

Abstract
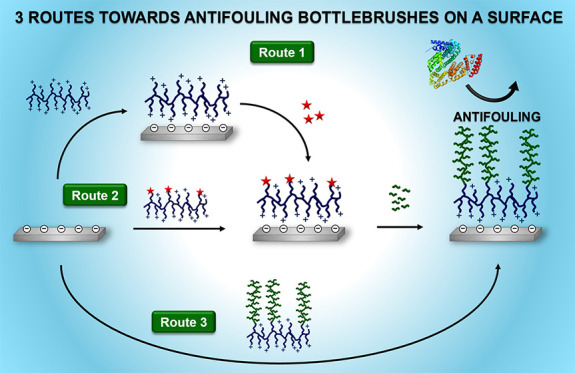
In this work, we compare three routes to prepare antifouling coatings that consist of poly(l-lysine)–poly(N-(2-hydroxypropyl)methacrylamide) bottlebrushes. The poly(l-lysine) (PLL) backbone is self-assembled onto the surface by charged-based interactions between the lysine groups and the negatively charged silicon oxide surface, whereas the poly(N-(2-hydroxypropyl)methacrylamide) [poly(HPMA)] side chains, grown by reversible addition–fragmentation chain-transfer (RAFT) polymerization, provide antifouling properties to the surface. First, the PLL–poly(HPMA) coatings are synthesized in a bottom-up fashion through a grafting-from approach. In this route, the PLL is self-assembled onto a surface, after which a polymerization agent is immobilized, and finally HPMA is polymerized from the surface. In the second explored route, the PLL is modified in solution by a RAFT agent to create a macroinitiator. After self-assembly of this macroinitiator onto the surface, poly(HPMA) is polymerized from the surface by RAFT. In the third and last route, the whole PLL–poly(HPMA) bottlebrush is initially synthesized in solution. To this end, HPMA is polymerized from the macroinitiator in solution and the PLL–poly(HPMA) bottlebrush is then self-assembled onto the surface in just one step (grafting-to approach). Additionally, in this third route, we also design and synthesize a bottlebrush polymer with a PLL backbone and poly(HPMA) side chains, with the latter containing 5% carboxybetaine (CB) monomers that eventually allow for additional (bio)functionalization in solution or after surface immobilization. These three routes are evaluated in terms of ease of synthesis, scalability, ease of characterization, and a preliminary investigation of their antifouling performance. All three coating procedures result in coatings that show antifouling properties in single-protein antifouling tests. This method thus presents a new, simple, versatile, and highly scalable approach for the manufacturing of PLL-based bottlebrush coatings that can be synthesized partly or completely on the surface or in solution, depending on the desired production process and/or application.
Introduction
The nonspecific adsorption of proteins on a surface, i.e., fouling, is an initial step in the process of accumulation of unwanted biomaterials on that surface. The adsorption of such biomolecules and biomaterials impairs the functions of biotechnological and biomedical devices, whose correct functioning is crucially dependent on the availability of a nonfouled surface.1 Surface modification by means of the application of antifouling coatings is advantageous for, e.g., manufacturing biosensors,2 implants,3,4 bioactive surfaces,5 and even big objects like the hull of a ship.6
Especially for sensing low concentrations of a target analyte in complex media, there is a demand for facile surface modifications to impart biosensors with antifouling properties to prevent nonspecific interactions and thereby enhance the signal-to-noise ratios.7 Antifouling coatings frequently consist of poly(ethylene glycol) (PEG)8,9 or zwitterionic polymers.10−15 More recently, however, also poly(N-(2-hydroxypropyl)methacrylamide) [poly(HPMA)] brushes grown by controlled radical polymerizations have been reported to result in stable and highly antifouling coatings, on par with and in some cases outperforming zwitterionic coatings.7,13,16−19 Although the antifouling properties of poly(HPMA) brushes are not entirely understood, the reported fouling levels are extremely low.16,18
These polymeric coatings can be created via a grafting-from approach, in which a polymer is grown from the surface.10,11,20 This is currently considered a highly promising route toward antifouling coatings in terms of long-term antifouling properties, as it leads to a high-density brush structure on the surface with tunable thickness.21−23 However, despite these advantageous properties of polymer brushes grown via the grafting-from method, there is a major hurdle to be overcome if these antifouling coatings are to be applied reproducibly on large, industrially relevant scales.24 Namely, these grafting-from polymer brushes are typically fabricated by surface-initiated, controlled living polymerization in the presence of a metal catalyst and in an oxygen-free environment.11,13,18,25,26 Since this is typically a rather critical technique, it is difficult to scale up and implement in, e.g., industrial production lines in a reproducible manner.20,23 For this reason, there is a need to investigate other macromolecular coatings that can potentially match the antifouling properties of these coatings but at the same time allow easy and reproducible fabrication.11,13,20,25 To this aim, one-step coatings using zwitterionic antifouling polymer brushes with a catechol end group were developed for grafting-to surface anchoring.27,28 In addition, Honda et al. created randomly composed block copolymers of zwitterionic antifouling groups, combined with triethoxy silanes for surface binding, to create antifouling coatings on glass.23
