

Abstract
Eliglustat is a first‐line oral therapy for adults with Gaucher disease type 1 (GD1) with extensive, intermediate, or poor CYP2D6‐metabolizer phenotypes (90% of patients). We report real‐world outcomes after 2 years of eliglustat therapy in the International Collaborative Gaucher Group Gaucher Registry (NCT00358943). As of January 2019, baseline and 2‐year data (±1 year) were available for 231 eliglustat‐treated GD1 patients: 19 treatment‐naïve (zero splenectomized) and 212 ERT patients who switched to eliglustat (36 splenectomized). Most patients (89%) were from the United States, where eliglustat was first approved. In treatment‐naïve patients, mean hemoglobin increased from 12.4 to 13.4 g/dL (P = .004, n = 18), mean platelet count increased from 113 to 156 × 109/L (P < .001, n = 17); mean spleen volume decreased from 7.4 to 3.5 multiples of normal (MN) (P = .02, n = 7); mean liver volume remained normal (n = 7), and median spine Z‐score was unchanged (−1.3 to −1.2, n = 6). In non‐splenectomized switch patients, mean hemoglobin remained stable/non‐anemic (n = 167); mean platelet count remained stable/normal (n = 165); mean spleen volume decreased from 3.3 to 2.8 MN (P < .001, n = 64); mean liver volume remained normal (n = 63), and median lumbar spine Z‐score improved from −0.7 to −0.4 (P = .014, n = 68). In splenectomized switch patients, mean hemoglobin remained stable/non‐anemic (n = 31); mean platelet count increased from 297 to 324 × 109/L (non‐significant, n = 29); mean liver volume remained normal (n = 13); median spine Z‐score improved from −0.8 to −0.6 (non‐significant, n = 11). Median chitotriosidase decreased in all groups (P < .01 for all). These real‐world results are consistent with eliglustat clinical trial results demonstrating long‐term benefit in treatment‐naïve patients and stability in ERT switch patients.
1. INTRODUCTION
Gaucher disease type 1 is a progressive, autosomal‐recessive disease characterized by splenomegaly, hepatomegaly, anemia, thrombocytopenia, and bone manifestations, which lead to major disability. 1 , 2 , 3 It is among the most common lysosomal storage disorders, affecting approximately 1:40000‐60 000 people worldwide, 4 but among people of Ashkenazi Jewish ancestry, it affects as many as 1:800 individuals. 5 The underlying defect is biallelic mutations in GBA (OMIM606463) that lead to deficiency of the lysosomal enzyme acid β‐glucosidase and, accumulation of its substrate, glucosylceramide, and its downstream metabolite, glucosylsphingosine, most conspicuously in macrophages. 4 The accumulating bioactive lipids cause marked metabolic inflammation involving myeloid cells, and result in induction of glucosylceramide synthase, which augments the metabolic defect. 6 The clinical presentation of Gaucher disease is heterogeneous, encompassing three classical disease subtypes. Type 1 is the most prevalent subtype in North America and Europe, and is characterized by lack of early onset neuronodegenerative disease. However, in older adult patients with Gaucher disease type 1, there is an increased life‐time risk of developing Parkinson disease 7 and higher incidence of peripheral neuropathy than the general population. 8 , 9 The historic standard of care for treatment of visceral, hematologic, and skeletal manifestations of Gaucher disease is recombinant macrophage‐targeted acid β‐glucosidase enzyme replacement therapy (ERT), which has been available since 1991. There are currently no treatment options for the neurologic manifestations of type 2 and type 3 Gaucher disease.
