

Abstract
Recent years have witnessed remarkable advances in radical reactions involving main‐group metal complexes. This includes the isolation and detailed characterization of main‐group metal radical compounds, but also the generation of highly reactive persistent or transient radical species. A rich arsenal of methods has been established that allows control over and exploitation of their unusual reactivity patterns. Thus, main‐group metal compounds have entered the field of selective bond formations in controlled radical reactions. Transformations that used to be the domain of late transition‐metal compounds have been realized, and unusual selectivities, high activities, as well as remarkable functional‐group tolerances have been reported. Recent findings demonstrate the potential of main‐group metal compounds to become standard tools of synthetic chemistry, catalysis, and materials science, when operating through radical pathways.
Keywords: bond formation, catalysis, main-group metals, organic and inorganic synthesis, radicals
Mainly main group: Remarkable progress has been made in the synthesis and investigation of main‐group metal complexes for radical reactions. This contribution highlights applications of such compounds for selective bond forming events in stoichiometric and catalytic reactions through radical pathways.

1. Introduction
The selective formation of new chemical bonds continues to be one of the major challenges in synthetic chemistry, especially when a high functional‐group tolerance is desired. For a long time, polar reactions between closed‐shell nucleophiles and electrophiles have been the main strategies for predictable bond‐forming events in organic and inorganic synthesis. In principle, radical reactions offer unique opportunities to generate a more versatile portfolio of reliable synthetic procedures that facilitate selective bond formations. This is due to the inherent characteristics of radical species, which are in certain respects complementary to the properties of reagents that follow polar reaction pathways: i) strongly polar functional groups may be tolerated,1, 2 ii) weakly polar functional groups can selectively be addressed in the presence of polar functional groups,3, 4 iii) active species can in many cases be generated by photochemical approaches,5 iv) different analytical techniques (most importantly: EPR spectroscopy) become available and are valuable tools in the identification of active species even in the presence of excess diamagnetic starting material(s), product(s), and by‐product(s).6 However, reliably inducing reactions along either polar or radical reaction pathways is not necessarily trivial. For instance, homolytic and heterolytic bond dissociation of a particular functional group can be very close in energy.7, 8, 9, 10 In addition, the polarity of the solvent can play a crucial role in favoring one or the other reaction profile, when polar or charged species are involved.11, 12 Thus, the development of strategies to generate radical species that follow predetermined reaction pathways remains an important challenge in synthetic chemistry.
The ability of transition‐metal complexes (especially those of 3 d metals) to engage in single‐electron‐transfer reactions was recognized early‐on and continues to generate valuable new synthetic protocols for radical reactions.13, 14, 15, 16, 17, 18, 19 In main‐group chemistry, research on radical reactions used to be largely focused on three different types of approaches. First, there was interest in alkali (and to a lesser extent alkaline‐earth) metals and their complexes for use as reducing agents.20, 21, 22, 23, 24, 25 Second, radical initiators such as azobis(isobutyronitrile) (AIBN) and benzoylperoxide (BPO) or stable radicals such as 2,2,6,6,‐tetramethyl‐piperidine‐N‐oxide (TEMPO) have been extensively exploited in organic synthesis and polymer chemistry.26 Third, tin compounds, especially [HSn(nBu)3] and [Sn(nBu)3]2, have found widespread use as H‐atom donors and mediators in radical chemistry.26
Continuous developments in radical chemistry during the last few decades have added important principles and new facets to this field of research. This includes the exploitation of the persistent‐radical effect (PRE),27, 28 progress in the understanding of solvent cages around pairs of radicals,29 the use of thiyl radicals in organic synthesis,30 the introduction of ligands such as cyclic (alkyl)(amino)carbenes to radical chemistry,31 and new perspectives on catalyzed radical reactions.32
With the above‐mentioned exceptions of s‐block reducing agents and toxic tin species,22, 23, 24, 25, 26 complexes of main‐group metals have traditionally played only a minor role in radical chemistry. Recent advances in main‐group chemistry have now changed this picture. Redox‐active ligands in the coordination sphere of main‐group metals have been investigated.33, 34, 35 Advances in the preparation and analysis of low‐valent and multiple‐bonded p‐block metal compounds have been achieved.36, 37, 38, 39 Access to species with weak M−M bonding has been forged,40 and unforeseen roles of classical bases or Grignard reagents in radical reactions have been uncovered.41 These developments have brought the complexes of s‐ and p‐block metals to the stage of selective bond formations through radical pathways.
In this contribution, recent advances in the radical chemistry of main‐group metal compounds42 that result in the selective formation of new chemical bonds are reviewed and put into perspective.
2. Strategies to Control Radical Reactions
A broad range of strategies—alone or in combination—have been applied for the exploitation of main‐group metal complexes in the context of selective bond formation in radical reactions. The most important examples are summarized in this chapter together with selected examples of how these strategies have been applied in practice.
2.1. Steric bulk
Steric bulk was key to success in the development of synthetic access to the first low‐valent main‐group metal complexes and has since then been established as a means to stabilize highly reactive compounds in this field of research.43 This strategy was again effective for the generation of species that pave the way for selective radical reactions with main‐group metal compounds. For instance, the sterically encumbered stannylene Sn(Ar)2 (1) allows access to the (thus far) non‐isolable radical species Sn(Ar)⋅ (2), which subsequently undergoes selective CH activation reactions with hydrocarbons (Scheme 1 a; Ar=2,6‐R2‐C6H3; R=2,6‐iPr2‐C6H3; also see section 3.4).44, 45 For the stannylene Sn((CH(SiMe3)2)2 (3) with its less bulky substituents, a different reaction pathway was observed, namely the formation of the SnIII radical species [Sn((CH(SiMe3)2)3]⋅ (4), along with further by‐products (Scheme 1 b).46, 47
Scheme 1.
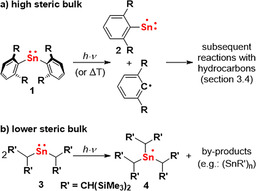
Impact of steric bulk on the reactivity of SnI radical species: a) subsequent reactions with hydrocarbons such as toluene (see section 3.4); R=2,6‐iPr2‐C6H3. b) formation of an SnIII radical (plus by‐products); R’=CH(SiMe3)2.
While the installation of bulky substituents around the metal center allowed for the generation of the detectable SnI radical 2, the same approach granted access to the first isolable bismuth radical compound: with a bulky diamide ligand, the BiII radical 5 was isolated and fully characterized (Scheme 2 a), setting the stage for investigations into the reactivity of well‐defined bismuth radical compounds (see section 3.5).48, 49 Notably, dimerization through Bi−Bi bond formation was observed when less bulky substituents were installed at the nitrogen atoms of a complex based on the same ligand platform (6→7; Scheme 2 b).50
Scheme 2.
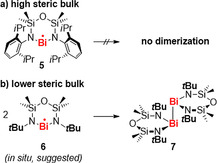
Fine‐tuning of the steric bulk around a bismuth(II) center leads to an isolable bismuth radical (a) or to the formation of a dibismuthane through Bi−Bi bond formation (b).
2.2. Delocalization of spin density in π‐electron systems
Radical species may be stabilized by delocalization of spin density in conjugated π‐electron systems.51 However, this strategy inherently generates multiple sites with considerable spin density, each of which acts as a potentially reactive site in subsequent transformations. A significantly higher spin density at a certain position may be sufficient to favor reactions at this site. Otherwise, additional approaches are necessary in order to realize selective reactions (e.g. combination with steric protection or chelation control, see sections 2.1, 2.6).
The spin‐density distribution of an unpaired electron in a delocalized π‐electron system can be affected by coordination to one or more metal atoms.52 This has been demonstrated, for instance, in persistent alkali‐metal aminotroponiminate (ATI) radicals, which were investigated by EPR spectroscopy and DFT calculations.53, 54 In the lithium compound 8, the spin density is predominantly localized at the C‐4 position of the ATI ligand backbone, which is simultaneously the only reactive site for subsequent C−C coupling (Scheme 3 a; also see section 3.1). Formal exchange of Li for Na gives radical 9, which shows a significantly altered spin‐density distribution with equal spin density in positions C‐4 (decreased from 0.36 to 0.29) and C‐7 (increased from 0.22 to 0.29; Scheme 3 b).53a, 54a Subsequent dimerization of 9 occurs exclusively at the C‐7 position, suggesting that phenomena such as aggregation/chelation control are also important when it comes to understanding selectivities of reactions with 9 (see sections 2.6, 3.1). Differences in spin‐density distributions of compounds such as 8 and 9 are ascribed to differences in the covalency and polarity of M−ligand bonding. These results exemplify that seemingly small differences such as the nature of the alkali metal acting as the central atom in a coordination entity can be decisive for the outcome of the reaction.
Scheme 3.
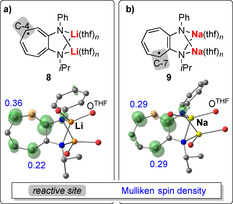
Lewis structures (top; one mesomeric form each) and Mulliken spin‐density plots (bottom) of aminotroponiminate (ATI) radical complexes of (a) lithium and (b) sodium. Reactive sites for dimerization are highlighted in grey; selected Mulliken spin densities are given in blue. Adapted from Ref. 53a and Ref. 54a with permission of The Royal Society of Chemistry and Wiley‐VCH, respectively.
In a related approach, the conjugated π‐electron system of a radical species (typically based on the elements C, N, O) is extended by a main‐group metal atom. That is, an empty or singly occupied metal‐centered orbital of sufficient energy and π‐symmetry overlaps with the orbitals of conjugated π‐electron system of the ligand framework. This allows for modulation of the spin density distribution by choice of the metal atom (and its additional supporting ligands, if present).
It has been shown that alkali‐metal radical complexes with diazadiene (DAD) ligands are isolable (compounds 10, 11; Scheme 4 a, top).55 EPR spectra of these compounds exhibit significant hyperfine coupling with the alkali metal (a(Li) of up to 20.30 mT and a(Na) of up to 3.50 mT). The incorporation of a metal‐centered orbital into the SOMO was demonstrated in the case of the lithium species 10 (Scheme 4 a, bottom). The same DAD ligand was also used for the synthesis of a [Ga2(DAD)2]⋅− radical anion (12, Scheme 4 b).56 EPR spectra of this compound suggest a significant spin density at the DAD ligands and the metal atoms.
Scheme 4.
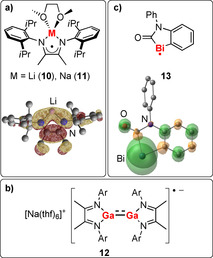
a) Lewis structures of alkali‐metal diazadiene radical compounds 10 and 11 (top; one mesomeric structure) and SOMO of the dme‐free lithium derivative [Li(DAD)] (bottom). b) Dinuclear gallium diazadiene radical complex 12 (Ar=2,6‐iPr2‐C6H3). c) Lewis structure (top; one mesomeric structure) and Mulliken spin‐density plot (bottom) of electrochemically generated bismuth radical compound 13. Adapted from Ref. 55 and Ref. 57 with permission of Wiley‐VCH and The Royal Society of Chemistry, respectively.
The construction of delocalized π‐electron systems from light elements (C, N) and heavy elements such as bismuth may be hampered by differences in size and energy of the relevant types of atomic orbitals. Nevertheless, DFT calculations on the electrochemically generated bismuth radical 13 show that the delocalization of spin density in molecular orbitals with contributions of both, light and heavy elements, is possible (Scheme 4 c).57 Although a large share of the spin density is localized on the bismuth atom (74 %), the lighter atoms C, N, and O also bear a significant amount of spin density (up to 5 % at an individual atom).
2.3. Redox‐active ligands
The use of redox‐active ligands in the coordination sphere of main‐group metals has previously been reviewed under different perspectives.33, 34, 35 The use of redox‐active ligands to control selective bond formations is in most cases closely related to the strategy described in section 2.2, in which the localization of spin density at certain positions of a delocalized π‐electron system is important and allows for bond formations at these positions. In this section, however, the focus is on redox‐active ligands changing their oxidation state while a bond formation takes place through radical pathways. These bond formations do not necessarily proceed at the position of highest spin density, and the exact spin‐density distribution within the redox‐active ligand is less important than the fact that it changes its overall oxidation state.
An example is given by a magnesium complex of a 1,2‐bis(arylimino)acenaphthene (BIAN) ligand, which can exist in a neutral diamagnetic, monoanionic radical, and a dianionic diamagnetic form. In the starting material 14, the ligand adopts its dianionic diamagnetic form, but is protonated resulting in an overall singly negative charge for the ligand (Scheme 5). Addition of benzophenone gives the insertion product 15 with release of H2. The BIAN ligand in 15 has adopted its monoanionic radical character. Thus, the C−C (and H−H) bond formation in the coordination sphere of magnesium is facilitated by a change in the oxidation state of the redox‐active ligand.
Scheme 5.
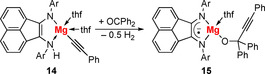
C−C coupling with concomitant release of H2 in the coordination sphere of Mg, facilitated by a redox‐active BIAN ligand (Ar=2,6‐iPr2‐C6H3).
2.4. Radicals from reversible M−M bond cleavage
In general, covalent bonds between heavy main‐group elements are relatively weak and are thus easily susceptible to homolytic dissociation. At the same time, the resulting metal‐centered radicals are stable enough to make radical recombination a feasible or even dominant reaction pathway. Taken together, this grants access to reversible homolytic bond dissociation reactions under mild reaction conditions, which is a highly attractive scenario for catalytic applications.
Recently, it was demonstrated that even Sn≡Sn triple bonds undergo reversible homolytic bond dissociation: in the temperature range of 25 to 94 °C, the distannyne [Sn(AriPr4)]2 ([2]2) is in equilibrium with the SnI radical Sn(AriPr4)⋅ (2) (Scheme 6 a).58 A reaction free enthalpy of ΔGdiss=7.84 kcal mol−1 at T=298 K was experimentally determined for this process using a van t'Hoff plot. A similar situation has been reported for the distibane 16‐Sb and the dibismuthane 16‐Bi, both bearing a sterically demanding ligand platform (Scheme 6 b).59 Considerable concentrations of the radical species 17‐Sb and 17‐Bi were detected at ambient temperature due to dissociation of 16‐Sb and 16‐Bi. Free reaction enthalpies of ΔGdiss=−0.69 and ΔGdiss=1.93 kcal mol−1 at T=298 K were associated with these homolytic bond dissociation reactions.60 The larger value for 16‐Bi was ascribed to larger Bi−C and Bi−Bi bond lengths, which decrease steric repulsion between the SiMe3 groups compared to those of 16‐Sb.
Scheme 6.
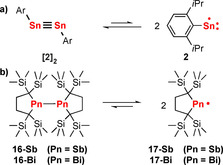
Generation of radical species from reversible M−M bond cleavage as reported for a distannyne (a) as well as a distibane and a dibismuthane (b).
2.5. Thermodynamic control of product formation
The homolytic bond‐dissociation energy of C−C single bonds in alkyl, vinyl, allyl, and aryl hydrocarbons is approximately in the range of 73–118 kcal mol−1.61 These values can be decreased significantly by destabilization of the diamagnetic species (e.g. through steric repulsion) or by stabilization of the radical (e.g. through delocalization of spin density). If conditions for the reversible homolytic bond dissociation of C−C bonds can be realized, this can be exploited for thermodynamic control of product formation in radical reactions.
In the reductive C−C coupling of guaiazulene (18) with elemental calcium, a mixture of the regioisomers 19 and 20 was initially obtained in a 60:40 ratio (Scheme 7 a).62 This ratio was modified by running the reaction at different temperatures.
Scheme 7.
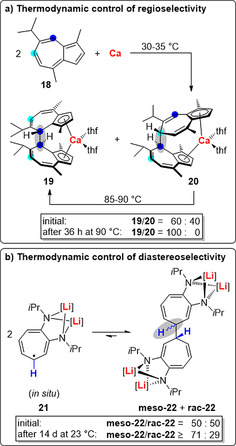
Thermodynamic control in (radical) C−C coupling reactions in order to address problems of regioselectivity (a) and diastereoselectivity (b). One mesomeric structure is shown for 21. [Li]=Li(thf)n.
Importantly, pure 20 could be fully converted to 19 (plus up to 10 % side products) by heating to 85–90 °C for 36 h. This is an example of thermodynamic control of regioselectivity through reversible C−C bond formation.63, 64 Furthermore, the same strategy also provides control over the diastereoselectivity of radical C−C coupling reactions. This was exemplified, for instance, by the dimerization of 21, which was generated in situ and detected by EPR spectroscopy (Scheme 7 b).54a Initially a 1:1 mixture of the meso‐ and the rac‐product (meso ‐22 and rac ‐22) was obtained. Crystallization from this mixture over a period of 14 d gave meso ‐22 in 71 % yield of isolated material. This indicated the slow conversion of rac ‐22 into meso ‐22, which was also observed by NMR and EPR spectroscopy in independent experiments. The free reaction enthalpy of the dimerization of alkali‐metal aminotroponiminate radicals such as 21 is sensitive to the choice of alkali metal and the nature of the substituents at N (Li vs. Na and iPr vs. Ph favor equilibrium conditions).
2.6. Control of radical reactions through chelation and aggregation effects
Salt‐free reduction of the dinuclear, dicationic germanium(II) compound 23 results in highly selective C−C bond formation through a (di‐)radical pathway and gives 24 after aqueous work‐up (Scheme 8).65 The dicationic diradical intermediate 23‐int‐1 was suggested based on EPR spectroscopic results. Intermediate 23‐int‐2 was isolated. The cis‐selectivity in the formation of 24 is unusual. In addition, reduction of the metal‐free di‐imine that is part of 23 by reducing agents such as Zn, Mn, or K did not give considerable yields of 24. These observations were ascribed to the germanium centers i) promoting reduction by Lewis acid‐activation of the di‐imine and ii) influencing the course of the reaction by chelation control, as suggested by the isolation of 23‐int‐2.
Scheme 8.
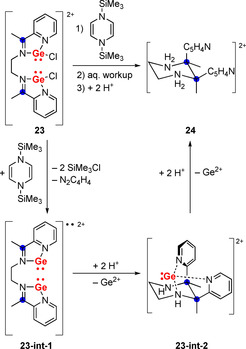
Selective radical C−C bond formation to give dicationic piperazine derivative 24. Positive charges are balanced by [O3SCF3]− counteranions.
Aggregation effects were suggested to also play a role in the dimerization of aminotroponiminate radical 9 (Scheme 3 b), because the spin‐density distribution alone (with equal spin density at two different positions) cannot account for the extremely high regioselectivity of this reaction.53a, 54a
2.7. Magnetic coupling through the main‐group metal atom
When two or more radical ligands coordinate to one metal center, magnetic coupling between the unpaired spins is possible. Thus, three situations can be envisaged: i) no coupling between the electrons, leading to two independent monoradicals; ii) ferromagnetic coupling leading to a triplet biradical; iii) antiferromagnetic coupling leading to a singlet biradical. All three situations can be expected to result in different physical and chemical properties of the compound. In other words, the reactivity of a complex containing two radical ligands can be controlled by fine‐tuning of the magnetic coupling between the unpaired spins.
Iminopyridine (IP) ligands, for instance, can adopt a neutral, a radical anionic, and a dianionic form. One‐electron reduction of AlCl(IP−)2 (25‐Cl) did not allow for the isolation of the targeted species Al(IP−)(IP2−) (26) with one mono‐ and one dianionic IP ligand (Scheme 9).66 Although the in situ formation of 26 was assumed, the product of a selective dimerization through C−C bond formation, [(IP2−)Al‐μ2‐(IP−IP)2−−Al(IP2−)], was isolated. It is important to note that compounds AlX(IP−)2 (25‐X) contain two IP ligands in their radical form, IP−, but are isolable and no dimerization of IP− radicals could be detected in these compounds (X=Cl, SO3CF3). In 25‐X, antiferromagnetic coupling of unpaired electrons through the aluminum center takes place as demonstrated by magnetic susceptibility and EPR spectroscopic measurements. This reduces the effective spin density at the ligands and was suggested as a reason for 25‐X being stable towards dimerization.
Scheme 9.
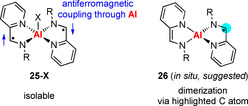
Aluminum complexes with two (left) or one (right) redox‐active iminopyridine (IP) ligand in its radical form. X=Cl, SO3CF3; R=2,6‐iPr2‐C6H3.
2.8. Radical reactions of frustrated Lewis pairs (FLPs)
Until recently, frustrated Lewis pairs (FLPs) were exclusively associated with polar reaction mechanisms due to the obvious assumption that the Lewis base would donate an electron pair to a substrate whereas the Lewis acid would accept an electron pair from the substrate.67 It has recently been demonstrated, however, that FLPs can enter radical reaction pathways, when the redox potentials of its two components are properly aligned. Thus, fine‐tuning of the redox‐potentials of FLPs is a valuable tool to selectively induce radical or polar mechanisms in the activation of substrates, which may ultimately lead to different outcomes of the reaction.68, 69, 70
For example, the reaction of the FLP P(tBu)3/2 Al(C6F5)3 with HSn(nBu)3 follows the expected polar pathway and yields the salt [(tBu)3P−Sn(nBu)3][μ2‐H‐(Al(C6F5)3)2] (Scheme 10 a).68 In contrast, the FLP P(Mes)3/Al(C6F5)3 gives strongly colored toluene solutions of the radical ion pair [P(Mes)3]⋅+[Al(C6F5)3]⋅−, as demonstrated by EPR spectroscopic detection of [P(Mes)3]⋅+. In the presence of an additional equivalent of Al(C6F5)3, reaction with the same substrate HSn(nBu)3 gave completely different products, namely [Sn(nBu)3]2 and the ion pair [HP(tBu)3][μ2‐H‐(Al(C6F5)3)2] (Scheme 10 b).
Scheme 10.
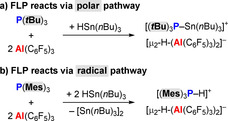
Reactions of frustrated Lewis pairs (FLPs) with HSn(nBu)3 through (a) polar or (b) radical pathways.
3. Selective Bond Formation Through Radical Pathways Aided by Main‐Group Metal Complexes
This section highlights examples of selective bond formations through radical pathways, in which main‐group metal compounds play a crucial role as starting materials, initiators, mediators or catalysts. It is structured according to the periodic table of the elements. The focus is on contributions, in which the participation of radical species was not only postulated, but also supported by experimental evidence.
3.1. Group 1 compounds
It has been demonstrated that alkali‐metal bases (especially KOtBu) can be key reagents in C−C coupling reactions through a homolytic aromatic substitution mechanism. Given that this area of research has lately been reviewed,41 only more recent results related to this field are included here. The KOtBu‐mediated coupling of aryl halides with benzene was known to be very sensitive to the presence of neutral donor ligands.71 Now it has been demonstrated that benzophenone and KOtBu undergo visible‐light‐induced radical chemistry.72 KOtBu and Ph2O form a complex which, after photoexcitation with visible light, yields a ketyl radical. This can be exploited for subsequent reactions: under photochemical conditions, coupling of 4‐iodo‐toluene with benzene to give 27 is strongly enhanced by the additive Ph2CO (Scheme 11). No such reactivity was observed for NaOtBu, which was ascribed to the complex of NaOtBu with Ph2CO showing absorption bands at lower wavelengths (318, 322 nm) according to time‐dependent DFT calculations. This is a rare example of rationalizing differences in the radical chemistry of alkali‐metal complexes, which (somewhat counterintuitively) have been reported to be quite dramatic in certain cases.54, 72
Scheme 11.
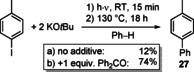
Photochemically induced formation of a ketyl radical, facilitating the C−C coupling of 4‐iodo‐toluene with benzene to give 27.
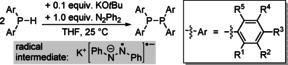
KOtBu (and to a lesser extent also LiOtBu, NaOtBu, and NaH) are effective catalysts for the dehydrogenative homo‐ and heterocoupling of phosphanes.73 Stoichiometric amounts of hydrogen acceptors such as benzophenone, azobenzene, trans‐stilbene, or N‐benzylidene‐aniline are necessary to facilitate these reactions. The acceptors dictate the mechanism of the transformations, and radical pathways were suggested for azobenzene, for which the radical K(N2Ph2)⋅ was detected in the course of P−P coupling reactions. The scope of P−P coupling with secondary phosphanes and azobenzene as a hydrogen acceptor is shown in Table 1. Primary phosphanes H2PPh and H2PCy2 give cyclo‐(PPh)5 and cyclo‐(PCy)4, respectively (Cy=cyclohexyl). The hetero dehydrogenative coupling of one primary and one secondary phosphane with an amine, an alcohol, and a thiol was realized with stoichiometric amounts of KOtBu and hydrazobenzene, (HNPh)2, as a hydrogen acceptor. The coupling products (PhHN)PPh2, (tBuO)2PPh, and (4‐Me‐C6H4‐S)2PPh, were obtained in 45–59 % yield.
Table 1.
KOtBu‐catalyzed dehydrogenative homocoupling of secondary phosphanes to the corresponding diphosphanes with N2Ph2 as a hydrogen acceptor.
|
| ||||||
|---|---|---|---|---|---|---|
|
Entry |
R1 |
R2 |
R3 |
R4 |
R5 |
Yield [%] |
|
|
|
|
|
|
|
|
|
1 |
H |
H |
H |
H |
H |
75 |
|
2 |
H |
H |
OMe |
H |
H |
40 |
|
3 |
H |
H |
F |
H |
H |
78 |
|
4 |
H |
H |
Cl |
H |
H |
87 |
|
5 |
OMe |
H |
H |
H |
H |
47 |
|
6 |
Me |
H |
H |
H |
H |
44 |
|
7 |
H |
Me |
H |
Me |
H |
72 |
|
8 |
Me |
H |
Me |
H |
Me |
59 |
Radicals obtained from the one‐electron reduction of aromatic systems can undergo reversible and selective dimerization reactions. In the case of alkali‐metal aminotroponiminates, it has been demonstrated that the choice of the alkali metal (Li vs. Na vs. K) and the substation pattern at the (hetero‐)aromatic core has a strong influence on regio‐ and diastereoselectivity.53, 54 This has been rationalized by analysis of spin density distributions in the radicals and the thermodynamic parameters of the dimerization (see sections 2.2, Scheme 3, and 2.4, Scheme 6 b).
Mixed‐metal compounds, containing an alkali metal and a second less electropositive metal, are little explored in radical chemistry to date. A Na/Zn compound, (TMEDA)Na(μ‐TMP)(μ‐tBu)Zn(tBu)] (28) has been reported to react with chalcone (29) in a C−C coupling reaction to give the tetranuclear compound 30 (Scheme 12; TMP=2,2,6,6‐tetramethylpiperidide). Based on experimental observations including reactivity studies of 28 with TEMPO, the formation of product 30 has been suggested to proceed though radical pathways.74
Scheme 12.
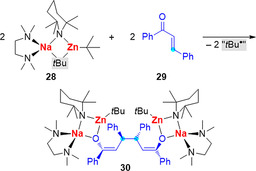
Mixed‐metal sodium zincate 28 reacts with chalcone 29 to give 30 through C−C coupling, which is suggested to proceed through radical pathways.
3.2. Group 2 compounds
Grignard reagents have been utilized for transition‐metal‐free aryl–aryl cross coupling reactions with aryl bromides and iodides (Scheme 13).41, 75 Although a mechanism involving a free aryl radical derived from the aryl halide, (ArR2)⋅, was discussed initially, aryl halide radical anions, (X−ArR2)⋅−, and biaryl radical anions, (ArR1−ArR2)⋅−, are currently assumed to be crucial intermediates.76 This conclusion was based on control reactions with radical‐clock substrates and with species that are known to act as sources of aryl radicals, such as phenylazo(triphenyl)methane. Even though a broad substrate scope is covered with this method,75 the yields of problematic substrates can be increased by addition of aryl radicals such as Li[4‐tBu‐C6H4]2 or in situ generated aminotroponiminate species.53a, 75
Scheme 13.
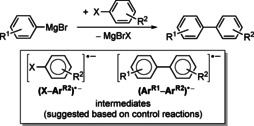
Cross coupling of aryl Grignard reagents with aryl halides (top) and suggested radical intermediates (bottom). X=Br, I.
The simple magnesium amide compound [Mg(N(SiMe3)2)2(thf)2] is catalytically active in Si−O bond formation between TEMPO and silanes with concomitant release of dihydrogen (Scheme 14 a).77 Mechanistically, the formation of a magnesium tempoxide, MgX(OTEMP), is suggested, followed by σ‐bond metathesis with SiPhH3 to give product 31 and a magnesium hydride, MgXH (X=monoanionic ligand; Scheme 14 b). Reaction of MgXH with TEMPO through single‐electron transfer would generate H2 and MgX(OTEMP), thereby closing the catalytic cycle. With 10 mol % catalyst loading, high yields of 77–99 % were obtained for the silanes SiPhH3, SiPh2H2, and SiPhMeH2 at 60–80 °C, although long reaction times (1–6 d) were required. A magnesium complex of the redox‐active 1,2‐bis(arylimino)acenaphthene (BIAN) ligand has also been reported to undergo single‐electron‐transfer reactions with TEMPO.78 In this case, the spin density is primarily delocalized in the ligand framework. Related magnesium BIAN complexes also undergo selective radical transformations such as ketone insertion with concomitant H2 release and pinacol coupling (see Scheme 5, section 2.3).79, 80
Scheme 14.
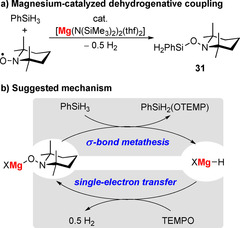
a) Dehydrogenative coupling of SiPhH3 with TEMPO, catalyzed by 10 mol % of [Mg(N(SiMe3)2)2(thf)2]. b) Suggested mechanism for the reaction shown in a); X=monoanionic ligand.
The generation of radical compounds from organometallic Group 2 complexes has also been reported for the heavier congener calcium. The calcoacene compound 32 undergoes homolytic C−C bond cleavage in solution to give radical 33 (Scheme 15 a).64, 81 Reactions of 32 with electrophiles such as ZrCl4 or SiMe3Cl lead to compounds of type 36 with newly formed C−E bonds (E=Zr, as in 36; Si). This has been ascribed to the disproportionation of radical 33 into neutral acene 34 and 35 with a dianionic acenyl ligand. Calcium guaiazulene complexes 19 and 20 are structurally related to 32 and also show reversible C−C bond cleavage/formation (see section 2.5, Scheme 7 a).62 The isolable CaI compound 37 undergoes radical CH activation with toluene to give the calcium benzyl complex 38, which is also accessible through salt metathesis (Scheme 15 b).82, 83 EPR spectroscopic reaction monitoring indicated the presence of an organic radical with an almost constant concentration, suggesting that the reduction equivalents for the generation of H2 and new Ca−C bonds originate from the CaI atoms. Recently, a dinuclear calcium hydride complex, [Ca‐μ2‐H(nacnac)]2, has been reported to react with naphthalene to give H2 and a trinuclear complex featuring the twofold reduced naphthalide as a bridging ligand, [Ca3‐μ3‐C10H8‐μ2‐H], demonstrating the strong reducing potency of the calcium hydride (nacnac=HC{(Me)CN‐2,6‐iPr2‐C6H3}2).84 EPR spectroscopic reaction monitoring indicated the [C10H8]⋅− radical to act as an intermediate in this reaction. Similar reactivities have been reported for related polyaromatic hydrocarbons.
Scheme 15.
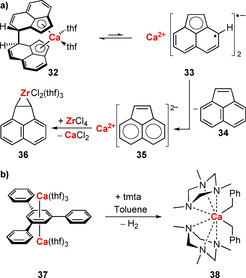
a) Equilibrium of homolytic C−C bond formation/cleavage in a calcium complex and reactivity of related compounds. b) Radical CH activation of toluene with a calcium(I) species. tmta=1,3,5‐trimethyl‐1,3,5‐triazinane.
3.3. Group 13 compounds
Indium metal‐mediated reactions in aqueous media have been used for the alkylation of electron‐poor unsaturated substrates such as imines and electron‐deficient alkenes, for pinacol couplings, and for the reductive coupling of alkyl halides. In addition, cyclization reactions and the exchange of halide atoms in substrates such as α‐halo‐carbonyl compounds for allyl, vinyl, and alkynyl groups by use of GaIII, In0, InI, and InIII reagents have been reported. These reactions have recently been reviewed and are not described here in detail.85, 86, 87, 88
In the search for less toxic alternatives to SnH(nBu)3 for use as reagents in radical reactions, halides of Group 13 metals have been in the focus of research efforts. In these approaches, MCl3 is reacted with a hydride source, which is suggested to generate a hydride species such as MHCl2 (M=Ga, In; Scheme 16). In some cases, a radical initiator such as BEt3 is also added in catalytic amounts. Thus, the combination of catalytic amounts of InCl3 with stoichiometric amounts of NaBH4 proved to be effective in the hydrodehalogenation of organo halides (Scheme 16 a).89 Alkyl bromides and iodides as well as aryl iodides could be reduced with high yields, whereas alkyl chlorides did not react. The radical nature of the reactions was established by using substrates with radical‐clock functionalities, for example compounds susceptible to radical cyclization reactions. In closely related reactions (e.g. the hydrodehalogenation of halo alkanes and the radical cyclization of halo acetals (halide=Br, I)), high yields were obtained with two different mixtures: i) GaCl3 (2 equiv)/ Red‐Al (1 equiv)/BEt3 (0.2 equiv) or ii) InCl3 (1.2 equiv)/AlH(iBu)2 (1.1 equiv)/BEt3 (0.1 equiv) (Red‐Al=Na[AlH2(OC2H4OMe)2].90, 91 For the hydroindation of alkynes and the radical cyclization of enynes, InCl3 (2 equiv) plus SiHEt3 (2 equiv) with catalytic amounts of BEt3 (0.1 equiv) was found to be an effective combination of reagents (Scheme 16 b–iv).92 NaBH4 had to be avoided in these cases because it liberates BH3 in the course of the reaction, which hydroborates alkynes in a side reaction.
Scheme 16.
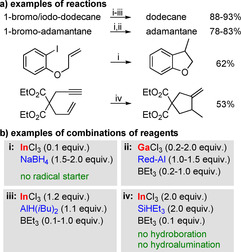
a) Reactions facilitated by in situ‐generated Group 13 hydride species and b) combination of regents used for their generation.
Selective bond formations have also been observed for Group 13 metal complexes with redox‐active ligands. Examples of selective C−C coupling involving an aluminum iminopyridine complex have been outlined above (see section 2.7, Scheme 9). A range of Group 13 BIAN complexes with the ligand in its radical monoanionic form have been reported (BIAN=1,2‐bis(arylimino)acenaphthene; for a Mg BIAN complex see Scheme 5). The compounds undergo selective reactions such as M−H, M−C, M−O, and M−F bond formations (M=Al, Ga).93, 94, 95 This occurs along with the activation of C−H (alkynes), Si−H (silanes), N−O (N2O), C−F (C6F5 groups), and P−F (PF6 −) bonds, as well as C−C multiple bonds. Furthermore, they can be applied as initiators for the polymerization of ϵ‐caprolactone.95 It has to be noted, however, that some of these reactions may well follow polar reaction pathways with the radical ligand acting as a spectator ligand or with the ligand changing its oxidation state without single‐electron‐transfer.
Reactions of FLPs such as P(Mes)3/Al(C6F5)3 react through radical pathways with HSn(nBu)3, resulting in Sn−Sn bond formation as discussed in detail in section 2.8 (Scheme 10).68
The formation of M−M bonds was investigated by addition of chelating bipy ligands to M[Al(ORF)4] (39‐M) (bipy=2,2’‐bipyridine; M=Ga, In; RF=C(CF3)3).96 While disproportionation of MI to M0 and MIII was observed in the case of M=Ga, the formation of cationic tri‐ and tetranuclear clusters such as [In3(bipy)6]3+ (403+) and [In4(bipy)6]4+ was reported for M=In. DFT calculations suggested that cluster formation proceeds via the triplet states of mononuclear building blocks, with the formation of relatively strong In−In bonds as an important driving force (Scheme 17).
Scheme 17.
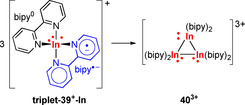
Formation of In−In bonds in the trimerization of 39+‐In in its triplet state to give 403+. Positive charges are balanced by [Al(OC(CF3)3)4]− anions.
3.4. Group 14 compounds
The Lewis pair formation between GeII complex 41 and B(C6F5)3 to give 43 has been reported to proceed through single‐electron‐transfer (SET) steps (Scheme 18).97 In a first step, the radicals 42‐HfIV and [B(C6F5)3]⋅− are formed. The second SET is delayed by an equilibrium between the redox‐isomers 42‐HfIV and 42‐HfIII, the latter of which does not undergo SET with [B(C6F5)3]⋅−. Thus, SET may not only be relevant in the chemistry of FLPs,68 but also in seemingly simple Lewis pair formations.