Another well-known example of combining antifouling groups with polydentate surface anchoring moieties are poly(l-lysine)-graft-poly(ethylene glycol) (PLL-g-PEG) polymers, which are known to assemble onto silicon oxide, metal oxide, and polymeric surfaces.29−34 PLL-g-PEG self-assembles on silicon oxide at pH > 2 through multiple electrostatic attractions between the negatively charged surface (SiO2 isoelectric point (IEP) ∼ 2.2)35 and the positively charged pendant amine groups present in PLL (IEP ∼9.5),36 leaving the PEG chains oriented away from the surface.34 Despite the ease of application and good antifouling properties of such PLL-g-PEG coatings, their use under certain circumstances can be limited due to properties inherent to PEG chains: PEG is known to undergo oxidative degradation, which may yield toxic compounds and lead to degradation of the coating, and has been shown to elicit antibody expression in vivo.37−41
In response to these limitations, Morgese et al. published in 2018 a study on polymers with PLL backbones that were grafted with other types of antifouling polymer brushes, such as poly(2-oxazines) and poly(2-oxazolines), to create biointerfaces that resist protein adsorption.31 However, the promising poly(HPMA) polymer was not included as a candidate in this study, and the authors only considered the grafting-to approach. Therefore, we aimed to develop and investigate a bottlebrush macromolecule with poly(HPMA)-grafted side chains for antifouling properties, together with a PLL backbone for multivalent surface interactions to achieve a strong surface anchoring to silicon oxide surfaces.28 Previously, PLL- and HPMA-based polymers have been combined in hybrid macromolecules for the synthesis of gene delivery agents42 or as transfection reagents with minimized toxicity.43 However, to the best of our knowledge, PLL–poly(HPMA) bottlebrushes have not yet been used for the creation of antifouling coatings. The overall goal of this project is to construct PLL bottlebrush coatings in an easy and highly scalable manner without the loss of antifouling performance, and the current paper is the first step in this approach for PLL–poly(HPMA) bottlebrushes as antifouling coatings.
In this study, silicon oxide was used as a model substrate because of its relevance in, e.g., biosensors44 and microfluidic devices.45 We explored three different routes toward such a coating with varying degrees of grafting-to and grafting-from components (Figure 1).
Route A: a coating was completely grafted from the surface. First, PLL was self-assembled on the surface. The reversible addition–fragmentation chain-transfer (RAFT) agent that allows for polymerization was subsequently reacted to the PLL coating, after which poly(HPMA) side chains were RAFT-polymerized from the PLL backbone.
Route B: a coating was partly grafted from the surface. The RAFT agent (RA) was coupled to PLL in solution to synthesize a PLL–RA macroinitiator. The PLL–RA was self-assembled on the surface, and HPMA was finally RAFT-polymerized from the RA-modified PLL coating.
Route C: it comprises a completely presynthesized grafted-to coating. HPMA was RAFT-polymerized from the PLL–RA macroinitiator in solution to create PLL–poly(HPMA) bottlebrushes. These bottlebrushes were then self-assembled on the surface in one single step.
Figure 1.

Schematic overview of the solution-based synthesis of the macroinitiator (PLL–RA) and PLL–poly(HPMA) bottlebrush structure (top). A–C denote the three investigated routes toward surface-immobilized PLL–poly(HPMA) bottlebrushes (bottom). PLL is poly(l-lysine), RA is the RAFT agent, HPMA is 2-hydroxypropyl methacrylamide.
For the growth of the poly(HPMA) brushes, a photoinduced electron transfer-reversible addition–fragmentation chain-transfer (PET-RAFT) technique was applied. This polymerization technique is oxygen-tolerant and metal-free, can be applied to polymerizations in water,46,47 works with an accessible and affordable organic photocatalyst (Eosin Y),45,46 and has been shown to be also applicable to surface-initiated polymerization of different monomers.48 Especially, this final feature makes PET-RAFT very suitable for our purpose, as it works well both in solution and from a surface. Subsequently, we determined and evaluated the various properties of the thus formed coatings in detail, including ease of synthesis, scalability, reproducibility, modularity, and ease of characterization. Finally, we performed a preliminary investigation of the antifouling performance obtained for these three coating approaches and provided a perspective on the use of such PLL–poly(HPMA) bottlebrush coatings for antifouling purposes.
Results and Discussion
We will first discuss the synthesis of the PLL–poly(HPMA) bottlebrush-based coatings on silicon oxide surfaces, as obtained via the three different routes presented in Figure 1. Afterward, the results of antifouling studies on the coatings prepared by these different routes will be discussed and evaluated.
Route A: PLL–Poly(HPMA) Coating via the Grafting-from Procedure
In route A, the poly(HPMA)-based coating was completely grafted from the surface. First, PLL was self-assembled on the surface, followed by coupling of the surface-bound PLL to the RAFT agent (RA), and finally the polymerization of the poly(HPMA) side chains from the PLL backbone (Figure 2).
Figure 2.

Schematic depiction of the construction of the PLL–RA coating via a grafting-from approach, followed by SI-PET-RAFT to grow poly(HPMA) brushes.
A1: Self-Assembly of Poly(l-lysine) on SiO2
PLL–poly(HPMA) bottlebrush-coated surfaces were prepared in this first route by building the layer from the surface upward. To this end, PLL (MW = 15–30 kDa) was self-assembled to form a monolayer by overnight immersion of freshly cleaned, negatively charged silicon oxide surfaces in a 0.1 mg/mL solution of PLL in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (following the procedure reported by Morgese et al.31). Upon modification of the silicon oxide surfaces with PLL, the presence of a thin layer of polymers on the surfaces was confirmed by analytical techniques. First, by X-ray photoelectron spectroscopy (XPS), we found signals corresponding to the presence of nitrogen (at 400 eV) and carbon (285 eV) on the surface (Figure 3, top). Furthermore, the C1s narrow-scan measurements showed the characteristic signals for the amide carbonyls (C=O, 288.4 eV) and for the C–N and C–C=O carbon atoms (both at 286.4 eV). Since these monolayers were too thin to be measured by ellipsometry (vide infra), we used the Si/C ratio in the XPS wide scan to calculate the average thickness of the layer,49,50 which was approximately 0.5 nm, which is in good agreement with values reported in the literature.51,52 The static water contact angle (SWCA) of the PLL-coated surfaces was <20°, displaying the very hydrophilic character of the coating due to the charges on the protonated terminal amines. These combined data suggest that PLL was deposited as a monolayer on the SiO2 surfaces.
Figure 3.

XPS wide-scan spectrum and C1s narrow-scan spectrum of self-assembled PLL on silicon oxide (top), surface-bound PLL, functionalized with RAFT agent (middle), and poly(HPMA) grafted from RAFT agent-modified silicon oxide (after 80 min polymerization) (bottom). On the left, the chemical structure of the analyzed surface is depicted.
A2: Surface Immobilization of the RAFT Agent on PLL-Modified SiO2
While a fraction of the pendant amine groups of PLL was involved in surface binding, the remaining free primary amines of the PLL coating can be used to immobilize a RAFT agent to allow the polymerization from the surface in the following step. To this end, an N-hydroxysuccinimide (NHS)-activated RAFT agent was reacted with the surface-bound PLL overnight in dry tetrahydrofuran (THF) (see Figure 2, step A2). In the XPS spectrum of the thus-prepared surface (Figure 3, middle), we found an expected increase in both the C1s (285 eV) and N1s (399 eV) signals in the wide scan since these elements are predominantly present in the RAFT agent. Moreover, indicatively, we detected sulfur (S2s, 227 eV), which confirms the presence of the RAFT agent. Based on the N/S ratio obtained from XPS wide spectra, roughly 40% of all PLL primary amines had reacted to hold a RAFT agent moiety (Figure S8). Taking into account that also a portion of the primary amines is involved in surface binding by electrostatic interactions, this conversion can be considered relatively high, compared to what would be maximally feasible for still strongly surface-bound PLL. The conversion was also confirmed in the C1s narrow scan, which showed that the carbonyl signal at 288 eV became more dominant, which can be attributed to the introduced carbonyl and thiocarbonyls. As expected, the thickness of the coating increased upon the addition of the RAFT agent to approximately 1.3 nm. The SWCA of the RAFT-modified surfaces increased to 42°, in line with the more hydrophobic nature of the RAFT agent.
A3: PET-RAFT Polymerization of HPMA on RAFT Agent-Modified SiO2
Poly(HPMA) side chains were grown from the RAFT-modified, PLL-covered silicon oxide surfaces by surface-initiated PET-RAFT in water using visible light and Eosin Y as an oxygen-tolerant photocatalyst, which allowed polymerization in air.46,48 Triethanolamine (TEOA) was used as a cocatalyst46 and also acted as a sacrificing electron donor to reduce oxygen in the polymerization system.48 The study of the polymer growth kinetics showed a linear growth for the first 40 min (Figure 4), which indicates the controlled nature of this surface-initiated polymerization. The leveling off after 40 min might indicate a reduced availability of the RAFT groups by either an increased steric hindrance or chemical degradation.
Figure 4.

Polymerization kinetics of both on-surface PET-RAFT polymerizations of HPMA. Route A3: polymerization on RAFT agent-modified surfaces that were obtained by reacting NHS-RAFT with surface-immobilized PLL (orange). Route B2: polymerization on RAFT agent-modified surfaces that were obtained by the self-assembly of a PLL–RA macroinitiator (blue). Thicknesses were measured in duplicate by ellipsometry.
The chemical composition of the grown poly(HPMA) brushes was studied using XPS, while ellipsometry was used to determine the layer thickness of the polymer brushes. After 80 min of polymerization, the coating reached a total thickness of 32.0 ± 0.2 nm. Similar growth rates have been reported for this HPMA monomer using PET-RAFT on silane anchoring layers.48 This implies that from a self-assembled polymeric PLL starting layer, the polymerization works equally well compared to well-defined silane monolayers. Figure 3 shows the XPS spectra for the polymer brushes that were grown for 80 min. In the wide scan, only three main peaks are observed, namely, O1s (531 eV), N1s (388 eV), and C1s (285 eV) in a 1.8:1:8.3 ratio. This is in reasonable agreement with the elemental composition of the poly(HMPA) structure (2:1:7) and with previously published papers on HPMA brushes, which report a ratio of 1.8:1:7.6,48 given the possibility of atmospheric contamination
In the C1s narrow scan, a clear, more intense signal around 286 eV could be discerned, compared to the RAFT agent-terminated surfaces from before the polymerization. This indicates the increase of C–heteroatom species, which is in agreement with the structure of the poly(HPMA) polymer. The SWCA of the polymer layers reached a stable value of ∼50° after 40 min of polymerization. Additionally, after 80 min of polymerization, the layers showed a low roughness (see Figure S12) as could be expressed by a root-mean-square roughness Rq = 2.44 ± 0.44 nm, as measured by AFM (see the Supporting Information). Overall, these data suggest a surface structure that is similar to that previously reported for poly(HPMA) polymer brushes grown by different surface-initiated controlled polymerization methods.13,18,48
Route B: Synthesis and Self-Assembly of a PLL–RA Macroinitiator and HPMA Polymerization from PLL–RA-Modified Surfaces
In route B, the coating was only partly grafted from the surface. The RA was first coupled to PLL in a solution to synthesize a PLL–RA macroinitiator. This PLL–RA macroinitiator was then self-assembled on the surface, after which HPMA was polymerized from the RA-modified PLL side chains (Figure 5).
Figure 5.

Schematic depiction of the build-up of the PLL–RA coating in a partly grafting-to approach, followed by SI-PET-RAFT to grow poly(HPMA) brushes.
B1: Synthesis and Self-Assembly of PLL–RA Macroinitiator on SiO2
Route B started with the synthesis of a RAFT agent-functionalized PLL polymer macroinitiator that could afterward be immobilized on the surface (Figure 5). By reacting a part of the amine end groups with an NHS-activated RA, we envisioned that enough lysine moieties would be left unreacted to achieve efficient binding of PLL to the silicon oxide surface in the subsequent self-assembly process. Therefore, we chose a 1:3 ratio of NHS-RA/lysine monomer in the synthesis of PLL–RA. The synthesized PLL–RA macroinitiator was first characterized by 1H NMR spectroscopy, also allowing for the determination of the actual achieved RA/lysine ratio, which was found to be 1:9.8 (see Figure S1). This ratio was also supported by the elemental composition of the PLL–RA after immobilization on a silicon oxide surface: By comparing the ratios between the N1s and S2s signals in XPS, we found the RAFT/lysine ratio to be 1:8.5, which is comparable to the ratio found by 1H NMR (see Figure S9).
Several reasons could account for the lower observed degree of RAFT agent incorporation. First, the reaction was stopped after 16 h, which might be before full conversion had been reached. The lower degree might have also been affected by the relatively low solubility of PLL and NHS-activated RAFT agent in a common solvent (HEPES buffer with 10 v/v% DMSO) or by partial hydrolysis of the NHS ester. Finally, partial protonation of the amine end groups might have lowered the conversion of the reaction.
Nevertheless, having approximately nine out of ten lysines available for surface anchoring likely leads to a more stable coating while still having enough initiation points for the growth of relatively long polymer brushes. This was demonstrated by the successful growth of poly(HPMA) brushes from silicon oxide surfaces that were coated by the PLL–RA macroinitiator, as discussed below. The PLL–RA macroinitiator formed a monolayer by self-assembly on a freshly cleaned silicon oxide surface using the same protocol as in route A1 (Figure 5). XPS analysis of this coating revealed signals for the elements N (at 400 eV), C (285 eV), and S (231 eV) on the surface. In addition, the C1s narrow scan showed the characteristic signals for the amide carbonyls (N–C=O, 288.2 eV) and the nitrogen- and carbonyl-bound carbon atoms (C–N and C–C=O), both at 286.2 eV (Figure 6).
Figure 6.

XPS wide-scan spectrum and C1s narrow-scan spectrum of self-assembled macroinitiator PLL–RA (top) and poly(HPMA) grafted from these surfaces (80 min polymerization) (bottom). On the left, the chemical structures of the analyzed surfaces are depicted.
The layer thickness of the PLL-RAFT macroinitiator coating was calculated using the Si2p/C1s ratio from the XPS wide scan and was found to be approximately 2.2 nm, which was thicker than the value obtained in route A (1.3 nm), in which an analogous coating was created by reacting the NHS-RA moiety on preassembled PLL. The assembled PLL–RA coating (in route B) had an SWCA of 36°, which is slightly less hydrophobic than the PLL–RA coating from route A (SWCA of 42°). This could be ascribed to the fact that the coupling of the RAFT agent to PLL in solution (step B1) occurs randomly on the entire polymer, while the coupling on the surface (step A2) predominantly occurs on the top (solution-exposed) part of the PLL coating. Therefore, in route B, a smaller fraction of the RAFT moieties could have an upward orientation compared to the PLL-functionalized polymer discussed in route A, which would explain the reduced increase in the hydrophobic character of the overall coating for route B.
B2: PET-RAFT Polymerization of HPMA on PLL–RA Macroinitiator-Modified SiO2
Once the PLL–RA macroinitiator was immobilized on the surface, poly(HPMA) brushes were grown using PET-RAFT conditions, as described in the previous paragraph (Figure 5). The layer thicknesses of the polymer coatings that were grown for different time intervals were measured using ellipsometry, allowing a comparison of the kinetics of the polymerization for routes A and B (Figure 4). The rate of polymerization is similar for the first 40 min, which implies that the amount of the RAFT agent and hydrophilicity of the surface are not rate-determining. After 40 min, the polymerization in route A seems to continue, while the route B polymerization seems to level off. This might be due to the different availability of RA on the surface as discussed in step B1.
Figure 6 shows the XPS data of a polymer brush that was grown for 80 min, and which had a thickness of 21 nm, as determined from ellipsometry. In the wide scan, we see three main peaks, namely, for the elements O1s (532 eV), N1s (400 eV), and C1s (285 eV) in a 2.3:1:9.1 ratio, which is in reasonable agreement with the ratios found in the full grafting-from procedure in route A (1.8:1:8.3). In this case, also, a very small signal is visible from the silicon oxide surface (Si2p, 102 eV), which confirms the slightly thinner coating as already measured by ellipsometry. Also, the C1s narrow scan gave a very similar spectrum as previously observed for the poly(HPMA) coating in route A. The SWCA stabilized after 30 min polymerization of HPMA to ∼50°. AFM topology measurements of the surfaces after 80 min of polymerization revealed a somewhat higher roughness Rq = 5.47 ± 0.75 nm compared to surface A3 (2.44 ± 0.44 nm). This is probably due to the more hydrophobic, hence less soluble, character of the initial PLL–RA. Overall, it can be concluded that the polymerization by PET-RAFT from the PLL–RA macroinitiator-modified surfaces was possible in only two surface modification steps. The coupling of the RAFT agent to the PLL polymer in solution did not significantly affect the final polymerization step, which implies that the surface modification procedure can be shortened by one step.
Route C: PLL–poly(HPMA) Coating via the Grafting-to Procedure
Route C comprises a completely presynthesized, grafted-to coating. HPMA was first polymerized from the PLL macroinitiator in solution to create PLL–poly(HPMA) bottlebrushes. These bottlebrushes were then self-assembled on the surface in one single step (Figure 7). Furthermore, bottlebrushes with carboxybetaine groups that offer the possibility for later biofunctionalization [PLL–poly(HPMA/CBMA)] were also synthesized and immobilized on a surface using the method presented in route C.
Figure 7.

Schematic depiction of the build-up of the PLL–poly(HPMA) coating in a completely grafting-to approach.
C1: Solution Synthesis and Surface Immobilization of PLL–poly(HPMA) by PET-RAFT Polymerization of HPMA from PLL–RA Macroinitiator
The previously discussed routes (A and B) required two or three consecutive surface modification steps to create a PLL–poly(HPMA) coating on silicon oxide. While route B is thus certainly attractive in terms of scalability, we considered it to be of even more interest to further decrease the number of surface modifications steps, so as to have a one-step procedure for the coating of silicon oxide surfaces by PLL–poly(HPMA) bottlebrush polymers. In such a route, the full polymer, a backbone polymer (PLL) with polymeric poly(HPMA) side chains, is presynthesized in solution, and only subsequently applied to the surface. To this aim, poly(HPMA) side chains were grown from the PLL–RA macroinitiator, which was already synthesized for route B, in solution (Figure 7). From the RAFT agent side groups of this macroinitiator, poly(HPMA) chains were grown by PET-RAFT polymerization in water using visible light, Eosin Y as an oxygen-tolerant photocatalyst, and triethanolamine (TEOA) as a cocatalyst, in line with the conditions used in route B to grow poly(HPMA).48 Extensive dialysis allowed the isolation of the PLL–poly(HPMA) bottlebrush that could then be characterized by 1H NMR spectroscopy (Figure 8). In the 1H NMR spectrum, peaks at δ 3.08 and δ 3.84 ppm confirm the presence of poly(HPMA) side chains of the bottlebrush. From the ratio between the 1H signal at δ 4.15 ppm from the PLL backbone and the 1H signal at δ 3.84 ppm from poly(HPMA), the ratio of the poly(HPMA) monomer to the lysine monomer was found to be 1.4:1. Combining this ratio, with the previously determined RA/lysine ratio of the PLL–RA macroinitiator, the average chain length of each poly(HPMA) side chain could be calculated to be roughly 14 repeating monomers, corresponding to approximately 2 kDa. The total weight of the PLL–poly(HPMA) bottlebrush was calculated to be 41 kDa (see calculations in the Supporting Information).
Figure 8.

1H NMR spectrum of the PLL–poly(HPMA) bottlebrush synthesis in solution (in D2O, at 400 MHz and 298 K).
The polymer molecular weight and polydispersity index (PDI) were determined by gel permeation chromatography (GPC) in water. Based on calibration by a set of poly(ethylene glycol) standards and a poly(HPMA) standard, a molecular weight of approximately 43 kDa was found, i.e., close to the NMR-derived value. Furthermore, from GPC, a polydispersity index (PDI) of 1.4 was determined. Further characterization by dynamic light scattering (DLS) revealed a narrow size distribution with an intensity peak maximum at a hydrodynamic radius of 77 nm in water (Figure S11).
The synthesized PLL–poly(HPMA) bottlebrushes were allowed to self-assemble on silicon oxide surfaces by overnight immersion in a 0.1 mg·mL–1 solution of PLL–poly(HPMA) in HEPES buffer. XPS analysis of this coating (Figure 9) showed signals for N (at 399 eV) and C (285 eV) on the silicon oxide surfaces, which is in agreement with the presence of a monolayer of PLL–poly(HPMA). The C1s narrow-scan measurements showed the expected signals for the amide carbonyls (C=O, 288.2 eV) and carbon–nitrogen and carbon–carbonyls (C–N and C–C=O), both at 286.2 eV. The layer thickness of the PLL–poly(HPMA) coating was calculated using the Si2p/C1s ratio from the XPS wide scan and was found to be approximately 0.9 nm. While this is a rather low layer thickness for a surface-immobilized bottlebrush polymer, it should be pointed out that the XPS thickness measurements were taken under ultrahigh-vacuum conditions, creating a collapsed polymer layer (which will expand upon immersion). Furthermore, the found dry thickness is in agreement with other types of surface-immobilized PLL-based bottlebrushes reported in the literature.29−31 The self-assembly of PLL–poly(HPMA) lead to the formation of a smooth layer (see Figure S12), as the reported roughness by AFM was Rq = 2.37 ± 0.05.
Figure 9.

XPS wide-scan spectrum and C1S narrow-scan spectrum of self-assembled PLL–poly(HPMA) (top) and PLL–poly(HPMA/CBMA) (bottom). On the left, the chemical structures of the analyzed surfaces are depicted.
The SWCA of this PLL–poly(HPMA) coating was approximately 20°. This hydrophilic character might be due to the polar (partially charged) amine groups of the PLL within the coating that are close to the solvent interface due to the relatively low layer thickness of the overall coating. However, the SWCA of the PLL–poly(HPMA) self-assembled coating is higher than for the PLL monolayer with a comparable thickness observed for step A1 (which has a reported SWCA of below 20° for a 0.5 nm surface-immobilized brush) due to the presence of poly(HPMA).
Carboxybetaine-Doped Poly(HPMA) Brushes for Biofunctionalization Purposes
We have further improved the concept of PLL–poly(HPMA) one-step antifouling coatings by incorporating the possibility for biomolecule immobilization, which is highly desirable for selective binding in, e.g., biosensors and tissue engineering.7,53,54 The polymerization of HPMA from the PLL–RA macroinitiator was also performed in the presence of a second antifouling monomer that contains a carboxylate group to allow for easy activation by conventional coupling strategies, e.g., NHS/EDC, to couple bioactive moieties. To this end, we selected a zwitterionic carboxybetaine (CBMA) monomer that was also used previously for surface functionalization.7,10,13,55 To this aim, 5% of CBMA monomer was used for this polymerization, keeping the conditions the same as described for the PET-RAFT solution polymerization. The obtained PLL–poly(HPMA/CBMA) polymer was analyzed by 1H NMR spectroscopy (see Figure S5). We observed the expected additional signals coming from the CBMA monomers, when compared to the PLL–poly(HPMA) bottlebrush spectrum. Based on 1H NMR integration, the content of CBMA was calculated to be 7.7% (see Figure S5). The somewhat higher incorporation of the CBMA monomer likely stems from the previously reported difference in reactivity between the monomers.48 The polymer molecular weight and PDI were approximated using GPC and gave a MW of approximately 53 kDa and a PDI of 1.6. In addition, DLS gave a narrow size distribution in water with a maximum at a hydrodynamic radius of 111 nm (see Figure S11).
The synthesized PLL–poly(HPMA/CBMA) bottlebrushes were allowed to self-assemble on silicon oxide surfaces under similar conditions as described above. XPS analysis of this coating (Figure 9) revealed signals for the elements N (at 400 eV) and C (285 eV) on the silicon oxide surfaces. The layer thickness of the PLL–poly(HPMA/CBMA) coating was calculated using the Si2p/C1s ratio from the XPS wide scan and was found to be approximately 1.3 nm, which is in accordance with the formation of a monolayer of PLL–poly(HPMA/CBMA). The C1s narrow-scan measurements showed the expected signals for the amide carbonyls (C=O, 288.2 eV), carbon–nitrogen and carbon–carbonyls (C–N and C–C=O, both at 286.1 eV). The higher amount of carbon–nitrogen and carbon−carbonyl signals compared to the PLL–poly(HPMA) layer (44 vs 38%) could be explained by the presence of the CBMA monomer that contains an additional carbonyl group. The layer thickness of the PLL–poly(HPMA/CBMA) coating was calculated using the Si2p/C1s ratio from the XPS wide scan and was found to be approximately 1.3 nm. This layer was thus slightly thicker than the PLL–poly(HPMA) monolayer, which might be explained by the longer side chains. The SWCA of the PLL–poly(HPMA/CBMA) coating was approximately 22°, which is very similar to the PLL–poly(HPMA) analogue. The self-assembly of the PLL–poly(HPMA)/CMBA lead to the formation of a smooth layer (see Figure S12), as the reported roughness by AFM was Rq = 1.16 ± 0.05.
Having successfully incorporated CBMA in the PLL–poly(HPMA) bottlebrushes, a broad and versatile platform for (bio)functionalization was created. For both poly(HPMA) and poly(CBMA), the antifouling properties are most probably related to the strong binding of water molecules.56 The combination of these polymers was previously utilized in polymer brush systems that showed good antifouling properties.14,57 By activation of the carboxylate groups—either in solution or on the surface—antibodies or other bioactive molecules can be installed for monitoring specific interactions in, e.g., biosensor platforms.7,14,58−60
Antifouling Properties of PLL–poly(HPMA) Coatings
Having successfully immobilized the three different PLL–poly(HPMA) bottlebrush coatings on the silicon oxide surface, a preliminary investigation of their antifouling properties was performed. We quantified the amount of protein adsorption by fluorescence microscopy, by exposing the PLL–poly(HPMA) bottlebrush coatings to fluorescently labeled protein solutions.13,23,28 This method allows for a limit of fluorescent protein detection of 300 pg·mm–1 and thus suffices for initial testing.48 For this study, lysozyme (LYS) and bovine serum albumin (BSA) were used as model proteins at concentrations of 0.1 mg·mL–1 in phosphate-buffered saline (PBS) and contacted with the surfaces for 15 min before washing with PBS. BSA was chosen since it is one of the most common proteins in blood plasma with an overall negative charge at pH 7.4 (PBS buffer).61 LYS is a relatively small, hydrophilic protein and is used because of its overall positive charge at pH 7.4 (PBS buffer).62 As controls for the protein adsorption experiments, we used bare silicon oxide surfaces, PLL-modified surfaces, and surfaces modified with commercially available PLL-PEG, which is known to have good antifouling properties.30
The unmodified silicon oxide surface showed high fluorescence intensities from both solutions (Figure 10), indicating significant fouling. PLL-coated silicon oxide was used as a control and already showed less fouling compared to the bare silicon oxide surface, probably due to the hydrophilic and charged nature of the sample, which contributes to the antifouling properties.20 However, there still was a significant amount of fouling by BSA visible on the PLL-coated silicon oxide, likely due to the oppositely charged nature of BSA and PLL. The PLL-PEG-coated silicon oxide control sample showed the expected low fluorescence intensities, which confirms the functioning of the procedure and antifouling behavior. The fluorescent intensities for PLL–poly(HPMA)-based antifouling coatings were observed on the background level. These data show that PLL–poly(HPMA) coatings that were synthesized in different ways (route A, B, or C) show antifouling properties close to the limit of detection.
Figure 10.

Fluorescence intensities of different uncoated and coated silicon oxide surfaces (see table) after exposure to a solution containing BSA-AF488 and LYS-FITC.
Comparing the Different Routes toward PLL–poly(HPMA) Bottlebrush Coatings
Coatings made by routes A, B, and C showed very similar antifouling properties in single-protein solutions, irrespective of any possible difference in their built-up, thickness, surface topology, and/or brush density. However, depending on the to-be-coated surface and application, one can foresee that a certain route might be preferred over the other two.
Both routes A and B lead to relatively thick and dense coatings since the HPMA was grafted from an initiator-modified surface. This could be beneficial for stability and long-term use since the underlying anchoring layer is better shielded from the environment.
Route A showed the straightforward application of PLL as a multivalent, amine-terminated anchoring layer, on which a polymerization agent can be attached (step A1). PLL could, therefore, be an alternative to the often used silanes.63−65 The follow-up steps are versatile with regard to the polymerization agent, technique, and monomer of choice.
In route B, the number of on-surface reactions is reduced to one. In solution, a macroinitiator is synthesized by a one-step coupling method. After that, the macroinitiator can be easily self-assembled on the surface, after which polymers can be grafted on the surface. This approach has also been used by Jain et al.,66 who modified a polymer with polymerization initiator groups and embedded this new polymer in a layer-by-layer assay on porous membranes to eventually grow polymer brushes from these membranes in a grafting-from approach. The presynthesis of the macroinitiator in solution allows for precise control over the amount of the embedded polymerization agent and can be specifically beneficial in cases were the anchoring layer (PLL) and the to-be-polymerized monomer are impossible to synthesize in solution because of opposite polarities (or other properties that are challenging to combine in synthesis).
Both route B and C might result in less long-term stable coatings compared to route A since the electrostatic interactions, that assure surface binding, are partly sacrificed by attaching the RAFT agent (route B) or poly(HPMA) side chains (route C) prior to surface binding.
Route C is the easiest to apply on a surface and by far the easiest to scale up because of the one-step self-assembly and lack of on-surface reactions. The complete synthesis in solution allows control and knowledge of the composition, molecular weight, and dimensions of the formed polymer. Also, this approach allows precise and quantifiable immobilization of (bio)molecules in solution, which typically requires smaller quantities of the (bio)molecule of interest in the overall coating process or on the surface. The synthesis procedure in solution takes time and requires purification. However, once synthesized, only very small amounts of PLL–poly(HPMA) (as little as 0.1 mg·mL–1) are needed to coat a surface, which makes it very desirable and cost-effective when a coating needs be applied to multiple or large surfaces.
Conclusions
In this work, we developed three different routes (A–C) to prepare effective antifouling coatings that consist of PLL–poly(HPMA) bottlebrushes. In these coatings, the poly(l-lysine) (PLL) backbone self-assembles onto a silicon oxide surface by charge-based interactions between the lysine groups and the negatively charged surface, whereas the poly(N-(2- hydroxypropyl)methacrylamide) [poly(HPMA)] side chains contribute to the antifouling properties.
The PLL–poly(HPMA) bottlebrush polymer coatings were produced using grafting-from techniques by polymerizing HPMA from the surface (route A and B) and grafting-to of a presynthesized PLL-poly(HPMA) bottlebrush (route C); the latter case—taking place fully under ambient conditions with only water as a solvent—is both very easy for repeated and/or large-scale use and allows detailed characterization of the final polymer in solution, while methods A and B have to rely on surface-sensitive analytical methods for characterization. Additionally, in route C, a bottlebrush was synthesized that contains 5% carboxybetaine (CB) in its side chains, which offers the possibility for further functionalization after an ester activation step.
Overall, all surface modification routes (A–C) yield coatings that show single-protein antifouling properties and are worthy of further, more detailed antifouling studies.
Acknowledgments
This project was supported by Netherlands Organization for Scientific Research (NWO; LIFT program, grant 731.015.042) with Surfix BV as a partner and the Science PPP Fund research program (project number 741.018.105), which is partly financed by NWO. The authors thank Hans Beijleveld, Remko Fokkink, and Lucas Teunissen for helpful discussions and instrumental support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.langmuir.0c01675.
Description of the materials and methods used; supporting NMR, XPS, IR, DLS, AFM, and GPC data and calculations; 1H NMR spectrum of macroinitiator PLL–RA, PLL–poly(HPMA), and PLL–poly(HPMA/CBMA), measured in D2O (400 MHz, 298 K) (Figure S1, S3, and S5, resp.); and 1H–13C-HSQC NMR spectrum of macroinitiator PLL–RA, PLL–poly(HPMA), and PLL–poly(HPMA/CBMA), measured in D2O (100 MHz, 298 K) (Figure S2, S4, and S6, resp.) (PDF)
Author Contributions
# E.R. and A.R.K. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Wisniewski N.; Reichert M. Methods for Reducing Biosensor Membrane Biofouling. Colloids Surf., B 2000, 18, 197–219. 10.1016/S0927-7765(99)00148-4. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Emmenegger C.; Avramenko O. A.; Brynda E.; Skvor J.; Alles A. B. Poly(HEMA) Brushes Emerging as a New Platform for Direct Detection of Food Pathogen in Milk Samples. Biosens. Bioelectron. 2011, 26, 4545–4551. 10.1016/j.bios.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Huang N.; Michel R.; Voros J.; Textor M.; Hofer R.; Rossi A.; Elbert D. L.; Hubbell Ja.; Spencer N. D. Poly(l-Lysine)-g-Poly (Ethylene Glycol) Layers on Metal Oxide Surfaces: Surface-Analytical Characterization and Resistance to Serum and Fibrinogen Adsorption Poly (l-Lysine)-g-Poly (Ethylene Glycol) Layers on Metal Oxide Surfaces: Surface-An. Langmuir 2001, 6, 489–498. 10.1021/la000736+. [DOI] [Google Scholar]
- Yu Y.; Cirelli M.; Li P.; Ding Z.; Yin Y.; Yuan Y.; de Beer S.; Vancso G. J.; Zhang S. Enhanced Stability of Poly(3-Sulfopropyl Methacrylate Potassium) Brushes Coated on Artificial Implants in Combatting Bacterial Infections. Ind. Eng. Chem. Res. 2019, 58, 21459–21465. 10.1021/acs.iecr.9b03980. [DOI] [Google Scholar]
- Thissen H.; Gengenbach T.; du Toit R.; Sweeney D. F.; Kingshott P.; Griesser H. J.; Meagher L. Clinical Observations of Biofouling on PEO Coated Silicone Hydrogel Contact Lenses. Biomaterials 2010, 31, 5510–5519. 10.1016/j.biomaterials.2010.03.040. [DOI] [PubMed] [Google Scholar]
- Rosenhahn A.; Schilp S.; Kreuzer H. J.; Grunze M. The Role of “Inert” Surface Chemistry in Marine Biofouling Prevention. Phys. Chem. Chem. Phys. 2010, 12, 4273–4274. 10.1039/c001968m. [DOI] [PubMed] [Google Scholar]
- Baggerman J.; Smulders M. M. J.; Zuilhof H. Romantic Surfaces: A Systematic Overview of Stable, Biospecific, and Antifouling Zwitterionic Surfaces. Langmuir 2019, 35, 1072–1084. 10.1021/acs.langmuir.8b03360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S.; Pellacani P.; van Beek T. A.; Zuilhof H.; Nielen M. W. F. Surface Characterization and Antifouling Properties of Nanostructured Gold Chips for Imaging Surface Plasmon Resonance Biosensing. Sens. Actuators, B 2015, 209, 505–514. 10.1016/j.snb.2014.11.133. [DOI] [Google Scholar]
- Kuzmyn A. R.; De Los Santos Pereira A.; Pop-Georgievski O.; Bruns M.; Brynda E.; Rodriguez-Emmenegger C. Exploiting End Group Functionalization for the Design of Antifouling Bioactive Brushes. Polym. Chem. 2014, 5, 4124–4131. 10.1039/C4PY00281D. [DOI] [Google Scholar]
- van Andel E.; de Bus I.; Tijhaar E. J.; Smulders M. M. J.; Savelkoul H. F. J.; Zuilhof H. Highly Specific Binding on Antifouling Zwitterionic Polymer-Coated Microbeads as Measured by Flow Cytometry. ACS Appl. Mater. Interfaces 2017, 9, 38211–38221. 10.1021/acsami.7b09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. T.; Baggerman J.; Paulusse J. M. J.; Rijn C. J. M.; Van; Zuilhof H. Stable Protein-Repellent Zwitterionic Polymer Brushes Grafted from Silicon Nitride. Langmuir 2011, 27, 2587–2594. 10.1021/la104657c. [DOI] [PubMed] [Google Scholar]
- Yang W.; Chen S.; Cheng G.; Vaisocherová H.; Xue H.; Li W.; Zhang J.; Jiang S. