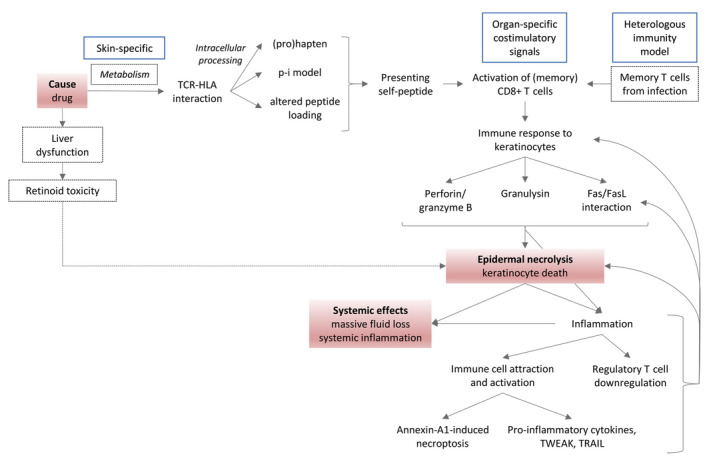
Integrated model of EN pathogenesis based on existing literature. The causal drug is thought to mediate a specific TCR‐HLA interaction involving a keratinocyte self‐peptide via the (pro)hapten, pharmacological interaction (p‐i) or altered peptide loading mechanism. (Memory) CD8 + T cells are activated and exhibit cytotoxicity against keratinocytes via the perforin/granzyme B and granulysin pathway and Fas/FasL interaction. In parallel, keratinocyte cytotoxicity can be induced by monocyte‐derived annexin A1 binding to the keratinocyte formyl peptide receptor 1. The subsequent keratinocyte death enhances local inflammation by attraction and activation of other immune cells producing pro‐inflammatory cytokines and death receptor ligands (TWEAK, TRAIL) and by downregulation of regulatory T cells. Local inflammation stimulates FasL expression on keratinocytes and perpetuation of keratinocyte cell death. Widespread epidermal necrolysis and inflammation can induce systemic effects. Skin‐specific drug metabolism, organ‐specific co‐stimulatory signals and local memory T cells from previous infections are thought to contribute to the skin‐specificity of this adverse reaction. An alternative to date less validated mechanism by which drugs could induce keratinocyte death is via liver dysfunction and retinoid toxicity. Red boxes: key events in pathogenesis; blue boxes: hypotheses on the localization of the adverse reaction in skin; dashed boxes/lines: hypotheses not (yet) generally accepted by EN research community.