Scheme 18.
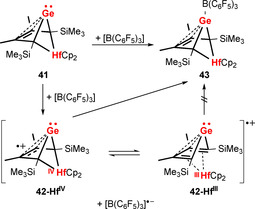
Lewis pair formation between 41 and B(C6F5)3 proceeding through single‐electron‐transfer steps.
Chelation control has been exploited in C−C bond formations in the coordination sphere of germanium for the synthesis of piperazine derivatives, as detailed in section 2.6 (Scheme 8).65 In addition, pinacol coupling has also been reported, starting from a redox‐active MIAN ligand (44) and Sn0 to give SnIV compound 45 (Scheme 19).98 The C−C coupling has been suggested to be reversible based on i) lower yields of 45 being obtained at elevated temperatures and ii) DFT calculations showing that the product of C−C bond cleavage (46) is energetically less favorable than 45 by only 5 kcal mol−1.
Scheme 19.
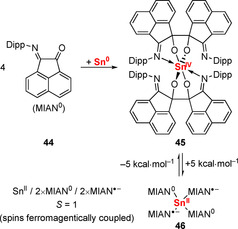
Reversible pinacol coupling of redox‐active MIAN ligand 44 in the coordination sphere of tin; Dipp=2,6‐iPr2‐C6H3.
Macrocyclic corrole ligands have been shown to stabilize germanium–TEMPO complex fragments.99, 100 Under photochemical conditions, Ge−O homolysis allows for the activation of N−H bonds of NH3, primary, and secondary amines, resulting in the formation of TEMPO−H and compounds of type [Ge(NR2)corrole] (R/R=H/H, H/alkyl, H/aryl, alkyl/alkyl). Proton‐coupled electron transfer has been suggested as a mechanistic pathway.
It has been outlined in section 2.1 that heating or irradiation of the stannylene Sn(Ar)2 (1) generates the radical species Sn(Ar)⋅ (2) (see Scheme 1; Ar=2,6‐R2‐C6H3; R=2,6‐iPr2‐C6H3). This radical reaction pathway has been suggested to be responsible for the generation of compounds 47, which are obtained in 43–62 % yield after heating 1 in an excess of toluene, m‐xylene, or mesitylene (Scheme 20). This is an extension of CH activation reactions with reactive species such as ”(MX2)2PhI“ which are generated in situ from MX2 and PhI, for which a radical pathway has been proposed (M=Ge, Sn; X=CH(SiMe3)2, N(SiMe3)2; or X2=[C(SiMe3)2CH2]2, [N(tBu)CH]2).101, 102
Scheme 20.
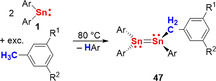
CH activation of toluene (R1/R2=H/H), m‐xylene (R1/R2=H/Me), and mesitylene (R1/R2=Me/Me) by compound 1 through radical intermediate Sn(Ar)⋅ (2); Ar=2,6‐R2‐C6H3; R=2,6‐iPr2‐C6H3; see Scheme 1.
3.5. Group 15 compounds
Only a small number of persistent and isolable antimony and bismuth radicals are known, including charged isolable species in the case of antimony (Schemes 2, 6b, 20a).48, 49, 59, 103, 104, 105 Most of these complexes have only recently been reported, and steric protection has been exploited as a strategy for the stabilization of these otherwise highly reactive compounds. Stoichiometric reactivity studies on the bismuth radicals 5 and 52 with P4 and S8 demonstrated the reversible activation of P−P bonds (5⇌53, Scheme 21 b)50 and the formation of unusual π*−π* (SOMO/SOMO) interactions of sulfur‐centered radicals in the solid state (54, Scheme 21 b).106 The reduction of a BiIII pyridine dipyrrolide complex, [Bi]I(thf)2, with organic reductants such as (CMe)4(NSiMe3)2 led to the ring opening of THF solvent molecules to give [Bi]−C4H8OSiMe3 ([Bi]=2,6‐((3‐Ph)(5‐Mes)C4HN)‐C5H3N).107 This reaction was suggested to proceed through radical pathways based on trapping experiments with TEMPO. Further examples of stoichiometric reactions with C−C, E−H, E−C, E−O, and Bi−Bi bond formations have been suggested to involve antimony or bismuth radical or biradicaloid species (E=Sb, Bi).105, 108, 109, 110, 111, 112
Scheme 21.
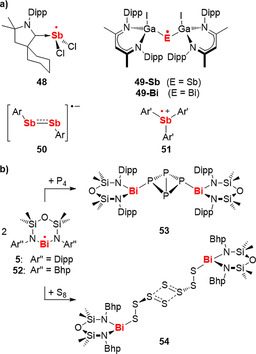
Persistent and isolable antimony and bismuth radicals (a) and examples of stoichiometric reactivity studies (b). Dipp=2,6‐iPr2‐C6H3; Ar=2,6‐[CH(SiMe3)2]2–4‐C(SiMe3)3‐C6H2; Ar’=2,4,6‐iPr3‐C6H2 or 2,6‐iPr2‐4‐OMe‐C6H2; Bhp=2,6‐(CHPh2)2–4‐tBu‐C6H2. Also see compounds 5 and 17 in Scheme 2 and Scheme 6 b, respectively.
Compounds ER2X and [Bi(η1‐C3H5)2(thf)2][B(C6H3Cl2)4] have been applied as mediators and initiators for the controlled living radical polymerization of activated olefins (E=Sb, Bi; R=Me, Ph; X=CMe2C(O)Me, CMe2C(O)Et, CN).113, 114, 115, 116, 117 The bismuth congeners of this small series of compounds were found to facilitate efficient polymerization catalysis without additional initiators such as AIBN. The radical character of these reactions has been supported by stoichiometric reactions of the bismuth compounds with TEMPO and HSn(nBu)3. Reversible homolytic E−C bond cleavage has been suggested as a key step in the polymerization reactions.
Recently, bismuth compounds have been introduced as catalysts for controlled radical reactions other than olefin polymerization. The isolable bismuth radical 5 (see Scheme 21 b) is active as a catalyst in the dehydrocoupling of TEMPO with SiPhH3 to give SiPhH2(TEMPO) (31) (i.e. the same reaction as in Scheme 14).118 Although the bismuth catalyst (70 °C, 15 d, 10 mol % catalyst loading, 75 % conversion) shows a much lower activity than the magnesium catalyst [Mg(N(SiMe3)2)2(thf)2] (60 °C, 1 d, 10 mol % catalyst loading, 99 % conversion; see section 3.2),77 it is the first heavy main‐group element complex to catalyze this type of transformation. Shortly after this report, transition‐metal bismuthanes such as [BiPh2(Mn(CO)5)] were shown to act as (pre‐)catalysts in the radical cyclo‐isomerization of δ‐iodo‐olefins (Table 2).119 In contrast to well‐established tin‐catalyzed reactions of this type,120 these transformations proceed via an intermediate with a new metal–carbon bond and without photochemical induction. Compared to more traditional (pre‐)catalysts or initiators based on Li,121 Sn,120 or B,1, 122 transition metal bismuthanes show complementary or even superior properties in terms of the type of initiation required (thermal vs. photochemical), toxicity, activity, and functional‐group tolerance.
Table 2.
Transition‐metal bismuthanes act as (pre‐)catalysts in thermally induced radical cyclo‐isomerizations of δ‐iodo‐olefins.
|
| ||||||
|---|---|---|---|---|---|---|
|
Entry |
E |
Z |
R |
R’ |
t [h] |
Yield [%] |
|
|
|
|
|
|
|
|
|
1 |
CH2 |
CH2 |
H |
H |
20 |
96 |
|
2 |
CH2 |
CH2 |
Me |
H |
3.5 |
>99[a] |
|
3[b] |
O |
CH2 |
H |
H |
20 |
83 |
|
4 |
O |
CH2 |
Me |
H |
10 |
>99[c] |
|
5 |
N(C3H5)2 |
CO |
H |
H |
4.5 |
>99 |
|
6[d] |
CH2 |
CH2 |
Me |
Me |
5 |
90 |
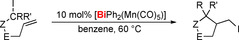
[a] cis/trans=3.4:1.0; [b] 20 mol % pre‐catalyst; [c] cis/trans=2.7:1.0; [d] T=23 °C.
4. Conclusions and Outlook
Controlled radical reactions of main‐group metal complexes have seen remarkable development in recent years. Various strategies have been applied for the isolation and thorough characterization of this important class of compounds. This includes well‐established approaches such as the use of steric bulk, chelation, and aggregation effects, the delocalization of spin density, coordination of redox‐active ligands, and the design of reversible reactions to enable thermodynamically controlled product formation. In addition, new strategies have been reported that allow exploitation of main‐group metal compounds in radical reactions. For example, frustrated Lewis pairs can be designed to undergo to single‐electron transfer. Furthermore, magnetic coupling through main‐group metal atoms and reversible homolytic M−X bond cleavage/formation can be realized (X=M, C, O, H). These strategies may be applied alone or in combination with each other. Based on the isolation and detailed characterization of unusual main‐group metal radicals, scientists have started to exploit their reactivity (and that of related species) in stoichiometric and catalytic transformations. This has granted access to new types of controlled bond formations through radical reactions pathways, including C−C, M−C, Si−O, P−O, P−N, and P−P bonds—a field that used to be predominantly associated with transition‐metal chemistry. In addition, it has been uncovered that even stereotypical examples of ”non‐radical reactions“ such as Lewis pair formation may proceed through single‐electron transfer steps. It is anticipated that the exciting reports on the unusual properties of main‐group metal complexes in radical processes will inspire chemists to develop new approaches for controlled radical reactions based on these species. This will create stoichiometric and catalytic methods for selective bond formations, complementing existing approaches in main‐group and transition‐metal chemistry. This will continue to stimulate the fields of radical chemistry, (in)organic and organometallic synthesis and catalysis, polymerization catalysis, and materials science.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Crispin Lichtenberg obtained his Diploma in chemistry under the supervision of Prof. J. Sundermeyer (University of Cambridge (UK) and Philipps‐Universität Marburg) and his Ph.D. under the guidance of Prof. J. Okuda (RWTH Aachen). He then carried out postdoctoral work with Prof. H. Grützmacher at the ETH Zurich (CH). Holding a Liebig‐ and an Emil Fischer fellowship, he obtained his venia legendi as a Junior Group Leader associated with the group of Prof. H. Braunschweig at the Julius‐Maximilians‐Universität Würzburg. His research interests are centered around tunable redox‐active ligands and non‐conventional (neutral and charged) bismuth compounds for synthesis and catalysis.

Acknowledgements
The author thanks Prof. Dr. Holger Braunschweig for continuous support and Dr. Rian Dewhurst for helpful discussions. Generous financial support by the Fonds der Chemischen Industrie, the DFG, the Universitätsbund Würzburg, and the University of Würzburg are gratefully acknowledged.
C. Lichtenberg, Chem. Eur. J. 2020, 26, 9674.
References
- 1. Yorimitsu H., Nakamura T., Shinokubo H., Oshima K., Omoto K., Fujimoto H., J. Am. Chem. Soc. 2000, 122, 11041–11047. [Google Scholar]
- 2. Akindele T., Yamada K.-I., Tomioka K., Acc. Chem. Res. 2009, 42, 345–355. [DOI] [PubMed] [Google Scholar]
- 3. Bosnidou A. E., Muñiz K., Angew. Chem. Int. Ed. 2019, 58, 7485–7489; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7564–7568. [Google Scholar]
- 4. Kurandina D., Yadagiri D., Rivas M., Kavun A., Chuentragool P., Hayama K., Gevorgyan V., J. Am. Chem. Soc. 2019, 141, 8104–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Riguet E., Hoffmann N., Synthetic Radical Photochemistry, in Encyclopedia of Radicals in Chemistry, Biology and Materials (Eds.: C. Chatgilialoglu, A. Studer), Wiley-VCH, Weinheim, 2012. [Google Scholar]
- 6. Goswami M., Chirila A., Rebreyend C., de Bruin B., Top. Catal. 2015, 58, 719–750. [Google Scholar]
- 7. Zhu X.-Q., Li H. R., Li Q., Ai T., Lu J.-Y., Yang Y., Cheng J.-P., Chem. Eur. J. 2003, 9, 871–880. [DOI] [PubMed] [Google Scholar]
- 8. Zhu X.-Q., Yang Y., Zhang M., Cheng J.-P., J. Am. Chem. Soc. 2003, 125, 15298–15299. [DOI] [PubMed] [Google Scholar]
- 9. Xian M., Zhu X.-Q., Lu J., Wen Z., Cheng J.-P., Org. Lett. 2000, 2, 265–268. [DOI] [PubMed] [Google Scholar]
- 10. Cheng J.-P., Xian M., Wang K., Zhu X., Yin Z., Wang P. G., J. Am. Chem. Soc. 1998, 120, 10266–10267. [Google Scholar]
- 11. Benayahoum A., Amira-Guebailia H., Houache O., Comput. Theor. Chem. 2014, 1037, 1–9. [Google Scholar]
- 12. Osuna S., Swart M., Baerends E. J., Bickelhaupt F. M., Solà M., ChemPhysChem 2009, 10, 2955–2965. [DOI] [PubMed] [Google Scholar]
- 13. Iqbal J., Bhatia B., Nayyar N. K., Chem. Rev. 1994, 94, 519–564. [Google Scholar]
- 14. Petrone D. A., Ye J., Lautens M., Chem. Rev. 2016, 116, 8003–8104. [DOI] [PubMed] [Google Scholar]
- 15. Clark A. J., Eur. J. Org. Chem. 2016, 2231–2243. [Google Scholar]
- 16. Demarteau J., Debuigne A., Detrembleur C., Chem. Rev. 2019, 119, 6906–6955. [DOI] [PubMed] [Google Scholar]
- 17. Wang W., Zhao J., Zhou N., Zhu J., Zhang W., Pan X., Zhang Z., Zhu X., Polym. Chem. 2014, 5, 3533–3546. [Google Scholar]
- 18. Clark A. J., Chem. Soc. Rev. 2002, 31, 1–11. [DOI] [PubMed] [Google Scholar]
- 19. Wang K., Kong W., Chin. J. Chem. 2018, 36, 247–256. [Google Scholar]
- 20. Connelly N. G., Geiger W. E., Chem. Rev. 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- 21. Bock H., Arad C., Näther C., Havlas Z., J. Chem. Soc. Chem. Commun. 1995, 2393–2394. [Google Scholar]
- 22. Scott T. A., Ooro B. A., Collins D. J., Shatruk M., Yakovenko A., Dunbar K. R., Zhou H.-C., Chem. Commun. 2009, 65–67. [DOI] [PubMed] [Google Scholar]
- 23. Castillo M., Metta-Magaña A. J., Fortier S., New J. Chem. 2016, 40, 1923–1926. [Google Scholar]
- 24. Rabideau P. W., Marcinow Z., The Birch Reduction of Aromatic Compounds, in Organic Reactions, Wiley-VCH ; 2004. [Google Scholar]
- 25. Engelhardt L. M., Harvey S., Raston C. L., White A. H., J. Organomet. Chem. 1988, 341, 39–51. [Google Scholar]
- 26.
- 26a.For examples from the more recent literature see: Rowlands G., Tetrahedron 2009, 65, 8603–8655; [Google Scholar]
- 26b. Rowlands G. J., Tetrahedron 2010, 66, 1593–1636. [Google Scholar]
- 27. Studer A., Chem. Eur. J. 2001, 7, 1159–1164. [DOI] [PubMed] [Google Scholar]
- 28. Leifert D., Studer A., Angew. Chem. Int. Ed. 2020, 59, 74–108; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 74–110. [Google Scholar]
- 29. Barry J. T., Berg D. J., Tyler D. R., J. Am. Chem. Soc. 2017, 139, 14399–14405. [DOI] [PubMed] [Google Scholar]
- 30. Dénès F., Pichowicz M., Povie G., Renaud P., Chem. Rev. 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- 31. Kundu S., Sinhababu S., Chandrasekhar V., Roesky H. W., Chem. Sci. 2019, 10, 4727–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Studer A., Curran D. P., Angew. Chem. Int. Ed. 2016, 55, 58–102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 58–106. [Google Scholar]
- 33. Berben L. A., Chem. Eur. J. 2015, 21, 2734–2742. [DOI] [PubMed] [Google Scholar]
- 34. Wei J., Diaconescu P. L., Acc. Chem. Res. 2019, 52, 415–424. [DOI] [PubMed] [Google Scholar]
- 35. Hanft A., Lichtenberg C., Eur. J. Inorg. Chem. 2018, 3361–3373. [Google Scholar]
- 36. Power P. P., Nature 2010, 463, 171–177. [DOI] [PubMed] [Google Scholar]
- 37. Weetman C., Inoue S., ChemCatChem 2018, 10, 4213–4228. [Google Scholar]
- 38. Power P. P., Chem. Rev. 2003, 103, 789–809. [DOI] [PubMed] [Google Scholar]
- 39. Lichtenberg C., Angew. Chem. Int. Ed. 2016, 55, 484–486; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 494–496. [Google Scholar]
- 40.
- 40a.Landmark contributions to the field of MgI species have recently been reviewed and are not covered in this contribution: Jones C., Stasch A., Top. Organomet. Chem. 2013, 45, 73–102; [Google Scholar]
- 40b. Stasch A., Jones C., Dalton Trans. 2011, 40, 5659–5672. [DOI] [PubMed] [Google Scholar]
- 41. Sun C.-L., Shi Z.-J., Chem. Rev. 2013, 113, 9219–9280. [DOI] [PubMed] [Google Scholar]
- 42.Complexes of the elements Li−Cs, Be−Ba, Al−Tl, Ge−Pb, and Sb−Bi are considered.
- 43.E.g.: Davidson P. J., Lappert M. F., J. Chem. Soc. Chem. Commun. 1973, 317a. [Google Scholar]
- 44. Spikes G. H., Peng Y., Fettinger J. C., Power P. P., Z. Anorg. Allg. Chem. 2006, 632, 1005–1010. [Google Scholar]
- 45. Lai T. Y., Fettinger J. C., Power P. P., J. Am. Chem. Soc. 2018, 140, 5674–5677. [DOI] [PubMed] [Google Scholar]
- 46. Davidson P. J., Hudson A., Lappert M. F., Lednor P. W., J. Chem. Soc. Chem. Commun. 1973, 829–830. [Google Scholar]
- 47. Hudson A., Lappert M. F., Lednor P. W., J. Chem. Soc. Dalton Trans. 1976, 2369–2375. [Google Scholar]
- 48. Schwamm R. J., Harmer J. R., Lein M., Fitchett C. M., Granville S., Coles M. P., Angew. Chem. Int. Ed. 2015, 54, 10630–10633; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10776–10779. [Google Scholar]
- 49.For the second example of an isolable bismuth radical compound see: Ganesamoorthy C., Helling C., Wölper C., Frank W., Bill E., G. E. Cutsail III , Schulz S., Nat. Commun. 2018, 9, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schwamm R. J., Lein M., Coles M. P., Fitchett C. M., Angew. Chem. Int. Ed. 2016, 55, 14798–14801; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15018–15021. [Google Scholar]
- 51. Kubo T., Molecules 2019, 24, 665. [Google Scholar]
- 52.
- 52a. Baker R. J., Farley R. D., Jones C., Mills D. P., Koth M., Murphy D. M., Chem. Eur. J. 2005, 11, 2972–2982; [DOI] [PubMed] [Google Scholar]
- 52b. Nishiguchi H., Nakai Y., Nakamura K., Ishizu K., Deguchi Y., Takai H., J. Chem. Phys. 1963, 38–39, 241–243; [Google Scholar]
- 52c. Atherton N. M., Weissman S. I., J. Am. Chem. Soc. 1961, 83, 1330–1334; [Google Scholar]
- 52d. Adam F. C., Weissman S. I., J. Am. Chem. Soc. 1958, 80, 1518–1519. [Google Scholar]
- 53.
- 53a. Lichtenberg C., Krummenacher I., Chem. Commun. 2016, 52, 10044–10047; [DOI] [PubMed] [Google Scholar]
- 53b. Lichtenberg C., Organometallics 2016, 35, 894–902. [Google Scholar]
- 54.
- 54a. Hanft A., Krummenacher I., Lichtenberg C., Chem. Eur. J. 2019, 25, 11883–11891; [DOI] [PubMed] [Google Scholar]
- 54b. Hanft A., Jürgensen M., Bertermann R., Lichtenberg C., ChemCatChem 2018, 10, 4018–4027. [Google Scholar]
- 55. Haeri H. H., Duraisamy R., Harmgarth N., Liebing P., Lorenz V., Hinderberger D., Edelmann F. T., ChemistryOpen 2018, 7, 701–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao Y., Liu Y., Li Q.-S., Su J.-H., Dalton Trans. 2016, 45, 246–252. [DOI] [PubMed] [Google Scholar]
- 57. Ramler J., Poater J., Hirsch F., Ritschel B., Fischer I., Bickelhaupt F. M., Lichtenberg C., Chem. Sci. 2019, 10, 4169–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lai T. Y., Tao L., Britt R. D., Power P. P., J. Am. Chem. Soc. 2019, 141, 12527–12530. [DOI] [PubMed] [Google Scholar]
- 59. Ishida S., Hirakawa F., Furukawa K., Yoza K., Iwamoto T., Angew. Chem. Int. Ed. 2014, 53, 11172–11176; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11354–11358. [Google Scholar]
- 60.The values for ΔGdiss were calculated from values for ΔHdiss (16-Sb: 54.3; 16-Bi: 54.5 kJ mol−1) and ΔGdiss (16-Sb: 191.9; 16-Bi: 155.8 J mol−1) as reported in Ref. [59].
- 61. Blanksby S. J., Ellison G. B., Acc. Chem. Res. 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- 62. Sinnema P.-J., Shapiro P. J., Höhn B., Twamley B., J. Organomet. Chem. 2003, 676, 73–79. [Google Scholar]
- 63.It has not formally been proved that the reaction of 18 with Ca to give 19 and 20 or the conversion of 20 to 19 proceeds through radical pathways. This may be assumed, however, based on comparison with related reactions of calcium with acene to give [(η5-C12H8)2Ca(thf)2] (see Ref. [64]).
- 64. Fedushkin I. L., Petrovskaya T. V., Bochkarev M. N., Dechert S., Schumann H., Angew. Chem. Int. Ed. 2001, 40, 2474–2477; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2540–2543. [Google Scholar]
- 65. Majumdar M., Raut R. K., Sahoo P., Kumar V., Chem. Commun. 2018, 54, 10839–10842. [DOI] [PubMed] [Google Scholar]
- 66. Myers T. W., Kazem N., Stoll S., Britt D., Shanmugam M., Berben L. A., J. Am. Chem. Soc. 2011, 133, 8662–8672. [DOI] [PubMed] [Google Scholar]
- 67.
- 67a. Welch G. C., Juan R. R. S., Masuda J. D., Stephan D. W., Science 2006, 314, 1124–1126; [DOI] [PubMed] [Google Scholar]
- 67b. Stephan D. W., Acc. Chem. Res. 2015, 48, 306–316; [DOI] [PubMed] [Google Scholar]
- 67c. Stephan D. W., J. Am. Chem. Soc. 2015, 137, 10018–10032; [DOI] [PubMed] [Google Scholar]
- 67d. Stephan D. W., Erker G., Angew. Chem. Int. Ed. 2015, 54, 6400–6441; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6498–6541. [Google Scholar]
- 68.
- 68a. Liu L. L., Cao L. L., Shao Y., Ménard G., Stephan D. W., Chem 2017, 3, 259–267; [Google Scholar]
- 68b. Liu L. L., Cao L. L., Zhu D., Zhou J., Stephan D. W., Chem. Commun. 2018, 54, 7431–7434. [DOI] [PubMed] [Google Scholar]
- 69.For a recent review article see: Liu L. L., Stephan D. W., Chem. Soc. Rev. 2019, 48, 3454–3463. [DOI] [PubMed] [Google Scholar]
- 70.
- 70a.For examples of (“non-metal“) FLPs in redox chemistry see: Sajid M., Stute A., Cardenas A. J. P., Bulotta B. J., Hepperle J. A. M., Warren T. H., Schirmer B., Grimme S., Studer A., Daniliuc C. G., Fröhlich R., Petersen J. L., Kehr G., Erker G., J. Am. Chem. Soc. 2012, 134, 10156–10168; [DOI] [PubMed] [Google Scholar]
- 70b. Longobardi L. E., Liu L., Grimme S., Stephan D. W., J. Am. Chem. Soc. 2016, 138, 2500–2503; [DOI] [PubMed] [Google Scholar]
- 70c. Longobardi L. E., Zatsepin P., Korol R., Liu L., Grimme S., Stephan D. W., J. Am. Chem. Soc. 2017, 139, 426–435; [DOI] [PubMed] [Google Scholar]
- 70d. Bamford K. L., Longobardi L. E., Liu L., Grimme S., Stephan D. W., Dalton Trans. 2017, 46, 5308–5319. [DOI] [PubMed] [Google Scholar]
- 71. Liu W., Cao H., Zhang H., Zhang H., Chung K. H., He C., Wang H., Kwong F. Y., Lei A., J. Am. Chem. Soc. 2010, 132, 16737–16740. [DOI] [PubMed] [Google Scholar]
- 72. Nocera G., Young A., Palumbo F., Emery K. J., Coulthard G., McGuire T., Tuttle T., Murphy J. A., J. Am. Chem. Soc. 2018, 140, 9751–9757. [DOI] [PubMed] [Google Scholar]
- 73. Wu L., Annibale V. T., Jiao H., Brookfield A., Collison D., Manners I., Nat. Commun. 2019, 10, 2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Armstrong D. R., Balloch L., Crawford J. J., Fleming B. J., Hogg L. M., Kennedy A. R., Klett J., Mulvey R. E., O'Hara C. T., Orr S. A., Robertson S. D., Chem. Commun. 2012, 48, 1541–1543. [DOI] [PubMed] [Google Scholar]
- 75. Shirakawa E., Hayashi Y., Itoh K.-I., Watabe R., Uchiyama N., Konagaya W., Masui S., Hayashi T., Angew. Chem. Int. Ed. 2012, 51, 218–221; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 222–225. [Google Scholar]
- 76. Uchiyama N., Shirakawa E., Hayashi T., Chem. Commun. 2013, 49, 364–366. [DOI] [PubMed] [Google Scholar]
- 77. Liptrot D. J., Hill M. S., Mahon M. F., Angew. Chem. Int. Ed. 2014, 53, 6224–6227; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6338–6341. [Google Scholar]
- 78. Fedushkin I. L., Morozov A. G., Chudakova V. A., Fukin G. K., Cherkasov V. K., Eur. J. Inorg. Chem. 2009, 4995–5003. [Google Scholar]
- 79. Fedushkin I. L., Khvoinova N. M., Skatova A. A., Fukin G. K., Angew. Chem. Int. Ed. 2003, 42, 5223–5226; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 5381–5384. [Google Scholar]
- 80. Fedushkin I. L., Skatova A. A., Cherkasov V. K., Chudakova V. A., Dechert S., Hummert M., Schumann H., Chem. Eur. J. 2003, 9, 5778–5783. [DOI] [PubMed] [Google Scholar]
- 81. Sinnema P.-J., Twamley B., Shapiro P. J., Acta Crystallogr. Sect. E 2001, 57, m438–m440. [Google Scholar]
- 82. Krieck S., Görls H., Yu L., Reiher M., Westerhausen M., J. Am. Chem. Soc. 2009, 131, 2977–2985. [DOI] [PubMed] [Google Scholar]
- 83. Krieck S., Görls H., Westerhausen M., J. Am. Chem. Soc. 2010, 132, 12492–12501. [DOI] [PubMed] [Google Scholar]
- 84. Wilson A. S. S., Dinoi C., Hill M. S., Mahon M. F., Maron L., Richards E., Angew. Chem. Int. Ed. 2020, 59, 1232–1237; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1248–1253. [Google Scholar]
- 85. Miyabe H., Naito T., Org. Biomol. Chem. 2004, 2, 1267–1270 and references therein. [DOI] [PubMed] [Google Scholar]
- 86. Takami K., Usugi S.-I., Yorimitsu H., Oshima K., Synthesis 2005, 5, 824–839. [Google Scholar]
- 87. Augé J., Lubin-Germain N., Uziel J., Synthesis 2007, 12, 1739–1764. [Google Scholar]
- 88. Shen Z.-L., Wang S.-Y., Chok Y.-K., Xu Y.-H., Loh T.-P., Chem. Rev. 2013, 113, 271–401. [DOI] [PubMed] [Google Scholar]
- 89. Inoue K., Sawada A., Shibata I., Baba A., J. Am. Chem. Soc. 2002, 124, 906–907. [DOI] [PubMed] [Google Scholar]
- 90. Mikami S., Fujita K., Nakamura T., Yorimitsu H., Shinokubo H., Matsubara S., Oshima K., Org. Lett. 2001, 3, 1853–1855. [DOI] [PubMed] [Google Scholar]
- 91. Takami K., Mikami S., Yorimitsu H., Shinokubo H., Oshima K., Tetrahedron 2003, 59, 6627–6635. [DOI] [PubMed] [Google Scholar]
- 92. Hayashi N., Shibata I., Baba A., Org. Lett. 2004, 6, 4981–4983. [DOI] [PubMed] [Google Scholar]
- 93. Sokolov V. G., Koptseva T. S., Moskalev M. V., Bazyakina N. L., Piskunov A. V., Cherkasov A. V., Fedushkin I. L., Inorg. Chem. 2017, 56, 13401–13410. [DOI] [PubMed] [Google Scholar]
- 94. Fedushkin I. L., Kazarina O. V., Lukoyanov A. N., Skatova A. A., Bazyakina N. L., Cherkasov A. V., Palamidis E., Organometallics 2015, 34, 1498–1506. [Google Scholar]
- 95. Dodonov V. A., Morozov A. G., Umyantsev R. V., Fukin G. K., Skatova A. A., Roesky P. W., Fedushkin I. L., Inorg. Chem. 2019, 58, 16559–16573 . [DOI] [PubMed] [Google Scholar]
- 96. Lichtenthaler M. R., Stahl F., Kratzert D., Heidinger L., Schleicher E., Hamann J., Himmel D., Weber S., Krossing I., Nat. Commun. 2015, 6, 8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Dong Z., Cramer H. H., Schmidtmann M., Paul L. A., Siewert I., Müller T., J. Am. Chem. Soc. 2018, 140, 15419–15424. [DOI] [PubMed] [Google Scholar]
- 98. Lukoyanov A. N., Univanova E. A., Razborov D. A., Khrizanforova V. V., Budnikova Y. H., Makarov S. G., Rumyantcev R. V., Ketkov S. Y., Fedushkin I. L., Chem. Eur. J. 2019, 25, 3858–3866. [DOI] [PubMed] [Google Scholar]
- 99. Fang H., Ling Z., Lang K., Brothers P. J., de Bruin B., Fu X., Chem. Sci. 2014, 5, 916–921. [Google Scholar]
- 100. Fang H., Jing H., Ge H., Brothers P. J., Fu X., Ye S., J. Am. Chem. Soc. 2015, 137, 7122–7127. [DOI] [PubMed] [Google Scholar]
- 101.
- 101a. Miller K. A., Bartolin J. M., O'Neill R. M., Sweeder R. D., Owens T. M., Kampf J. W., Banaszak Holl M. M., Wells N. J., J. Am. Chem. Soc. 2003, 125, 8986–8987; [DOI] [PubMed] [Google Scholar]; Kavara A., T. T. Boron III , Ahsan Z. S., Banaszak Holl M. M., Organometallics 2010, 29, 5033–5039. [Google Scholar]
- 102.
- 102a.Also see: Bartolin J. M., Kavara A., Kampf J. W., Banaszak Holl M. M., Organometallics 2006, 25, 4738–4740; [Google Scholar]
- 102b. Kavara A., Cousineau K. D., Rohr A. D., Kampf J. W., Banaszak Holl M. M., Organometallics 2008, 27, 1041–1043; [Google Scholar]
- 102c. Kavara A., Kampf J. W., Banaszak Holl M. M., Organometallics 2008, 27, 2896–2897; [Google Scholar]
- 102d. Walker R. H., Miller K. A., Scott S. L., Cygan Z. T., Bartolin J. M., Kampf J. W., Banaszak Holl M. M., Organometallics 2009, 28, 2744–2755. [Google Scholar]
- 103. Kretschmer R., Ruiz D. A., Moore C. E., Rheingold A. L., Bertrand G., Angew. Chem. Int. Ed. 2014, 53, 8176–8179; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8315–8318. [Google Scholar]
- 104. Sasamori T., Mieda E., Nagahora N., Sato K., Shiomi D., Takui T., Hosoi Y., Furukawa Y., Takagi N., Nagase S., Tokitoh N., J. Am. Chem. Soc. 2006, 128, 12582–12588. [DOI] [PubMed] [Google Scholar]
- 105. Li T., Wie H., Fang Y., Wang L., Chen S., Zhang Z., Zhao Y., Tan G., Wang X., Angew. Chem. Int. Ed. 2017, 56, 632–636; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 647–651. [Google Scholar]
- 106. Schwamm R. J., Lein M., Coles M. P., Fitchett C. M., J. Am. Chem. Soc. 2017, 139, 16490–16493. [DOI] [PubMed] [Google Scholar]
- 107. Turner Z., Inorg. Chem. 2019, 58, 14212–14227. [DOI] [PubMed] [Google Scholar]
- 108. Hering-Junghans C., Schulz A., Thomas M., Villinger A., Dalton Trans. 2016, 45, 6053–6059. [DOI] [PubMed] [Google Scholar]
- 109.
- 109a. Casely I. J., Ziller J. W., Fang M., Furche F., Evans W. J., J. Am. Chem. Soc. 2011, 133, 5244–5247; [DOI] [PubMed] [Google Scholar]
- 109b. Hanna T. A., Rieger A. L., Rieger P. H., Wang X., Inorg. Chem. 2002, 41, 3590–3592. [DOI] [PubMed] [Google Scholar]
- 110. Bresien J., Hinz A., Schulz A., Villinger A., Dalton Trans. 2018, 47, 4433–4436. [DOI] [PubMed] [Google Scholar]
- 111. Knispel C., Limberg C., Tschersich C., Chem. Commun. 2011, 47, 10794–10796. [DOI] [PubMed] [Google Scholar]
- 112.
- 112a. Balázs G., Breunig H. J., Lork E., Organometallics 2002, 21, 2584–2586; [Google Scholar]
- 112b. Balázs G., Breunig H. J., Lork E., Z. Naturforsch. B 2005, 60, 180–182. [Google Scholar]
- 113. Yamago S., Ray B., Iida K., Yoshida J.-I., Tada T., Yoshizawa K., Kwak Y., Goto A., Fukuda T., J. Am. Chem. Soc. 2004, 126, 13908–13909. [DOI] [PubMed] [Google Scholar]
- 114. Yamago S., J. Polym. Sci. Part A 2006, 44, 1–12. [Google Scholar]
- 115. Yamago S., Kayahara E., Kotani M., Ray B., Kwak Y., Goto A., Fukuda T., Angew. Chem. Int. Ed. 2007, 46, 1304–1306; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 1330–1333. [Google Scholar]
- 116. Kayahara E., Yamago S., J. Am. Chem. Soc. 2009, 131, 2508–2513. [DOI] [PubMed] [Google Scholar]
- 117. Lichtenberg C., Pan F., Spaniol T. P., Englert U., Okuda J., Angew. Chem. Int. Ed. 2012, 51, 13011–13015; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 13186–13190. [Google Scholar]
- 118. Schwamm R. J., Lein M., Coles M. P., Fitchett C. M., Chem. Commun. 2018, 54, 916–919. [DOI] [PubMed] [Google Scholar]
- 119. Ramler J., Krummenacher I., Lichtenberg C., Angew. Chem. Int. Ed. 2019, 58, 12924–12929; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13056–13062. [Google Scholar]
- 120. Curran D. P., Kim D., Tetrahedron Lett. 1986, 27, 5821–5824. [Google Scholar]
- 121.
- 121a. Bailey W. F., Carson M. W., J. Org. Chem. 1998, 63, 361–365; [Google Scholar]
- 121b. Bailey W. F., Carson M. W., J. Org. Chem. 1998, 63, 9960–9967. [Google Scholar]
- 122.
- 122a. Yorimitsu H., Nakamura T., Shinokubo H., Oshima K., J. Org. Chem. 1998, 63, 8604–8605; [Google Scholar]
- 122b. Fusano A., Sumino S., Nishitani S., Inouye T., Morimoto K., Fukuyama T., Ryu I., Chem. Eur. J. 2012, 18, 9415–9422. [DOI] [PubMed] [Google Scholar]