

Abstract
ATP-binding cassette (ABC)-transporters protect tissues by pumping their substrates out of the cells in many physiological barriers, such as the blood–brain barrier, intestine, liver, and kidney. These substrates include various endogenous metabolites, but, in addition, ABC transporters recognize a wide range of compounds, therefore affecting the disposition and elimination of clinically used drugs and their metabolites. Although numerous ABC-transporter inhibitors are known, the underlying mechanism of inhibition is not well characterized. The aim of this study is to deepen our understanding of transporter inhibition by studying the molecular basis of ligand recognition. In the current work, we compared the effect of 44 compounds on the active transport mediated by three ABC transporters: breast cancer resistance protein (BCRP and ABCG2), multidrug-resistance associated protein (MRP2 and ABCC2), and P-glycoprotein (P-gp and ABCB1). Eight compounds were strong inhibitors of all three transporters, while the activity of 36 compounds was transporter-specific. Of the tested compounds, 39, 25, and 11 were considered as strong inhibitors, while 1, 4, and 11 compounds were inactive against BCRP, MRP2, and P-gp, respectively. In addition, six transport-enhancing stimulators were observed for P-gp. In order to understand the observed selectivity, we compared the surface properties of binding cavities in the transporters and performed structure–activity analysis and computational docking of the compounds to known binding sites in the transmembrane domains and nucleotide-binding domains. Based on the results, the studied compounds are more likely to interact with the transmembrane domain than the nucleotide-binding domain. Additionally, the surface properties of the substrate binding site in the transmembrane domains of the three transporters were in line with the observed selectivity. Because of the high activity toward BCRP, we lacked the dynamic range needed to draw conclusions on favorable interactions; however, we identified amino acids in both P-gp and MRP2 that appear to be important for ligand recognition.
Keywords: drug transporter, efflux, inhibitor, stimulator, modulator
Introduction
ATP-binding cassette (ABC) transporters are found in tissues and at physiological barriers throughout the body, where they hydrolyze ATP to pump their substrates out of the cells, thus limiting the access and exposure of toxic compounds. Their substrates range from endogenous metabolites and hormones to drugs and xenobiotics. Breast cancer resistance protein (BCRP or ABCG2), multidrug-resistance associated protein 2 (MRP2, ABCC2), and P-glycoprotein (P-gp, ABCB1) are among the most studied drug transporters. Originally, these transporters were associated with drug resistance in cancer, but later considerable interest has been directed toward their physiological role and impact on drug therapies.1−3
BCRP is abundantly present in the intestine, where it hinders the permeation of drugs such as rosuvastatin, atorvastatin, and sulfasalazine.4 As a consequence of drug–drug interactions (DDIs), the peak plasma concentration and exposure of a drug substrate may be significantly elevated by the inhibition of BCRP.5 BCRP is found in many other tissues as well, for instance the blood–brain barrier, placenta, liver, and kidney. Hepatic BCRP is located on the canalicular membrane of hepatocytes and confers the excretion of estrone sulfate into bile6 but other estrogen conjugates are substrates as well.7 In the kidney, BCRP faces the lumen of the urinary tract and is responsible for translocating urate into primary urine. Patients with nonfunctional variants of BCRP are more susceptible to hyperuricemia, which increases the risk of gout.8
MRP2 is expressed mainly in the intestine, kidney, and liver. The main physiological function of hepatic MRP2 is pumping bile salts as well as glucuronide, glutathione, and to a smaller extent sulfate conjugates to the bile. Patients with a dysfunctional variant of MRP2 suffer from the Dubin–Johnson syndrome, where bilirubin-glucuronide excretion and bile flow may be disrupted.9,10 This leads to elevated serum levels of conjugated bilirubin, but otherwise the syndrome is benign. Currently, no clinically significant DDIs are known for MRP2, which may be due to other MRP transporters with overlapping substrate specificity acting as compensatory pumps. However, animal studies suggest that MRP2 may limit oral bioavailability of its substrate,11,12 and it is suggested that drugs inhibiting multiple MRP transporters may lead to iatrogenic hepatotoxicity or nephrotoxicity.2,13
P-gp affects the pharmacokinetics of many drugs, including digoxin,14 fexofenadine,15 doxorubicin,16 vinblastine,17 and loperamide.18 In addition, several DDIs involving P-gp are reported in the literature. For instance, oral bioavailability and plasma levels of digoxin and dabigatran increase significantly upon P-gp inhibition.19,20 At the blood–brain barrier, the P-gp inhibitor quinidine promotes the permeation of loperamide to the central nervous system, causing depressed respiratory activity.21 P-gp is well expressed in the placenta, adrenal glands, liver, and kidney as well.22,23
Because of the localization and ability to affect pharmacokinetics of many drugs and drug metabolites, alteration of a transporter’s function may cause accumulation of its substrates and lead to adverse events. The Food and Drug Administration (FDA) and European Medicines Agency (EMA) consider BCRP and P-gp as clinically relevant transporters and recommend studying them during drug development to characterize potential DDIs and reveal underlying mechanisms of action.24,25 In addition, the International Transporter Consortium recommends that new drug candidates are investigated for MRP2 inhibition, if signs of cholestasis or conjugated hyperbilirubinemia surface in clinical trials.2
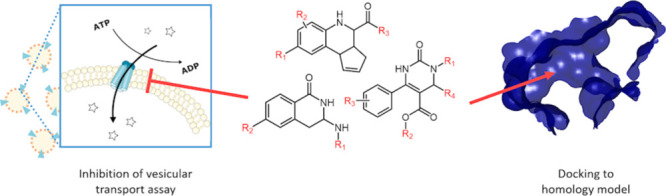
The interaction between the inhibitor and transporter can be explored with various in vitro and in silico methods. Cell assays are recommended for substrate studies and may provide kinetic parameters for inhibition as well. However, inhibitors concentrations inside the cell cannot be controlled, which is a drawback in the case of efflux transporters. However, this limitation can be circumvented in the vesicular transport assay with inverted membrane vesicles, where the studied inhibitors are not required to penetrate into the cell but have direct access to transporters. Several structure–activity relationship (SAR) analyses, mechanistic hypotheses and pharmacophore models have been reported based on in vitro inhibition data for BCRP and P-gp (reviewed by Gandhi & Morris26 and Wang et al.(27)) or MRP2.28−30 Matsson and co-workers31 compared the inhibitory activity of 122 registered drugs for BCRP, MRP2, and P-gp inhibition. They identified several specific inhibitors in the set, and found that inhibitors generally had a larger molecular size, lipophilicity, and aromaticity than noninhibitors but did not provide structural explanation for the observed selectivity. In ligand-based methods, the information about ligand–protein interactions is assumed to lie in the chemical structure and properties of the ligand in contrast to structure-based in silico docking methods, where the three-dimensional protein structure is considered as well. For instance, molecular docking scores describe the physicochemical interaction between a ligand and protein, which can be used to predict relative substrate affinities32 but also to visualize the binding of ligands and to map the binding sites.33 The structure-based approach relies heavily on the quality of the used structure. This restriction has limited its application, as, until recently, only few experimentally determined structures of ABC transporters were available.
In this work, we studied in vitro and in silico the interaction of 44 compounds with BCRP, MRP2, and P-gp in order to investigate the molecular basis of their selectivity to these transporters. These compounds belong to three different scaffolds for which we have previously studied the SAR for MRP2.29,30 Our in silico study points to amino acid residues in the transporters that may play an important role in the interaction with these compounds.
Materials and Methods
Materials
Spodoptera frugiperda (Sf9) insect cells were obtained from the American Type Culture Collection (ATCC, USA). The cells were cultured in HyClone SFX Insect medium (GE Healthcare, USA) supplemented with the fetal bovine serum (Gibco). Lucifer yellow (LY, Sigma-Aldrich, USA), 5(6)-carboxy-2′,7′-dichlorofluorescein (CDCF, Sigma-Aldrich, USA), and N-methyl-quinidine (NMQ, Solvo Biotechnology, Hungary) were used as substrate probes. The 44 tested inhibitors were provided by the University of Pittsburgh Chemical Methodologies and Library Development Center (UPCMLD, USA) and were described previously.29,30 The remainder of the chemicals were purchased from Sigma-Aldrich (USA) unless stated otherwise.
Vesicle Preparation
Using a Bac-to-Bac Baculovirus expression system (Invitrogen Life Technologies, USA), we generated BCRP, MRP2, and P-gp (corresponding to Uniprot entries Q9UNQ0, Q92887, and P08183, respectively) encoding baculoviruses in Sf9 cells. Inside-out membrane vesicles containing investigated transporters were prepared as described previously.34 In short, Sf9 cells were transfected with an amplified virus solution and cultivated for approximately 60 h, where after the cells were harvested and washed with harvest buffer (50 mM Tris-HCl, 300 mM d-mannitol). Then, the cells were lysed and homogenized with a Dounce tissue homogenizator with pestle B (Sigma-Aldrich) in membrane buffer (50 mM Tris-HCl, 50 mM d-mannitol, 2 mM ethylene glycol tetraacetic acid) on ice. The supernatant of the lysate was centrifuged at 100,000g for 75 min to collect the crude membrane. This membrane pellet was resuspended in membrane buffer, passed through a 27-gauge needle 20 times, and measured for protein concentration before storing at −80 °C until use.
The BCRP and P-gp membrane vesicles were loaded with cholesterol to improve their activity in the VT-assay.35 Membranes were incubated on ice with a cholesterol-RAMED (randomly methylated-β-cyclodextrin) complex (Cyclolab, Hungary). During the incubation, the concentration of cholesterol was set to 2.5 mM. After 20 min of incubation, the excess cholesterol was removed and vesicles were resuspended in membrane buffer. Finally, the newly acquired suspension was passed through a 27-gauge needle 20 times before storing at −80 °C.
Vesicular Transport Assay
The vesicular transport assay was performed as described previously.34 As substrate probes for BRCP, MRP2, and P-gp, we used 50 μM LY, 5 μM CDCF, and 2 μM NMQ, respectively. Each investigated compound was tested as triplicates in the presence and absence of ATP using a concentration of 80 μM. Membrane vesicles in assay buffer (40 mM pH 7.0 MOPS-tris, 55 mM KCl, and 6 mM MgCl2) were first incubated at 37 °C with the substrate probe and test compounds for 10 min. In MRP2 assays, additionally, 2 mM glutathione was included. The transport was started by adding a prewarmed 4 mM ATP-solution or assay solution. The total reaction time depended on the substrate probe and was 10 min for LY and NMQ, and 30 min for CDCF. At the end of incubation, the reaction was terminated with ice-cold assay buffer (40 mM pH 7.0 MOPS-tris and 70 mM KCl). After termination, the membrane solution was quickly transferred and filtered on glass fiber filter plates (MultiScreenHTS-FB, Millipore, USA) and then washed five times with ice cold assay buffer. Finally, depending on the probe, either 0.1 M NaOH (LY, CDCF) or 3:1 MeOH/H2O + 0.1% formic acid (NMQ) was added to break down the vesicles and release the probe.
LY samples in NaOH were treated with an equal volume of 0.1 M HCl before they were fluorometrically detected with a Varioskan Flash (Thermo Fisher Scientific, Finland) using excitation and emission wavelengths of 430 and 538 nm, respectively. The corresponding wavelengths for CDCF were 510 and 535 nm. The analysis of NMQ was performed with Agilent 110 series high-performance liquid chromatography (Agilent Technologies, USA) using a Poroshell 120 EC-C18 column with a size of 4.6 mm × 100 mm and 2.7 μm particle size (Agilent Technologies, USA). The temperature of the column was kept at 40 °C. The following method was used for analysis: 0–1 min(15% B), 1–3 min(15–35% B), 3–4 min (90% B), and 4–6.5 min (15% B), where eluent A was 0.1% formic acid and eluent B was acetonitrile. The flow rate of the eluent was 1 mL/min. The injection volume of samples was 10 μL, and the retention time for NMQ was 2.6 min. A fluorescence detector was used for detection with the excitation and emission at 248 and 442 nm, respectively.
Interference by the aggregation of the studied compounds in the assay were evaluated with a Nepheloskan Ascent nephelometer (Thermo Fisher Scientific, USA). In the aggregation control test, the turbidity by light scattering of a vesicular transport assay solution, which contained 80 μM of the test compound, was measured.
Transport Data Analysis
The ATP-dependent transport was determined by subtracting the substrate probe transport in the absence of ATP from the transport in the presence of ATP (eq 1). Relative inhibitory activity from the vesicular transport assay was calculated by comparing the ATP-dependent transport in the presence of the tested compound with a control containing vehicle (eq 2).
| 1 |
| 2 |
The statistical analysis whether docking scores or ligand interactions correlated with in vitro activity was determined by Graph Pad Prism 6.07 using an F-test (GraphPad Software Inc., USA).
Homology Modeling
Homology models were generated using MODELLER (v. 9.18)36 (see Supporting Information 1 for more details). MRP2 was modeled on the template of bovine MRP1 bound to leukotriene C4 (PDB ID 5UJA). Alignment of MRP2 to MRP1 was fetched from the Ensembl database (release 89),37 where we selected the gene tree containing MRP1, MRP2, and MRP3 genes. The tree had around 200 sequences, which were curated manually, resulting in a tree of 196 sequences. We generated 200 models using a MODELLER’s slow refinement protocol and selected the best model as assessed by the global DOPE score.38 P-gp had multiple mouse structures available in the PDB in different conformations (apo and substrate-bound). We selected three templates upon which to model human P-gp: PDB IDs 3G5U, 4Q9K, and 5KO2. Alignment was done with Clustal Omega (v.1.2.4).39 One hundred models were generated with a standard MODELLER protocol, and the best one was selected by the global DOPE score. For illustrations of the binding cavity, the electrostatic surfaces were calculated with the APBS plugin40 in PyMOL version 2.1 (Schrödinger LLC, USA).
Docking Simulations
To characterize the active site residues and predict binding modes for the scaffold 1, 2B, 2C, and 3 compounds, we conducted docking simulations to the human BCRP structure in the inward-facing translocation pathway conformation (PDB ID 5NJ3) and homology models generated for MRP2 and P-gp using GLIDE software (Schrödinger LLC, USA).41 The proteins were prepared using Maestro (Schrödinger LLC, USA), applying the OPLS-2005 force field. The protein GRID was calculated for a box that was defined to cover the substrate binding cavity (SBC) or the nucleotide binding site. A standard box size of 10 Å from each side was used. Neither positional nor hydrogen bonding constraints were applied. The compounds were prepared for docking using the LigPrep module (Schrödinger LLC, USA). For docking simulations, the extra precision (XP) parameters of GLIDE were selected with a flexible ligand and flexible receptor routine for identifying the best molecular interaction pose of the ligand with the target protein. We generated up to one million poses during the docking run and then selected 100 best poses for post-docking minimization. Ten docking poses were saved for each molecule, the docking being terminated if two consecutive solutions were within a root-mean-square deviation (rmsd) of 0.5 Å.
The interaction fingerprint program (Schrödinger LLC, USA), which implements a variant of the method described by Deng et al.,42 was used to compute interaction fingerprints between a protein and ligands. The R statistic program was used to analyse the protein–ligand interaction matrix and identify key binding site residues and interactions.43
Results
Vesicular Transport Assay
We have previously investigated the inhibitory activity of the 44 compounds in this study with MRP2,30 but now also tested their capacity to inhibit transport by BCRP and P-gp, as well as retested them with MRP2. The set contains compounds from three main scaffolds (Figure 1). Scaffold 2 is further divided into three sub-scaffolds based on the R1 substituent.29 The results from the vesicular transport assays are presented in Figure 2 and Tables 1–5 (and summarized in Supporting Information 2). The data of MRP2 are in line with our previous studies (Supporting Information 3), with every scaffold except 2A being highly active toward MRP2. Out of 44 compounds, 39, 25, and 11 inhibited strongly (>75% inhibition) BCRP, MRP2, and P-gp, respectively, while 8 of these compounds exerted inhibition toward all studied transporters. In contrast, only 1, 4, and 11 compounds were considered inactive (inhibitory activity between 25 and −25%) against BCRP, MRP2, and P-gp, respectively. Transport enhancing compounds, stimulators, were observed only for P-gp. In total, 6 compounds stimulated P-gp-mediated transport by more than 25% (inhibitory activity −25% or less). In addition, the P-gp stimulators were spread across different scaffolds. No intrinsic fluorescence of the test compounds was found in our previous study.30 To rule out false inhibition caused by aggregation, we examined the assay interference potential of each compound. Only three compounds (2ab, 2g, and 2h, the compounds in scaffold 2A) exhibited turbidity in the assay environment (Supporting Information 4).
Figure 1.

Markush structures of the three main scaffolds in the set of compounds.
Figure 2.

Heatmap of the interactions between test compounds and transporters. Intensity of the color stands for the degree of interaction. Red color indicates inhibition, while blue color describes stimulation. SARs within the scaffolds.
Table 1. Inhibitory Activity and Docking Scores in Scaffold 1a.

In every cell, below the name of the compound are first given the relative inhibitory activities and then the docking scores for BCRP, MRP2, and P-gp, respectively. R4 in all compounds is 2-cyclopentene.
Table 5. Inhibitory Activity and Docking Scores of Scaffold 3a.
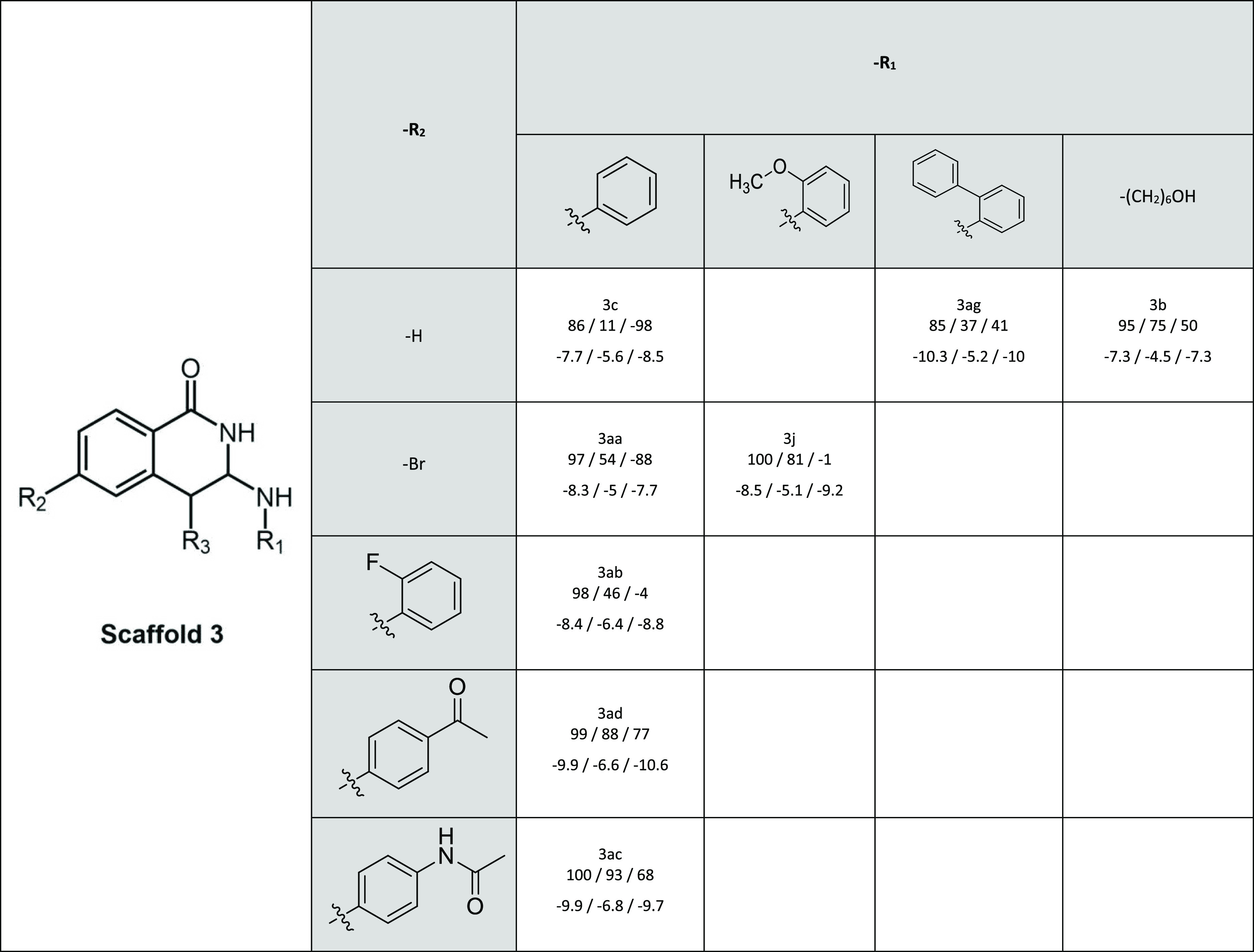
In every cell, below the name of the compound are first given the relative inhibitory activities and then the docking scores for BCRP, MRP2, and P-gp, respectively. R3 in all compounds is a hydrogen (−H)
Compounds of scaffold 1 were highly active toward all transporters (Table 1). The positive impact of the carboxylic acid in the R3-position on MRP2 inhibition was observed earlier.30 For P-gp, the introduction of a carboxylic acid in the R3-position (e.g. 1a vs 1b and 1e vs 1f) or fluorine (1k vs 1i) in the R1-position weakened inhibition, while compound 1g with fluorine in the R2-position was a good inhibitor of P-gp.
Scaffold 2A was the least active scaffold in interacting with MRP2 in our previous studies and therefore only three compounds (2ab, 2g, 2h), all with an ethyl group in the R2-position, were selected (Table 2). All three compounds were good BCRP and P-gp inhibitors. These compounds lack a carboxyl group in position R1, which was present in scaffold 2B and 2C and was previously suggested to be connected with the weak MRP2 interaction.30
Table 2. Inhibitory Activity and Docking Scores of Scaffold 2Aa.

In every cell, below the name of the compound are first given the relative inhibitory activities and then the docking scores for BCRP, MRP2, and P-gp, respectively. R1 and R4 are free amide (−H) and ethylbenzene, respectively.
According to our previous findings, halogen substituents in position R2 are important for MRP2 inhibition in scaffold 2B (Table 3). Similarly, as for MRP2, aromatic and halogen groups at R2 are beneficial for the inhibition of BCRP by scaffold 2B compounds. Interestingly, benzyl at R2 may cause P-gp stimulation as demonstrated by 2ag and 2ah, but the effect is disrupted by two chlorine substituents in the aromatic ring of the R2 substituent in compound 2al.
Table 3. Inhibitory Activity and Docking Scores of Scaffold 2Ba.

In every cell, below the name of the compound are first given the relative inhibitory activities and then the docking scores for BCRP, MRP2, and P-gp, respectively. R1 in all compounds is a butyric acid and R4 a methyl group.
Scaffold 2C contains inhibitors for all three transporters. An aromatic ring in the R2 position (2ax, 2bb, 2av, and 2az) is connected with markedly better inhibitory activity compared to the ethyl substituent (2p, 2o, 2ao, and 2an) (Table 4). Previously, we have already observed that scaffold 2C had an overall higher inhibitory activity toward MRP2 than scaffold 2B.30 Both scaffolds 2B and 2C contain a carboxylic acid group in position R1, butyric acid in 2B and hexanoic acid in 2C. The longer chain seems to increase the inhibitory activity also for P-gp and BCRP. This can especially be seen for P-gp in compounds 2al (scaffold 2B) and 2bb (scaffold 2C), and for BCRP in 2af (scaffold 2B) and 2ao (scaffold 2C) that differ only by the length of the carbonyl chain in the R1 substituent.
Table 4. Inhibitory Activity and Docking Scores of Scaffold 2Ca.
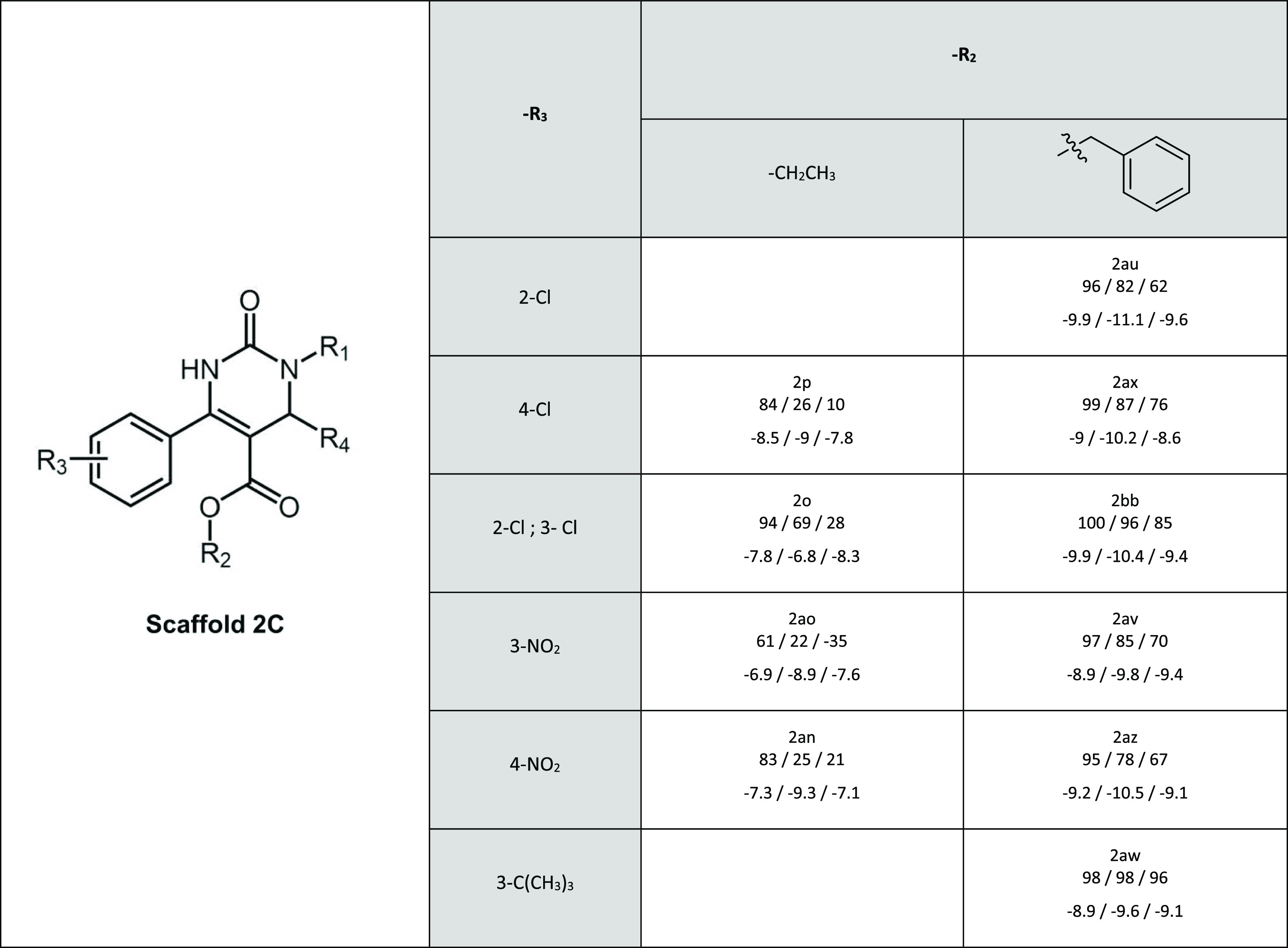
In every cell, below the name of the compound are first given the relative inhibitory activities and then the docking scores for BCRP, MRP2, and P-gp, respectively. R1 in all compounds is a hexanoic acid and R4 a methyl group.
Bulkiness and aromaticity of scaffold 3 compounds, predominantly in the R2 position, were previously reported to improve MRP2 inhibition (Table 5).30 The size of the R2 position appears to have a great impact on the activity of P-gp as well but with the opposite effect, as compounds with smaller substituents (3c, 3aa) stimulate P-gp transport while the bulkiest compounds (3ad, 3ac) inhibit P-gp.
Binding Site Analysis
Known ABC transporters share a large SBC that is located half-way across the membrane and can recognize multiple compounds, with a stoichiometry that can be more than 1. We also focus the study on the SBC, the most likely binding site, although binding to the nucleotide binding domains (NBDs) or to other allosteric sites may not be excluded. The electrostatic surfaces, the SBC of BCRP, MRP2, and P-gp, are illustrated in Figure 3, and we can observe clear differences in the surface properties of the binding cavities. The SBC of BCRP is relatively nonpolar, but there is a small positively charged area that may interact with polar ligands. The binding cavity of P-gp is also mostly nonpolar with minor polar regions but predominantly with a negative charge. In contrast, the MRP2 SBC is strongly polar, containing large positively charged and small negatively charged regions.
Figure 3.
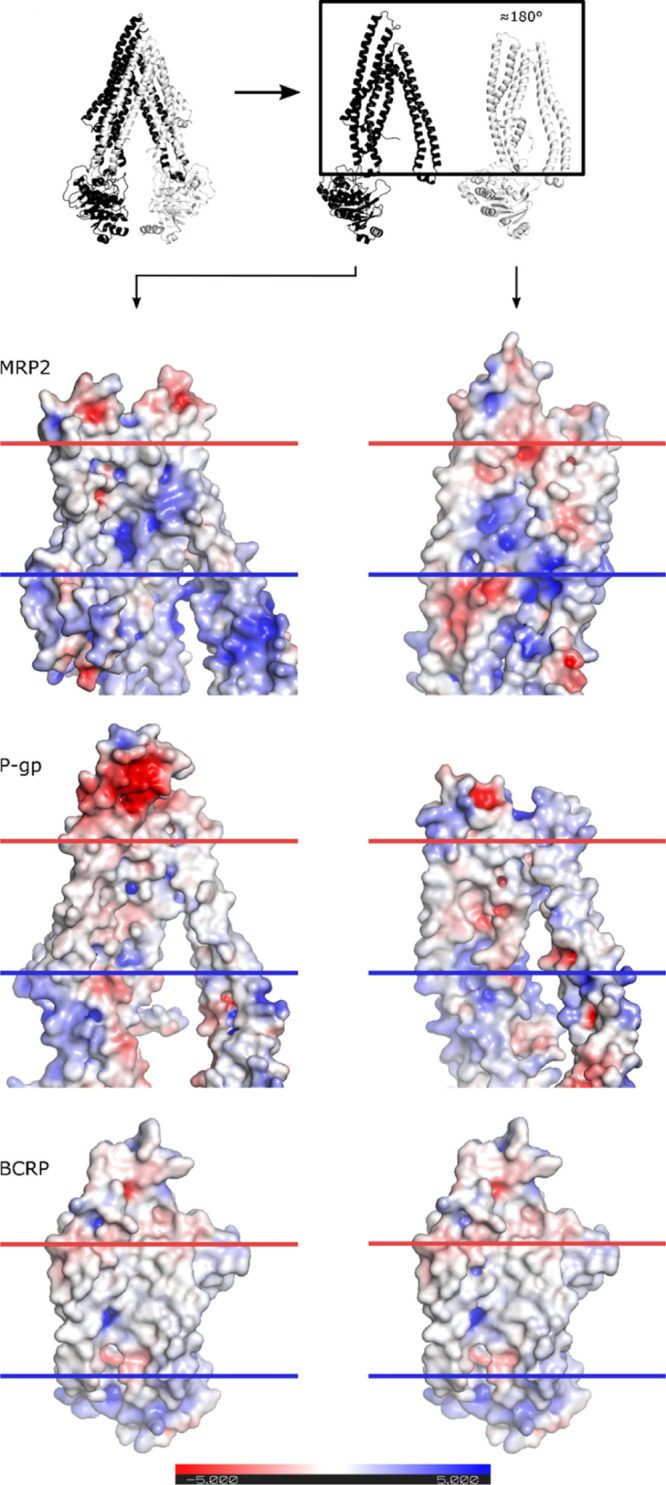
Transporters were separated into halves and rotated to show the inner part, as depicted in the schematic. Electrostatic surfaces were calculated with APBS plugin in PyMOL version 2.1. The negatively and positively charged surfaces are illustrated by red and blue colors, respectively. The plasma membrane region is indicated with horizontal blue and red lines.
In addition to the transmembrane binding cavity, the ABC transporters contain binding sites for ATP in the NBDs. The NBDs are structurally similar, with rmsd values ranging from 2.1 to 3 Å between transporters (Supporting Information 5), even though the sequence identity is rather low, only 20–33%. The ATP-binding site is formed by several conserved motifs such as the Walker A (GXXGXGK(S/T)), Walker B (ϕϕϕϕDE, where ϕ is a hydrophobic residue), the H-motif, as well as the ABC signature motif LSGGQ. Therefore, it is possible that binding to the conserved ATP-binding site provides a means to the nonselective inhibition by compounds that inhibited all three transporters.
Ligand-Residue Interaction Analysis
In order to investigate the molecular basis of the specificity of the tested compounds to BCRP, MRP2, and P-gp, we docked the test compounds and substrates to the crystal structure of BCRP and the homology models of MRP2 and P-gp. All test compounds were docked to both the SBC in the transmembrane domain and the ATP-binding site in the NBD of each transporter. The substrates (LY, CDCF, and NMQ) were docked to the SBC, and ATP was docked to the NBD of each transporter. The docking scores for the highest ranked pose of the substrates in BCRP were −5.3 (LY) and −9.5 (ATP), in MRP2 -6.8 (CDCF) and −8.2 (ATP), and in P-gp −8.1 (NMQ) and −7.7 (ATP). The docking of the test compounds was performed at both sites, as interaction at either site could interfere with transport and it is not possible to conclude from the results in the vesicle transport assays where the compounds bind. Eight compounds (1b, 1f, 1g, 1i, 2aw, 2ax, 2bb, and 3ad) were considered to be nonselective inhibitors as they showed inhibitory activity of 75% or more toward all studied transporters. The docking scores for these compounds to the NBD were not different from the more specific or noninteracting compounds (Supporting Information 2) and markedly higher than for ATP (higher docking scores indicate lower binding affinity). In general, the docking scores to the NBD were much higher than to the TMD (Supporting Information 2). It should be noted that while it is fair to compare docking scores relatively across a set of compounds (for a given binding site, as indicator of experimental binding affinity), comparing docking scores across binding sites is more likely to be biased, for example, by binding site composition, and appears to be a very speculative exercise. Thus, we decided to focus the theoretical part of this study on the SBC, the most likely binding site. The three substrates docked to the SBC of the transporters are visualized in Supporting Information 6. The lowest single pose SBC docking score of each compound is listed in Tables 1–5, and the docking scores are compared to the in vitro inhibitory activity in Supporting Information 7. In general, MRP2 compounds with good (low) docking scores are found in scaffolds 2B and 2C. In BCRP and P-gp, the compounds with the lowest docking scores are found in scaffolds 2A, 2C, and 3, yet the difference between scaffolds is less marked than for MRP2.
In scaffold 1, the impact of the decreased inhibition of P-gp by carboxyl groups is reflected by the docking scores. The effect of the trifluoromethyl group at the R1 position on probe transport cannot be connected to the docking scores (Table 1). In contrast, the pattern of halogen substitutions in scaffold 2B compounds for MRP2 somewhat followed the docking score (Table 3). In scaffold 2C, the aromatic ring at the R2 position lowered docking scores considerably compared to an ethyl substituent (Table 4). This was in the line with the in vitro data, where the introduction of an aromatic ring improved MRP2 and P-gp inhibition. The scaffold 3 compounds with hexanol at the R1 position had higher docking scores for both MRP2 and P-gp; yet, the in vitro activity was similar or even greater compared to the aromatic substituents (Table 5).
As there were considerable differences in the specificity of the tested compounds in vitro, we further investigated in more detail the molecular interactions between the tested compounds and transporter residues in the SBC, with the intention to identify key interactions for the observed differences in activity. First, the number of and types of interactions that each tested compound had with the transporters was compared to the in vitro activity (Supporting Information 8). For instance, a higher number of polar interactions with MRP2 in scaffold 2C compounds and hydrophobic interactions with P-gp by the compounds of scaffold 3 correlated with in vitro activity. Then, we examined the interactions in order to identify specific residues that were associated with modulated transport activity (Table 6). For instance, in MRP2 where scaffold 2C compounds with high activity had several polar interactions, particularly interactions with Ser594 distinguished weak inhibitors from strong ones. On the other hand, in P-gp the hydrophobic and aromatic residues Phe72, Phe957, and Phe978 appeared to be important for stimulation of transport. These residues are located near the extracellular side of the TMD (Figure 4). In scaffold 2C, the only compound (2ao) that stimulated transport was also the only one that interacted with Phe72 and Phe957, while the two stimulating compounds in scaffold 3 (3c and 3aa) were not interacting with Phe978, unlike the inhibitors and noninteracting compounds in that scaffold. Based on the docking results, the binding sites of NMQ, the substrate used in our in vitro assay and the tested compounds partially overlap, but NMQ does not interact with Phe978 or with Phe72 or Phe957.
Table 6. Selected Residues in the Transporter Proteins Interacting with Inhibitors Based on the Docking Analysisa.

The impact of tested compounds on MRP2 and P-gp transport is described with an activity value. A positive value indicates inhibition, while a negative value stands for stimulation. The interaction of tested compounds with each residue is defined with the – (no interaction) or + (interaction) sign. The type of interaction is described above the name of the residue.
Figure 4.
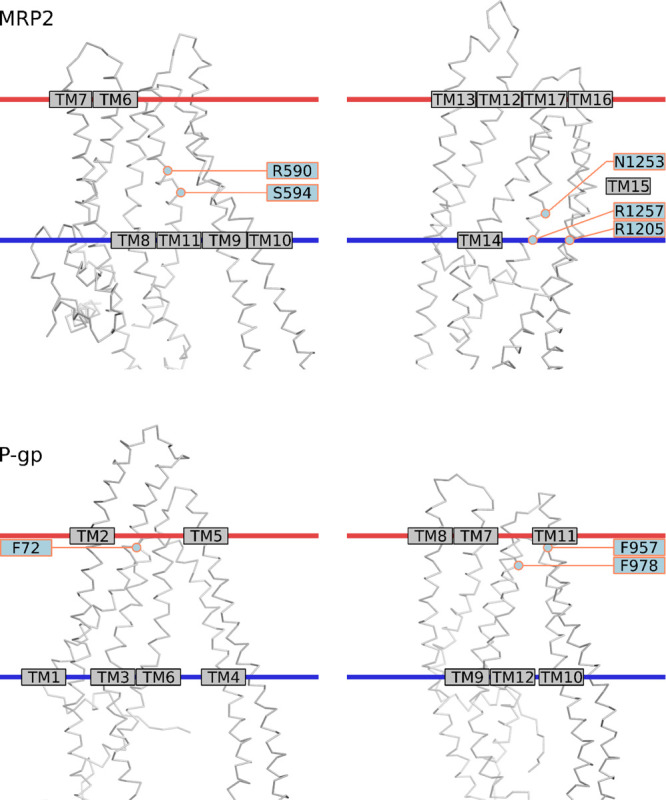
C alpha-trace of MRP2 and P-gp models. Selected residues in the binding cavity are labeled. The membrane approximation (horizontal red and blue lines) was fetched from OPM database (Lomize et al. 2012) (PDB ID 5UJA for MRP2 and 5KO2 for P-gp).
Because of the high correlation of polar interactions for compounds in scaffold 2C and MRP2 inhibition (Table 6), the binding mode was studied in more detail in the MRP2 homology model. The carboxyl group orients the compounds by interacting to either Arg1205, Arg1257, and Asn1253 or to Arg590 (Figure 5, Supporting Information 9). These residues are located in the inner leaflet of the plasma membrane (Figure 4). Ser594 lies in the end of a small sub-pocket and the inhibitors interacting with the residues in this pocket (Asn477, Val546, Ser594, Met595, Met598) have either a phenyl ring or tert-butyl group reaching into the sub-pocket, while the inactive compounds or the CDCF substrate do not reach into the sub-pocket (Figure 6).
Figure 5.

Binding site residues in MRP2 interacting with 2au and 2av—two inhibitors belonging to scaffold 2C.
Figure 6.

Binding pocket in MRP2. (A) Inhibitors in scaffold 2 reach into the sub-pocket formed by Asn477, Val546, Ser594, Met595, and Met598, while the substrate, CDCF (in yellow) or (B) noninteracting compounds in scaffold 2 do not extend into the sub-pocket.
Discussion
We compared the ability of 44 compounds to inhibit the in vitro activity of the active transport mediated by BCRP, MRP2, and P-gp. We found that eight compounds were strong inhibitors of all three transporters at the tested concentration, while the activity of the other 36 compounds was transporter specific. We then aimed to find an explanation on a molecular level for the observed similarities and differences in activity toward the transporters. Visualization of the surface properties of the SBC showed that MRP2 has the most polar surface with both negatively and positively charged regions, while P-gp and BCRP have mostly nonpolar SBC surfaces, with small negatively and positively charged regions, respectively. We chose to focus the in silico characterization of binding interactions within the SBC in the TMD.
Almost all tested compounds were able to inhibit BCRP to some degree, as only one compound (2af) was inactive and four were weak inhibitors while the 39 remaining strong inhibitors were able to decrease the transport by >75%. Unfortunately, the strong inhibition of BCRP by almost all compounds in the study reduced the dynamic range of analysis, thus limiting the conclusions we could draw. The docking results were in line with the in vitro results, as the docking score for the LY substrate (−5.3) was similar to or higher than for the test compounds (average −8.2). The high BCRP activity was unexpected, even though BCRP has been reported to be more susceptible for inhibition by natural compounds and drugs compared to MRP2.31,34,44 Furthermore, the large difference in the inhibition profile of BCRP compared to P-gp was surprising. P-gp and BCRP are reported to have a considerable overlap in inhibitors,31 but within our set of compounds, we found that the activity patterns were rather distinctive. Only 11 of the tested compounds were strong inhibitors of P-gp, while, in contrast, six compounds were able to stimulate the active transport of P-gp by >25%. A likely explanation for the observed differences in activity of BCRP and P-gp is that the narrow chemical space of the compound library, which was selected based on inhibition toward MRP2, favored BCRP inhibition. For instance, many tested compounds contained a negative charge, which is present in many conjugated metabolites transported by BCRP and therefore should interact more favorably with BCRP than with P-gp.7,45,46 The negative charge in these compounds also favors binding to the positively charged regions found in the SBC of BCRP, compared to the negatively charged regions in P-gp that would repel these compounds. The substrates and competitive inhibitors of P-gp are generally hydrophobic and positively charged,47,48 providing for favorable interactions with negatively charged regions in the SBC in P-gp. Our observations that bulkiness and aromaticity, in the inhibitors, increase the activity are in line with these findings. Halogen substitutions are thought to increase the lipophilicity and membrane permeation and favor interactions with electron-rich aromatic and negatively charged oxygen containing residues.49−51 However, the aliphatic halogenated compounds in our study showed decreased activity toward P-gp. Particularly in scaffold 1, the replacement of hydrogens (compounds 1f, 1ad, 1i) with fluorine (1j, 1ag, 1k) in the R1 position reduced inhibition. The size of the R2 substitution in scaffold 3 affected the activity of P-gp, where the compounds with the smallest substituents (3aa, 3c) were in fact stimulating active transport. Our docking studies suggest that these stimulators in scaffold 3 were not interacting with Phe978, in contrast to the inhibitors and inactive compounds in that scaffold. Phe978 is located in TM12 (Figure 4) and has been previously been found to play a key role in substrate recognition and drug resistance in P-gp expressing cells.52
MRP2 is well known as an organic anion transporter, pumping out numerous negatively charged drugs and conjugates. Therefore, it was expected that the ligand-binding cavity in the MRP2 model is highly positively charged and that a negative charge in the tested compounds was beneficial for inhibition in our assay. With the help of docking studies, we suggest several amino acids that may have a key role in the binding of active compounds. The carboxyl group in scaffold 2C compounds interacts with Arg1205 (located in transmembrane helix 16, TM16), Asn1253, and Arg1257 (TM17) or alternatively with Arg590 (TM11) (Figure 4). Arg1257 has previously been identified to participate in the binding of substrates, as the Arg1257Ala mutant had decreased transport activity, while the Arg590Ala mutant did not have altered transport properties.53 These three arginines are conserved in MRP1 (Arg593, Arg1197, and Arg1249), and the mutation of any of them to a differently charged amino acid affects the transport activity significantly.54,55 The transmembrane helices corresponding to TM11 and TM17 in MRP2 have previously been identified to be involved in substrate binding in MRP1, MRP2, and MRP4.56−61 We identified a sub-pocket of the MRP2 binding site that is particularly interesting as the interaction is associated with high inhibitory activity for the scaffold 2C compounds in our study. Interestingly, the MRP2 substrate CDCF that was used in the in vitro assay, does not reach into this pocket, which support that these interactions are important for inhibiting the transporter. It is, however, still unclear which of the interactions with the residues (Asn477, Val546, Ser594, Met595, and Met598) in the binding site sub-pocket can affect inhibition. This region is not highly conserved and could therefore play a role in ligand specificity. Mutagenesis studies are required to reveal the importance of the individual residues in the sub-pocket for the activity of inhibitors.
Conclusions
Our study revealed unexpected differences in the inhibitor recognition of BCRP, MRP2, and P-gp. The selectivity of the inhibitors was partially explained by the different surface charges in the SBCs of the three transporters. We identified a sub-pocket and three conserved arginines in the MRP2 binding site that potentially have a key role in inhibitor binding.
Acknowledgments
We want to thank Tuomas Tepponen and Erkka Järvinen for technical assistance. Also, we would like to thank University of Helsinki, Academy of Finland (292779), Magnus Ehrnrooth Foundation, Finnish Cultural Foundation and Finnish Pharmacists’ Society for funding of this research. Furthermore, L.G. gratefully acknowledges the support of the Drug Discovery and Chemical Biology Network of Finland. The CSC—IT Center for Science Ltd. is thanked for organizing computational resources.
Glossary
Abbreviations
- ABC
ATP-binding cassette.
- BCRP
breast cancer resistance protein (ABCG2).
- CDCF
5(6)-carboxy-2′,7′-dichlorofluorescein.
- DDI
drug–drug interaction.
- EMA
European Medicines Agency.
- FDA
The Food and Drug Administration.
- LY
Lucifer yellow.
- MRP
multidrug resistance associated protein (ABCC).
- NBD
nucleotide-binding domain.
- NMQ
N-methyl-quinidine.
- P-gp
P-glycoprotein (ABCB1).
- SBC
substrate-binding cavity.
- TM
transmembrane helix.
- TMD
transmembrane domain.
- VT
vesicular transport.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.0c00155.
Homology modeling protocol, in vitro activity and docking score values, comparison of MRP2 assay results, nephelometer results, rmsd value matrix, visualization of docked substrates, correlation of activity and docking score, correlation of activity and number of interactions, and MRP2 interaction to 2aw, 2ax, 2az, 2bb, and 2o (PDF)
Author Present Address
∥ Department of Clinical Pharmacology, Faculty of Medicine, University of Helsinki, P.O. Box 56, 00014 University of Helsinki, Helsinki, Finland.
Author Contributions
⊥ F.D. and L.G., equal contribution by the co-authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Giacomini K. M.; Huang S. M.; Tweedie D. J.; Benet L. Z.; Brouwer K. L.; Chu X.; Dahlin A.; Evers R.; Fischer V.; Hillgren K. M.; Hoffmaster K. A.; Ishikawa T.; Keppler D.; Kim R. B.; Lee C. A.; Niemi M.; Polli J. W.; Sugiyama Y.; Swaan P. W.; Ware J. A.; Wright S. H.; Yee S. W.; Zamek-Gliszczynski M. J.; Zhang L. Membrane transporters in drug development. Nat. Rev. Drug Discovery 2010, 9, 215–236. 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillgren K. M.; Keppler D.; Zur A. A.; Giacomini K. M.; Stieger B.; Cass C. E.; Zhang L. Int Transporter, C., Emerging Transporters of Clinical Importance: An Update From the International Transporter Consortium. Clin. Pharmacol. Ther. 2013, 94, 52–63. 10.1038/clpt.2013.74. [DOI] [PubMed] [Google Scholar]
- Szakács G.; Paterson J. K.; Ludwig J. A.; Booth-Genthe C.; Gottesman M. M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discovery 2006, 5, 219–234. 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Mao Q.; Unadkat J. D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport--an update. AAPS J. 2015, 17, 65–82. 10.1208/s12248-014-9668-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsby R.; Martin P.; Surry D.; Sharma P.; Fenner K. Solitary Inhibition of the Breast Cancer Resistance Protein Efflux Transporter Results in a Clinically Significant Drug-Drug Interaction with Rosuvastatin by Causing up to a 2-Fold Increase in Statin Exposure. Drug Metab. Dispos. 2016, 44, 398–408. 10.1124/dmd.115.066795. [DOI] [PubMed] [Google Scholar]
- Imai Y.; Asada S.; Tsukahara S.; Ishikawa E.; Tsuruo T.; Sugimoto Y. Breast cancer resistance protein exports sulfated estrogens but not free estrogens. Mol. Pharmacol. 2003, 64, 610–618. 10.1124/mol.64.3.610. [DOI] [PubMed] [Google Scholar]
- Järvinen E.; Deng F.; Kidron H.; Finel M. Efflux transport of estrogen glucuronides by human MRP2, MRP3, MRP4 and BCRP. J. Steroid Biochem. Mol. Biol. 2018, 178, 99–107. 10.1016/j.jsbmb.2017.11.007. [DOI] [PubMed] [Google Scholar]
- Matsuo H.; Nakayama A.; Sakiyama M.; Chiba T.; Shimizu S.; Kawamura Y.; Nakashima H.; Nakamura T.; Takada Y.; Oikawa Y.; Takada T.; Nakaoka H.; Abe J.; Inoue H.; Wakai K.; Kawai S.; Guang Y.; Nakagawa H.; Ito T.; Niwa K.; Yamamoto K.; Sakurai Y.; Suzuki H.; Hosoya T.; Ichida K.; Shimizu T.; Shinomiya N. ABCG2 dysfunction causes hyperuricemia due to both renal urate underexcretion and renal urate overload. Sci. Rep. 2015, 4, 3755. 10.1038/srep03755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen P. L. M.; Peters W. H.; Lamers W. H. Hereditary chronic conjugated hyperbilirubinemia in mutant rats caused by defective hepatic anion transport. Hepatology 1985, 5, 573–579. 10.1002/hep.1840050408. [DOI] [PubMed] [Google Scholar]
- Paulusma C. C.; van Geer M. A.; Evers R.; Heijn M.; Ottenhoff R.; Borst P.; Oude Elferink R. P. J. Canalicular multispecific organic anion transporter/multidrug resistance protein 2 mediates low-affinity transport of reduced glutathione. Biochem. J. 1999, 338, 393–401. 10.1042/bj3380393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamek-Gliszczynski M. J.; Bedwell D. W.; Bao J. Q.; Higgins J. W. Characterization of SAGE Mdr1a (P-gp), Bcrp, and Mrp2 knockout rats using loperamide, paclitaxel, sulfasalazine, and carboxydichlorofluorescein pharmacokinetics. Drug Metab. Dispos. 2012, 40, 1825–1833. 10.1124/dmd.112.046508. [DOI] [PubMed] [Google Scholar]
- Dietrich C. G.; de Waart D. R.; Ottenhoff R.; Bootsma A. H.; van Gennip A. H.; Oude Elferink R. P. J. O. Mrp2-deficiency in the rat impairs biliary and intestinal excretion and influences metabolism and disposition of the food-derived carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Carcinogenesis 2001, 22, 805–811. 10.1093/carcin/22.5.805. [DOI] [PubMed] [Google Scholar]
- Izzedine H.; Launay-Vacher V.; Isnard-Bagnis C.; Deray G. Drug-induced Fanconi’s syndrome. Am. J. Kidney Dis. 2003, 41, 292–309. 10.1053/ajkd.2003.50037. [DOI] [PubMed] [Google Scholar]
- Drescher S.; Glaeser H.; Murdter T.; Hitzl M.; Eichelbaum M.; Fromm M. F. P-glycoprotein-mediated intestinal and biliary digoxin transport in humans. Clin. Pharmacol. Ther. 2003, 73, 223–231. 10.1067/mcp.2003.27. [DOI] [PubMed] [Google Scholar]
- Cvetkovic M.; Leake B.; Fromm M. F.; Wilkinson G. R.; Kim R. B. OATP and P-glycoprotein transporters mediate the cellular uptake and excretion of fexofenadine. Drug Metab. Dispos. 1999, 27, 866–871. [PubMed] [Google Scholar]
- Woodahl E. L.; Crouthamel M. H.; Bui T.; Shen D. D.; Ho R. J. Y. MDR1 (ABCB1) G1199A (Ser400Asn) polymorphism alters transepithelial permeability and sensitivity to anticancer agents. Cancer Chemother. Pharmacol. 2009, 64, 183–188. 10.1007/s00280-008-0906-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F.; Horie K.; Borchardt R. T. Are MDCK cells transfected with the human MRP2 gene a good model of the human intestinal mucosa?. Pharm. Res. 2002, 19, 773–779. 10.1023/a:1016192413308. [DOI] [PubMed] [Google Scholar]
- Skarke C.; Jarrar M.; Schmidt H.; Kauert G.; Langer M.; Geisslinger G.; Lötsch J. Effects of ABCB1 (multidrug resistance transporter) gene mutations on disposition and central nervous effects of loperamide in healthy volunteers. Pharmacogenetics 2003, 13, 651–660. 10.1097/00008571-200311000-00001. [DOI] [PubMed] [Google Scholar]
- Fenner K. S.; Troutman M. D.; Kempshall S.; Cook J. A.; Ware J. A.; Smith D. A.; Lee C. A. Drug-drug interactions mediated through P-glycoprotein: clinical relevance and in vitro-in vivo correlation using digoxin as a probe drug. Clin. Pharmacol. Ther. 2009, 85, 173–181. 10.1038/clpt.2008.195. [DOI] [PubMed] [Google Scholar]
- Delavenne X.; Ollier E.; Basset T.; Bertoletti L.; Accassat S.; Garcin A.; Laporte S.; Zufferey P.; Mismetti P. A semi-mechanistic absorption model to evaluate drug-drug interaction with dabigatran: application with clarithromycin. Br. J. Clin. Pharmacol. 2013, 76, 107–113. 10.1111/bcp.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeque A. J. M.; Wandel C.; He H.; Shah S.; Wood A. J. J. Increased drug delivery to the brain by P-glycoprotein inhibition. Clin. Pharmacol. Ther. 2000, 68, 231–237. 10.1067/mcp.2000.109156. [DOI] [PubMed] [Google Scholar]
- Thiebaut F.; Tsuruo T.; Hamada H.; Gottesman M. M.; Pastan I.; Willingham M. C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 7735–7738. 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara I.; Kataoka I.; Morishita Y.; Hamada H.; Tsuruo T.; Itoyama S.; Mori S. Tissue Distribution of P-Glycoprotein Encoded by a Multidrug-Resistant Gene as Revealed by a Monoclonal-Antibody, Mrk-16. Cancer Res. 1988, 48, 1926–1929. [PubMed] [Google Scholar]
- FDA . In Vitro Metabolism and Transporter Mediated Drug-Drug Interaction Studies Guidance for Industry, 2017. [Google Scholar]
- EMA . Guideline on the Investigation of Drug Interactions. CPMP/EWP/560/95/Rev. 1 Corr. 2**, 2012. [Google Scholar]
- Gandhi Y. A.; Morris M. E. Structure-activity relationships and quantitative structure-activity relationships for breast cancer resistance protein (ABCG2). AAPS J. 2009, 11, 541–552. 10.1208/s12248-009-9132-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. B.; Kuo C. L.; Lien L. L.; Lien E. J. Structure–activity relationship: analyses of p-glycoprotein substrates and inhibitors. Int. J. Clin. Pharmacol. Ther. 2003, 28, 203–228. 10.1046/j.1365-2710.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- Pedersen J. M.; Matsson P.; Bergström C. A. S.; Norinder U.; Hoogstraate J.; Artursson P. Prediction and identification of drug interactions with the human ATP-binding cassette transporter multidrug-resistance associated protein 2 (MRP2; ABCC2). J. Med. Chem. 2008, 51, 3275–3287. 10.1021/jm7015683. [DOI] [PubMed] [Google Scholar]
- Wissel G.; Kudryavtsev P.; Ghemtio L.; Tammela P.; Wipf P.; Yliperttula M.; Finel M.; Urtti A.; Kidron H.; Xhaard H. Exploring the structure-activity relationships of ABCC2 modulators using a screening approach. Bioorg. Med. Chem. 2015, 23, 3513–3525. 10.1016/j.bmc.2015.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissel G.; Deng F.; Kudryavtsev P.; Ghemtio L.; Wipf P.; Xhaard H.; Kidron H. A structure-activity relationship study of ABCC2 inhibitors. Eur. J. Pharm. Sci. 2017, 103, 60–69. 10.1016/j.ejps.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsson P.; Pedersen J. M.; Norinder U.; Bergström C. A. S.; Artursson P. Identification of Novel Specific and General Inhibitors of the Three Major Human ATP-Binding Cassette Transporters P-gp, BCRP and MRP2 Among Registered Drugs. Pharm. Res. 2009, 26, 1816–1831. 10.1007/s11095-009-9896-0. [DOI] [PubMed] [Google Scholar]
- Palestro P. H.; Gavernet L.; Estiu G. L.; Bruno Blanch L. E. Docking applied to the prediction of the affinity of compounds to P-glycoprotein. BioMed Res. Int. 2014, 2014, 358425. 10.1155/2014/358425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- László L.; Sarkadi B.; Hegedus T. Jump into a New Fold-A Homology Based Model for the ABCG2/BCRP Multidrug Transporter. PLoS One 2016, 11, e0164426 10.1371/journal.pone.0164426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöstedt N.; Deng F.; Rauvala O.; Tepponen T.; Kidron H. Interaction of Food Additives with Intestinal Efflux Transporters. Mol. Pharm. 2017, 14, 3824–3833. 10.1021/acs.molpharmaceut.7b00563. [DOI] [PubMed] [Google Scholar]
- Telbisz Á.; Müller M.; Özvegy-Laczka C.; Homolya L.; Szente L.; Váradi A.; Sarkadi B. Membrane cholesterol selectively modulates the activity of the human ABCG2 multidrug transporter. Biochim. Biophys. Acta 2007, 1768, 2698–2713. 10.1016/j.bbamem.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Šali A.; Blundell T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. 10.1016/S1357-4310(95)91170-7. [DOI] [PubMed] [Google Scholar]
- Zerbino D. R.; Achuthan P.; Akanni W.; Amode M. R.; Barrell D.; Bhai J.; Billis K.; Cummins C.; Gall A.; Girón C. G.; Gordon L.; Haggerty L.; Haskell E.; Hourlier T.; Izuogu O. G.; Janacek S. H.; Juettemann T.; To J. K.; Laird M. R.; Lavidas I.; Liu Z.; Loveland J. E.; Maurel T.; McLaren W.; Moore B.; Mudge J.; Murphy D. N.; Newman V.; Nuhn M.; Ogeh D.; Ong C. K.; Parker A.; Patricio M.; Riat H. S.; Schuilenburg H.; Sheppard D.; Sparrow H.; Taylor K.; Thormann A.; Vullo A.; Walts B.; Zadissa A.; Frankish A.; Hunt S. E.; Kostadima M.; Langridge N.; Martin F. J.; Muffato M.; Perry E.; Ruffier M.; Staines D. M.; Trevanion S. J.; Aken B. L.; Cunningham F.; Yates A.; Flicek P. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M.-y.; Sali A. Statistical potential for assessment and prediction of protein structures. Curr. Protein Pept. Sci. 2006, 15, 2507–2524. 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Söding J.; Thompson J. D.; Higgins D. G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurrus E.; Engel D.; Star K.; Monson K.; Brandi J.; Felberg L. E.; Brookes D. H.; Wilson L.; Chen J.; Liles K.; Chun M.; Li P.; Gohara D. W.; Dolinsky T.; Konecny R.; Koes D. R.; Nielsen J. E.; Head-Gordon T.; Geng W.; Krasny R.; Wei G.-W.; Holst M. J.; McCammon J. A.; Baker N. A. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Deng Z.; Chuaqui C.; Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein– ligand binding interactions. J. Med. Chem. 2004, 47, 337–344. 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- R Core Team . R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017.
- Sjöstedt N.; Holvikari K.; Tammela P.; Kidron H. Inhibition of Breast Cancer Resistance Protein and Multidrug Resistance Associated Protein 2 by Natural Compounds and Their Derivatives. Mol. Pharm. 2017, 14, 135–146. 10.1021/acs.molpharmaceut.6b00754. [DOI] [PubMed] [Google Scholar]
- Suzuki M.; Suzuki H.; Sugimoto Y.; Sugiyama Y. ABCG2 transports sulfated conjugates of steroids and xenobiotics. J. Biol. Chem. 2003, 278, 22644–22649. 10.1074/jbc.m212399200. [DOI] [PubMed] [Google Scholar]
- Zamek-Gliszczynski M. J.; Nezasa K.-i.; Tian X.; Kalvass J. C.; Patel N. J.; Raub T. J.; Brouwer K. L. R. The important role of Bcrp (Abcg2) in the biliary excretion of sulfate and glucuronide metabolites of acetaminophen, 4-methylumbelliferone, and harmol in mice. Mol. Pharmacol. 2006, 70, 2127–2133. 10.1124/mol.106.026955. [DOI] [PubMed] [Google Scholar]
- Li D.; Chen L.; Li Y.; Tian S.; Sun H.; Hou T. ADMET evaluation in drug discovery. 13. Development of in silico prediction models for P-glycoprotein substrates. Mol. Pharm. 2014, 11, 716–726. 10.1021/mp400450m. [DOI] [PubMed] [Google Scholar]
- Seelig A.; Blatter X. L.; Wohnsland F. Substrate recognition by P-glycoprotein and the multidrug resistance-associated protein MRP1: a comparison. Int. J. Clin. Pharmacol. Ther. 2000, 38, 111–121. 10.5414/cpp38111. [DOI] [PubMed] [Google Scholar]
- Bois F.; Beney C.; Boumendjel A.; Mariotte A.-M.; Conseil G.; Di Pietro A. Halogenated chalcones with high-affinity binding to P-glycoprotein: potential modulators of multidrug resistance. J. Med. Chem. 1998, 41, 4161–4164. 10.1021/jm9810194. [DOI] [PubMed] [Google Scholar]
- Sirimulla S.; Bailey J. B.; Vegesna R.; Narayan M. Halogen interactions in protein-ligand complexes: implications of halogen bonding for rational drug design. J. Chem. Inf. Model. 2013, 53, 2781–2791. 10.1021/ci400257k. [DOI] [PubMed] [Google Scholar]
- Gerebtzoff G.; Li-Blatter X.; Fischer H.; Frentzel A.; Seelig A. Halogenation of drugs enhances membrane binding and permeation. Chembiochem 2004, 5, 676–684. 10.1002/cbic.200400017. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. Functional consequences of phenylalanine mutations in the predicted transmembrane domain of P-glycoprotein. J. Biol. Chem. 1993, 268, 19965–19972. [PubMed] [Google Scholar]
- Ryu S.; Kawabe T.; Nada S.; Yamaguchi A. Identification of basic residues involved in drug export function of human multidrug resistance-associated protein 2. J. Biol. Chem. 2000, 275, 39617–39624. 10.1074/jbc.m005149200. [DOI] [PubMed] [Google Scholar]
- Situ D.; Haimeur A.; Conseil G.; Sparks K. E.; Zhang D.; Deeley R. G.; Cole S. P. C. Mutational analysis of ionizable residues proximal to the cytoplasmic interface of membrane spanning domain 3 of the multidrug resistance protein, MRP1 (ABCC1): glutamate 1204 is important for both the expression and catalytic activity of the transporter. J. Biol. Chem. 2004, 279, 38871–38880. 10.1074/jbc.m403832200. [DOI] [PubMed] [Google Scholar]
- Haimeur A.; Conseil G.; Deeley R. G.; Cole S. P. C. Mutations of charged amino acids in or near the transmembrane helices of the second membrane spanning domain differentially affect the substrate specificity and transport activity of the multidrug resistance protein MRP1 (ABCC1). Mol. Pharmacol. 2004, 65, 1375–1385. 10.1124/mol.65.6.1375. [DOI] [PubMed] [Google Scholar]
- Ito K.; Oleschuk C. J.; Westlake C.; Vasa M. Z.; Deeley R. G.; Cole S. P. Mutation of Trp1254 in the multispecific organic anion transporter, multidrug resistance protein 2 (MRP2) (ABCC2), alters substrate specificity and results in loss of methotrexate transport activity. J. Biol. Chem. 2001, 276, 38108–38114. 10.1074/jbc.M105160200. [DOI] [PubMed] [Google Scholar]
- Ito K.-i.; Olsen S. L.; Qiu W.; Deeley R. G.; Cole S. P. C. Mutation of a single conserved tryptophan in multidrug resistance protein 1 (MRP1/ABCC1) results in loss of drug resistance and selective loss of organic anion transport. J. Biol. Chem. 2001, 276, 15616–15624. 10.1074/jbc.m011246200. [DOI] [PubMed] [Google Scholar]
- Zhang D.-W.; Cole S. P. C.; Deeley R. G. Identification of a nonconserved amino acid residue in multidrug resistance protein 1 important for determining substrate specificity: evidence for functional interaction between transmembrane helices 14 and 17. J. Biol. Chem. 2001, 276, 34966–34974. 10.1074/jbc.m105063200. [DOI] [PubMed] [Google Scholar]
- Zhang D.-W.; Nunoya K.; Vasa M.; Gu H.-M.; Cole S. P. C.; Deeley R. G. Mutational analysis of polar amino acid residues within predicted transmembrane helices 10 and 16 of multidrug resistance protein 1 (ABCC1): effect on substrate specificity. Drug Metab. Dispos. 2006, 34, 539–546. 10.1124/dmd.105.007740. [DOI] [PubMed] [Google Scholar]
- Koike K.; Conseil G.; Leslie E. M.; Deeley R. G.; Cole S. P. C. Identification of proline residues in the core cytoplasmic and transmembrane regions of multidrug resistance protein 1 (MRP1/ABCC1) important for transport function, substrate specificity, and nucleotide interactions. J. Biol. Chem. 2004, 279, 12325–12336. 10.1074/jbc.m311435200. [DOI] [PubMed] [Google Scholar]
- El-Sheikh A. A. K.; van den Heuvel J. J. M. W.; Krieger E.; Russel F. G. M.; Koenderink J. B. Functional role of arginine 375 in transmembrane helix 6 of multidrug resistance protein 4 (MRP4/ABCC4). Mol. Pharmacol. 2008, 74, 964–971. 10.1124/mol.107.043661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.