Eliglustat (Cerdelga, Sanofi Genzyme) is a substrate reduction therapy first approved in the United States in 2014 as a first‐line treatment for adults with Gaucher disease type 1, who have compatible CYP2D6‐metabolizer phenotypes (>90% of patients 10 ). 11 Currently, the drug is approved in more than 55 countries. The eliglustat clinical trial program is the largest to date in Gaucher disease, spanning 12 years and involving 393 eliglustat‐treated patients from 29 countries in one Phase 2 and three Phase 3 trials. Two trials in treatment‐naïve patients (Phase 2 and Phase 3 ENGAGE) showed eliglustat monotherapy resulted in clinically significant reductions in spleen and liver volume, increases in hemoglobin concentration and platelet counts, improved bone mineral density, and commensurate decline of biomarkers of Gaucher disease. 12 , 13 Two Phase 3 trials in switch or predominantly switch patients (ENCORE and EDGE) demonstrated long‐term stability and/or modest improvements in patients switching from ERT to eliglustat. 14 , 15 In the clinical trials, eliglustat was dosed by plasma drug level; however, pharmacokinetic analysis of clinical trial data demonstrated that the CYP2D6 metabolizer phenotype was the major determinant of plasma drug exposure. Therefore, as recommended in the eliglustat label, the standard of care is a dose regimen of 84 mg twice daily, for patients who are intermediate or extensive CYP2D6 metabolizers, and 84 mg once daily for patients who are poor CYP2D6 metabolizers. Eliglustat is not recommended for CYP2D6 ultra‐rapid metabolizers (URMs) due to insufficient data for this uncommon metabolizer subtype, and concerns that very high rate of metabolism could diminish eliglustat efficacy. 11
Following drug approval, it is prudent to evaluate “real‐world” safety and effectiveness outside of the clinical trial setting, where patients are selected based on specific inclusion and exclusion criteria and treatment compliance is generally very high. Accordingly, we analyzed efficacy outcome data from eliglustat‐treated patients with Gaucher disease type 1 enrolled in the International Collaborative Gaucher Group (ICGG) Gaucher Registry, to determine how these real‐world data compare with what was observed in the eliglustat clinical trials. Our objectives were to determine whether treatment‐naïve patients achieved clinically significant improvements of indicators of disease activity after 2 years of eliglustat treatment, and whether previously ERT‐treated patients maintained (or improved) their clinical status 2 years after switching to eliglustat treatment.
2. METHODS
Initiated in 1991, the ICGG Gaucher Registry (NCT00358943) is an observational, longitudinal, international database of clinical, biochemical, and therapeutic characteristics of patients with Gaucher disease, regardless of disease severity, treatment status, or treatment choice. It is the largest Gaucher disease registry in the world, with data from more than 6000 patients in 60 countries. The Registry is supported by Sanofi Genzyme and is governed by a collaborative group of international physician experts in Gaucher disease.
Herein, the analysis included all patients with Gaucher disease type 1 enrolled in the Registry as of January 2019, who had diagnosis and treatment dates documented. Also, all patients received eliglustat treatment for at least 1 year, and had baseline and follow‐up data for at least one of the following clinical parameters: spleen volume and liver volume (in multiples of normal [MN] organ size: 0.2% and 2.5% of body weight in kilograms, respectively), hemoglobin concentration, platelet count, and bone pain or bone crisis. Two windows of time were evaluated: the baseline window encompassed 12 months before to 1 month after beginning eliglustat, and the 2‐year window encompassed 1 year to 3 years after beginning eliglustat. We also performed an analysis of change in Short Form Health Survey (SF‐36) scores from baseline to 2 years of eliglustat treatment. For bone disease, we examined lumbar spine Z‐scores before and after eliglustat, presence or absence of bone pain or bone crises since last assessment, and presence or absence of bone lesions (avascular necrosis, Erlenmeyer flask deformity, fractures, bone infarction, lytic lesions, and bone marrow infiltration). Levels of the Gaucher disease biomarker, chitotriosidase, were also evaluated before and after eliglustat treatment. For switch patients, mean time on prior treatment was determined with a start date cutoff of 1989 (approximate year that investigational ERT first became available for Gaucher disease).
Patients were divided into three cohorts for analysis: treatment‐naïve patients, splenectomized switch patients, and non‐splenectomized switch patients. Treatment‐naïve patients (all were non‐splenectomized) had no prior Gaucher disease treatment before starting eliglustat therapy. Switch patients (whether splenectomized or not) had been treated with at least one other Gaucher disease therapy (primarily ERT) before switching to eliglustat. If a patient was taking eliglustat and switched to an alternate Gaucher disease therapy during the analysis period, no data after the alternative therapy switch date were included in the analysis.
2.1. Statistical analysis
A paired t test was used to compare baseline mean values with 2‐year values for hemoglobin, platelet count, spleen volume, liver volume and SF‐36 scores. The Wilcoxon signed‐rank test was used to compare baseline median values with 2‐year values for chitotriosidase and lumbar spine Z‐scores, because the data distributions were not normal. For parameters with two levels of response, McNemarʼs test was used to compare baseline values with 2‐year values for anemia (yes, no), thrombocytopenia for splenectomized patients (present, none), and all bone parameters (yes, no) except lumbar spine Z‐scores. For parameters with more than two levels of response, the Wilcoxon exact test was used to compare baseline values with 2‐year values for thrombocytopenia for non‐splenectomized patients (none, mild, moderate, severe), hepatomegaly (none, mild, moderate, severe) and splenomegaly (none, mild, moderate, severe).
3. RESULTS
3.1. Patients
Among 6341 patients enrolled in the ICGG Gaucher Registry as of January 2019, 466 had been treated with eliglustat. Among eliglustat‐treated patients, 231 met the following criteria: known eliglustat treatment dates, confirmed Gaucher disease type 1 with a reported diagnosis date, known splenectomy status (including date of splenectomy if splenectomized), and baseline and 2‐year data while on eliglustat only for at least one of the key clinical parameters (hemoglobin concentration, platelet count, liver volume, spleen volume, bone pain, or bone crisis). All treatment‐naïve patients in this analysis were continuing eliglustat therapy at the time of database lock (January 2019). Among switch patients, 22/212 patients (10%) in this analysis discontinued eliglustat after at least 1 year of treatment: 18/176 were non‐splenectomized patients and 4/36 were splenectomized patients. Reasons for discontinuation are not captured in the Registry. Overall, 15 patients who discontinued were women, of whom seven were younger than 50 years of age (range: 22‐48 years).
Demographic characteristics and genotype distribution are summarized in Table 1. Overall, the splenectomized switch patients appeared to have the most severe underlying disease, reflected by the younger age at diagnosis, proportionally fewer patients who were N370S homozygous, and proportionally more L444P heteroallelic patients (Table 1).
TABLE 1.
Demographic characteristics and Gaucher genotype distribution by eliglustat treatment group
| Parameter | Treatment‐Naïve a (N = 19) | Switch (N = 212) | |
|---|---|---|---|
| No splenectomy (N = 176) | Splenectomy (N = 36) | ||
| Sex, n (%) | |||
| Male | 8 (42) | 88 (50) | 10 (28) |
| Female | 11 (58) | 88 (50) | 26 (72) |
| Age at diagnosis in years, mean ± SD | 26 ± 12.5 | 22 ± 15.4 | 15 ± 13.5 |
| Age at initiation of eliglustat in years, mean ± SD | 42 ± 18.0 | 42 ± 15.2 | 52 ± 9.7 |
| Duration of previous treatment in years, mean ± SD | 0 | 14.2 ± 6.6 b | 17.0 ± 6.5 |
| CYP2D6‐metabolizer phenotype, n (%) | |||
| Ultra‐rapid | 1 (5) | 4 (2) | 1 (3) |
| Extensive | 12 (63) | 117 (67) | 29 (81) |
| Intermediate | 4 (21) | 37 (21) | 0 |
| Poor | 1 (5) | 7 (4) | 1 (3) |
| Indeterminate/unknown | 0 | 1 (1) | 1 (3) |
| Not reported | 1 (7) | 10 (6) | 4 (11) |
| Geographic distribution, n (%) | |||
| United States | 17 (90) | 157 (89) | 31 (86) |
| Canada and Europe | 2 (10) | 18 (10) | 4 (11) |
| Other | 0 | 1 (<1) | 1 (3) |
| Genotype, n (%) | |||
| N370S/N370S (p.Asn409Ser/p.Asn409Ser) | 11 (58) | 64 (36) | 2 (6) |
| N370S/L444P (p.Asn409Ser/p.Leu483Pro) | 1 (5) | 25 (14) | 9 (25) |
| N370S/Rare Allele (p.Asn409Ser/Rare Allele) | 0 | 30 (17) | 3 (8) |
| N370S/84GG (p.Asn409Ser/p.Leu29AlafsTer18) | 1 (5) | 13 (7) | 4 (11) |
| N370S/? (p.Asn409Ser/?) | 0 | 10 (6) | 3 (8) |
| Rare Allele/Rare Allele | 1 (5) | 8 (4) | 6 (17) |
| L444P/Rare Allele (p.Leu483Pro/Rare Allele) | 0 | 4 (2) | 3 (8) |
| N370S/IVS2+1 (p.Asn409Ser/IVS2+1G>A) | 0 | 6 (3) | 0 |
| N370S/D409H (p.Asn409Ser/p.Asp448His) | 2 (10) | 4 (2) | 1 (3) |
| 84GG/Rare Allele (p.Leu29AlafsTer18/Rare allele) | 0 | 1 (<1) | 0 |
| 84GG/? (p.Leu29AlafsTer18/?) | 0 | 1 (<1) | 0 |
| L444P/? (p.Leu483Pro/?) | 0 | 0 | 1 (3) |
| Rare Allele/? | 0 | 1 (<1) | 1 (3) |
| L444P/L444P (p.Leu483Pro/p.Leu483Pro) | 0 | 0 | 1 (3) |
| Unknown | 3 (16) | 9 (5) | 2 (6) |
Note: Percentages are based on the number of patients in the population, not the number of patients with data.
All naïve patients had intact spleens.
One patient who had a treatment start date prior to 1989 was excluded from the treatment duration analysis.
Among non‐splenectomized switch patients, prior treatment was primarily ERT. Overall, 43% of non‐splenectomized switch patients and 58% of splenectomized switch patients had been on more than one previous therapy, which included imiglucerase/alglucerase (94% of all switch patients), velaglucerase alfa (40%), taliglucerase alfa (1%), miglustat (11%), and unidentified investigational therapy (8%). No patient had been on miglustat exclusively as a prior therapy. Mean time on previous therapies was 14 years for non‐splenectomized patients and 17 years for splenectomized patients (Table 1). Six patients with a CYP2D6 URM phenotype were prescribed eliglustat (not approved in the drug label) and were included in this real‐world analysis.
3.2. Hematologic and visceral outcomes
Among treatment‐naïve patients, after 2 years of eliglustat therapy, mean hemoglobin concentration improved by 1 g/dL (P = .004), platelet count improved by 38% (P < .001), spleen volume decreased by 53% (P = .02), and liver volume decreased by 8% (not statistically significant) (Figure 1A). This response to eliglustat was within long‐term therapeutic goal thresholds established for Gaucher patients on ERT 16 , 17 for all four parameters. Individual values for all treatment‐naïve patients are shown in Figure 2.
FIGURE 1.
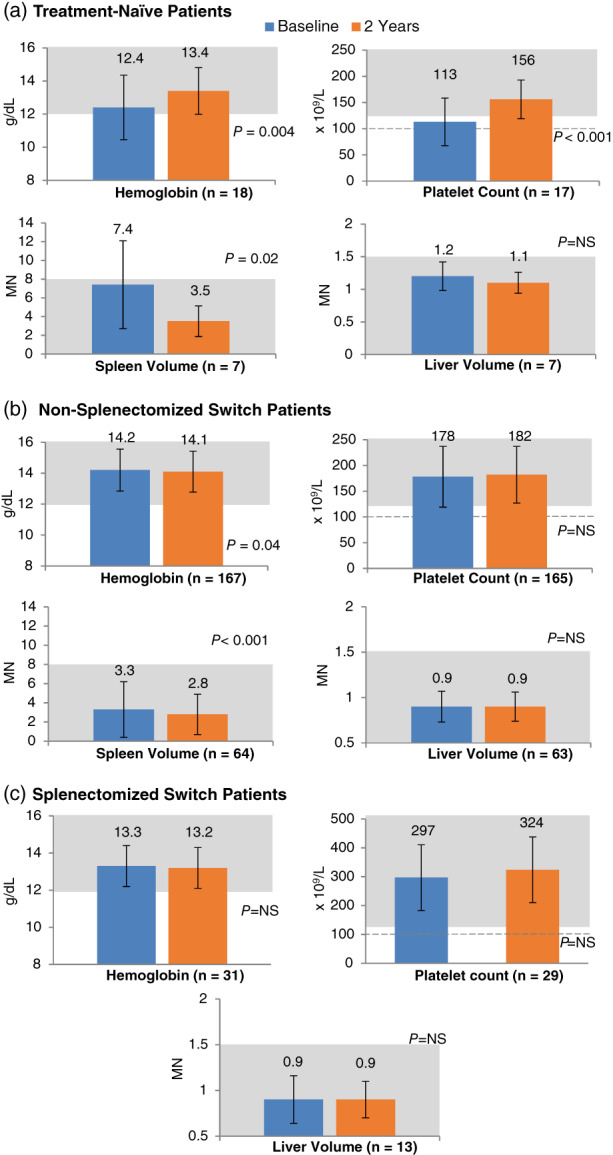
Mean hematologic and visceral values at baseline and after 2 years of eliglustat treatment. The P values are from paired t tests, comparing 2‐year parameters to baseline. MN, multiples of normal organ size; NS, not significant. Error bars denote standard deviations. Shaded areas represent long‐term therapeutic goal thresholds established for patients on long‐term ERT. 16 Dotted line represents updated platelet therapeutic goal threshold 17
FIGURE 2.
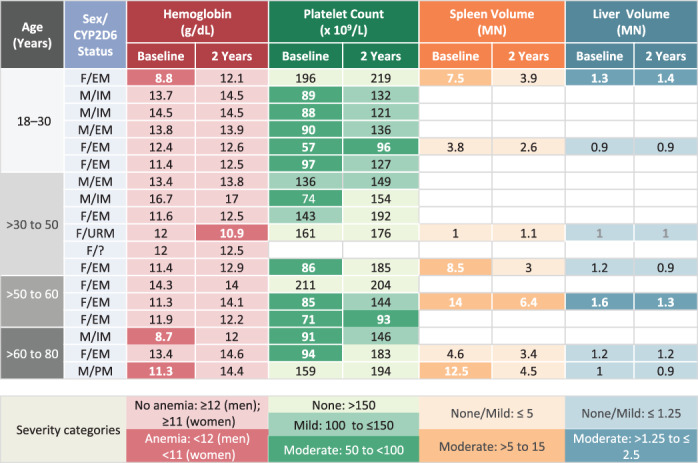
Disease severity categories at baseline and after 2 years of eliglustat therapy in treatment‐naïve patients. EM, extensive metabolizer; F, female; IM, intermediate metabolizer; M, male; MN, multiples of normal organ size; PM, poor metabolizer; URM, ultra‐rapid metabolizer
Mean baseline values among both non‐splenectomized and splenectomized switch patients were within therapeutic goal thresholds, and remained stable and within these thresholds after 2 years of eliglustat (Figure 1B,C). In non‐splenectomized switch patients, eliglustat therapy resulted in further reversal of splenomegaly: spleen volume decreased by 15% (P < .001) (Figure 1B). At baseline most switch patients did not have anemia, had no or mild splenomegaly and hepatomegaly, and no or mild thrombocytopenia. This status was largely maintained after 2 years of eliglustat therapy, with no statistically significant changes (Figure 3A,B).
FIGURE 3.

Proportion of switch patients with anemia, thrombocytopenia, splenomegaly, and hepatomegaly at baseline and after 2 years of eliglustat therapy. The P values are from Wilcoxon signed‐rank tests comparing 2‐year parameters to baseline, except where McNemarʼs test was used for anemia (non‐splenectomized and splenectomized switch patients) and thrombocytopenia in splenectomized switch patients. MN, multiples of normal organ size; NS, not significant
In both treatment‐naïve and switch patients, the vast majority of patients who had normal baseline clinical values maintained these normal values after 2 years of eliglustat, and most shifts in severity category were from more severe to less severe. All patients maintained or improved their splenomegaly and hepatomegaly severity category with eliglustat treatment, 98% of patients maintained or improved their anemia status, and 93% of patients maintained or improved their thrombocytopenia category. Among the small subset of patients who had a downward shift in either anemia or thrombocytopenia category, there were no precipitous declines in either variable, no patient had a downward shift in both variables, and no patient discontinued eliglustat. Four patients (1.7% overall), all female ranging in age from 28 to 63 years old (one treatment‐naïve, two non‐splenectomized switch, and one splenectomized switch) who were not anemic at baseline had anemia after 2 years of eliglustat; the lowest hemoglobin value among these patients was 10.6 g/dL, just below the anemia threshold of <11 g/dL for women. Fourteen patients had a shift from no thrombocytopenia (platelet count >150 × 109/L) at baseline to mild thrombocytopenia (platelet count of 100‐150 × 109/L) after 2 years of eliglustat. The lowest value among these patients was 104 × 109/L, above the updated long‐term therapeutic goal threshold for platelets of 100 × 109/L established by the European Working Group on Gaucher Disease. 17 One patient with a baseline platelet count of 60 × 109/L signifying moderate thrombocytopenia (platelet count of 50 to <100 × 109/L) moved to the severe thrombocytopenia category (platelet count <50 × 109/L) after 2 years of eliglustat with a value of 48 × 109/L.
3.3. Outcomes in ultra‐rapid metabolizers (URMs)
Among the six patients who were CYP2D6 URMs, one was treatment‐naïve with very mild baseline disease (Figure 2), four were non‐splenectomized switch patients, and one was a splenectomized switch patient. For four of the six patients, the most recent recorded eliglustat dose was 84 mg eliglustat three times daily; for the remaining two patients, the most recent recorded dose was 84 mg twice daily (the recommended dose for extensive or intermediate CYP2D6 metabolizers). In four of the six URM patients, hemoglobin concentration and platelet counts remained stable within the therapeutic goal range after 2 years of therapy. The treatment‐naïve URM patient (a female whose most recent recorded eliglustat dose was 84 mg three times daily) shifted from no anemia (12 g/dL) to anemia (10.9 g/dL). And, one non‐splenectomized switch patient (a female whose most recent dose was 84 mg twice daily) shifted from no thrombocytopenia (181 × 109/L) to moderate thrombocytopenia (104 × 109/L). Organ volumes remained stable within the therapeutic goal range after 2 years in the three URM patients with available liver volume measurements, and two patients with spleen volume measurements. One URM patient discontinued eliglustat; this male splenectomized switch patient maintained normal hemoglobin, platelet count, and liver volume values after switching to eliglustat.
3.4. Bone outcomes
Bone data were limited in all three cohorts. Lumbar spine Z‐score data showed no significant changes in treatment‐naïve patients (n = 6) or splenectomized switch patients (n = 11). In non‐splenectomized switch patients (n = 68), lumbar spine Z‐score improved from a median (25th, 75th) of −0.7 (−1.2, 0.0) to −0.4 (−1.0, 0.3) (P = .014). Bone lesion data were especially limited, with between two and eight treatment‐naïve patients, 13 and 64 non‐splenectomized switch patients, and one and 10 splenectomized switch patients, having either a yes or no value for any one category of bone lesion (avascular necrosis, Erlenmeyer flask deformity, fractures, infarction, lytic lesions, or marrow infiltration). There were no significant changes after eliglustat therapy in any cohort. Among the three treatment‐naïve patients, 35 non‐splenectomized switch patients and three splenectomized patients with data, no new episodes of avascular necrosis were reported after 2 years of eliglustat treatment. Bone pain and bone crisis data, available for 10 treatment‐naïve patients, 124 non‐splenectomized switch patients (121 patients for bone crisis), and 23 splenectomized switch patients, also did not show significant changes after eliglustat treatment in any cohort.
3.5. Biomarker response
Median chitotriosidase values decreased from baseline to those at 2 years of eliglustat treatment by 78% in treatment‐naïve patients, 24% in non‐splenectomized switch patients, and 38% in splenectomized switch patients (P < .01 for all, Figure S1). Individual chitotriosidase values varied markedly, likely reflecting chitotriosidase genotype status and baseline disease severity. Chitotriosidase genotype data are not collected in the Registry. Therefore, patients who were homozygous for the common CHIT polymorphism and are likely to have no chitotriosidase activity could not be excluded. Likewise, chitotriosidase values for patients who were heterozygous for this variant, and thus expected to have half of the normal amount of chitotriosidase activity, could not be doubled as was done in the eliglustat clinical trials to normalize chitotriosidase values. Six patients had chitotriosidase values less than 30 nmol/mL/h at baseline, likely reflecting no chitotriosidase enzyme activity due to homozygosity of this common variant.
3.6. Quality of life
The SF‐36 data were available for a subset of patients. Among non‐splenectomized (n = 62) and splenectomized (n = 20) switch patients with SF‐36 data, all domain scores remained in the normal range (>47) before and after eliglustat. 18
4. DISCUSSION
In this real‐world experience in the worldʼs largest Gaucher disease registry, eliglustat treatment in patients with Gaucher disease type 1 resulted in clinically meaningful and statistically significant improvements in hematologic parameters and organ volumes for treatment‐naïve patients, and stability or modest improvement for ERT‐switch patients. In all patients, 2‐year mean values after eliglustat treatment for these indicators of disease activity were within long‐term therapeutic goal thresholds established for ERT. 16 , 17 Median chitotriosidase values also decreased significantly in all three cohorts, suggesting reversal of the metabolic inflammation and aberrant macrophage polarization that underpins Gaucher disease pathophysiology. Bone data were limited, but in the largest patient cohort (non‐splenectomized switch patients), the median lumbar spine Z‐score increased significantly after eliglustat treatment.
These findings from a real‐world registry cohort parallel those reported in the eliglustat clinical trials. While the treatment‐naïve cohort had less severe baseline disease than treatment‐naïve patients in the Phase 2 and Phase 3 ENGAGE clinical trials, it showed comparable improvements with eliglustat treatment, 12 , 13 achieving responses within therapeutic goal thresholds. Importantly, the largest therapeutic responses were seen in patients with the worst baseline disease status 19 (Figure 2). Similarly, improvements in switch patients paralleled those in the ENCORE switch trial of stable patients (34% of whom had a partial or total splenectomy 3 or more years before enrollment) at predefined therapeutic goals who switched to eliglustat after a mean of 10 years on ERT. 20 In both switch patient cohorts, improvements in spleen volume and lumbar spine bone mineral density were comparable and statistically significant. In our non‐splenectomized switch cohort, which had even longer prior exposure to ERT (mean 14 years), mean spleen volume decreased by 15% after 2 years of eliglustat treatment vs a 13% least‐square mean decrease after 4 years in ENCORE. In both cohorts, lumbar spine Z‐score improved by a similar margin (−0.7 to −0.4 in this analysis vs −0.3 to 0.04 in ENCORE). Median chitotriosidase values showed highly significant commensurate reduction with eliglustat treatment in both cohorts (P ≤ .005): 24% in non‐splenectomized switch patients and 38% in splenectomized switch patients after 2 years in this real‐world cohort compared to 63% after 4 years in ENCORE. 20
The safety of daily oral eliglustat cannot be assessed in the Registry, because adverse event data and reasons for discontinuing treatment are not captured. However, eliglustatʼs general tolerability is supported by the small proportion of Registry patients who discontinued treatment (22/231, 9%) as well as in the eliglustat clinical trials. Discontinuation could be attributable to adverse events, preference for ERT, limited commercial availability or reimbursement for eliglustat, or, in the seven women of childbearing age who discontinued treatment, because of pregnancy or desire to become pregnant, as eliglustat is not recommended for women during pregnancy. In a pooled analysis of adverse events from all four eliglustat clinical trials representing a mean of 3.6 years on treatment, 83% of patients remained in their trial until they were switched to commercial eliglustat (patients living in the United States) or the trial was completed, with 2.3% of the combined trial population discontinuing treatment due to an adverse event that was considered related to eliglustat. 21
This Registry analysis has several limitations. The ICGG Gaucher Registry is a voluntary observational database, and most patients did not have data for all variables of interest, particularly organ volumes, chitotriosidase, SF‐36 scores, and bone data. Clinical trial participation is also generally not recorded, and since data can be entered retrospectively, some data may have been collected during a patientʼs time in a clinical trial. However, the majority of data were collected in a real‐world setting, as 78% of patients began eliglustat treatment after the final patient was enrolled in an eliglustat clinical trial on 12 November 2012. Approximately 90% of patients in this analysis were from the United States, the first country to approve eliglustat. In addition, there was a preponderance of genotypes generally associated with less severe disease, especially among treatment‐naïve and non‐splenectomized switch patients. The dose of ERT prior to switching to eliglustat was not evaluated, and compliance with eliglustat or ERT is not captured in the Registry. The current analysis has no independent comparator group and does not capture the time course of treatment response, as only baseline and 2‐year (± 1 year) data were evaluated. As additional data accumulate in the real‐world setting, it will be possible to evaluate response to eliglustat treatment longitudinally. Although this analysis included six URM patients, safety and efficacy data in such patients are very limited and the drug label recommends against prescribing eliglustat for URMs.
In conclusion, eliglustat treatment outcomes in the real‐world setting of the ICGG Gaucher Registry are consistent with those reported in the pivotal clinical trials, demonstrating long‐term benefit in treatment‐naïve patients and ERT switch patients, in keeping with established therapeutic goals for Gaucher disease type 1. 16
CONFLICT OF INTEREST
This study was sponsored by Sanofi Genzyme. L.H.U. is an employee of Sanofi Genzyme and M.R.M. was an employee of Sanofi Genzyme during the time this manuscript was developed. All other authors (P.K.M., M.B., J.C., P.K., C.N.) are members of regional advisory boards of the ICGG Gaucher Registry and/or are principal investigators in clinical trials sponsored by Sanofi Genzyme, and have received research funding, educational grants, honoraria, consulting fees, and/or travel reimbursement from Sanofi Genzyme. J.C. has also served on advisory boards for Amicus and BioMarin; received support for multicenter trials from Shire, Amicus, BioMarin, Protalix; and honoraria for speaking from Shire and the Fabry Support and Information Group. C.N. has also received honoraria for scientific talks, grants, and advice from AbbVie, Alexion, Biogen, BMS, Boehringer, Falk, Gilead, Janssen, MSD, Roche, and Takeda‐Shire.
AUTHOR CONTRIBUTIONS
M.R.M. designed the analyses and reviewed all data for accuracy. P.K.M., M.B., J.C., C.N., L.H.U., and M.R.M. reviewed and interpreted the data. Laurie LaRusso (medical writer) drafted the manuscript with L.H.U. All authors reviewed early and final drafts of the manuscript and were fully responsible for the content and editorial decisions related to this manuscript.
Supporting information
Figure S1. Median chitotriosidase values at baseline and after 2 years of eliglustat therapy. Upper limit of normal for chitotriosidase: 120‐181 nmol/h/mL. 1 , 2 P values are from Signed‐Rank test (nonparametric) comparing 2‐year parameters to baseline. Chitotriosidase genotype data are not collected in the ICGG Gaucher Registry; thus, patients with a homozygous CHIT mutation could not be excluded and values for patients with a heterozygous mutation could not be doubled.
ACKNOWLEDGMENTS
The authors thank the patients with Gaucher disease, their physicians and health‐care personnel who submit data to the ICGG Gaucher Registry and the Gaucher Registry support team at Sanofi Genzyme. We thank Elizabeth Singer (Sanofi Genzyme) for programming support and Laurie LaRusso (Chestnut Medical Communications) for medical writing support paid for by Sanofi Genzyme.
Mistry PK, Balwani M, Charrow J, et al. Real‐world effectiveness of eliglustat in treatment‐naïve and switch patients enrolled in the International Collaborative Gaucher Group Gaucher Registry. Am J Hematol. 2020;95:1038–1046. 10.1002/ajh.25875
REFERENCES
- 1. Grabowski GA, Petsko GA, Kolodny EH. Gaucher disease. In: Valle D, Beaudet AL, Vogelstein B, et al., eds. OMMBID: The Online Metabolic and Molecular Bases of Inherited Disease New York, NY: McGraw‐Hill; 2013. http://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225546056. Accessed August 22, 2019.
- 2. Grabowski GA, Andria G, Baldellou A, et al. Pediatric non‐neuronopathic Gaucher disease: presentation, diagnosis and assessment. Consensus Statements. Eur J Pediatr. 2004;163(2):58‐66. [DOI] [PubMed] [Google Scholar]
- 3. Charrow J, Andersson HC, Kaplan P, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160(18):2835‐2843. [DOI] [PubMed] [Google Scholar]
- 4. Mistry PK, Weinthal JA, Weinreb NJ. Disease state awareness in Gaucher disease: a Q&A expert roundtable discussion. Clin Adv Hematol Oncol. 2012;10(6 suppl 8):1‐16. [PubMed] [Google Scholar]
- 5. Grabowski GA. Phenotype, diagnosis, and treatment of Gaucherʼs disease. Lancet. 2008;372(9645):1263‐1271. [DOI] [PubMed] [Google Scholar]
- 6. Pandey MK, Burrow TA, Rani R, et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature. 2017;543(7643):108‐112. [DOI] [PubMed] [Google Scholar]
- 7. Bultron G, Kacena K, Pearson D, et al. The risk of Parkinsonʼs disease in type 1 Gaucher disease. J Inherit Metab Dis. 2010;33(2):167‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Halperin A, Elstein D, Zimran A. Are symptoms of peripheral neuropathy more prevalent in patients with Gaucher disease? Acta Neurol Scand. 2007;115(4):275‐278. [DOI] [PubMed] [Google Scholar]
- 9. Biegstraaten M, Mengel E, Marodi L, et al. Peripheral neuropathy in adult type 1 Gaucher disease: a 2‐year prospective observational study. Brain. 2010;133(10):2909‐2919. [DOI] [PubMed] [Google Scholar]
- 10. Hicks JK, Swen JJ, Thorn CF, et al. Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin Pharmacol Ther. 2013;93(5):402‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.CERDELGA (eliglustat) [package insert]. Waterford, Ireland: Genzyme Corporation, A Sanofi Company; 2018. http://www.cerdelga.com/pdf/cerdelga_prescribing_information.pdf. Accessed August 22, 2019.
- 12. Lukina E, Watman N, Arreguin EA, et al. A phase 2 study of eliglustat tartrate (Genz‐112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood. 2010;116(6):893‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mistry PK, Lukina E, Ben Turkia H, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA. 2015;313(7):695‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cox TM, Drelichman G, Cravo R, et al. Eliglustat compared with imiglucerase in patients with Gaucherʼs disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open‐label, non‐inferiority trial. Lancet. 2015;385(9985):2355‐2362. [DOI] [PubMed] [Google Scholar]
- 15. Charrow J, Fraga C, Gu X, et al. Once‐versus twice‐daily dosing of eliglustat in adults with Gaucher disease type 1: the phase 3, randomized, double‐blind EDGE trial. Mol Genet Metab. 2018;123(3):347‐356. [DOI] [PubMed] [Google Scholar]
- 16. Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004;41(4 suppl 5):4‐14. [DOI] [PubMed] [Google Scholar]
- 17. Biegstraaten M, Cox TM, Belmatoug N, et al. Management goals for type 1 Gaucher disease: an expert consensus document from the European working group on Gaucher disease. Blood Cells Mol Dis. 2018;68:203‐208. [DOI] [PubMed] [Google Scholar]
- 18. Ware JE, Sherbourne CD. Userʼs Manual for the SF‐36v2 Health Survey. 2nd ed. QualityMetric Incorporated: Lincoln, RI; 2007:81‐84. chap 7. [Google Scholar]
- 19. Lukina E, Watman N, Dragosky M, et al. Outcomes after 8 years of eliglustat therapy for Gaucher disease type 1: final results from the phase 2 trial. Am J Hematol. 2019;94:29‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cox TM, Drelichman G, Cravo R, et al. Eliglustat maintains long‐term clinical stability in patients with Gaucher disease type 1 stabilized on enzyme therapy. Blood. 2017;129(17):2375‐2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peterschmitt MJ, Freisens S, Underhill LH, Foster MC, Lewis G, Gaemers SJM. Long‐term adverse event profile from four completed trials of oral eliglustat in adults with Gaucher disease type 1. Orphanet J Rare Dis. 2019;14(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Median chitotriosidase values at baseline and after 2 years of eliglustat therapy. Upper limit of normal for chitotriosidase: 120‐181 nmol/h/mL. 1 , 2 P values are from Signed‐Rank test (nonparametric) comparing 2‐year parameters to baseline. Chitotriosidase genotype data are not collected in the ICGG Gaucher Registry; thus, patients with a homozygous CHIT mutation could not be excluded and values for patients with a heterozygous mutation could not be doubled.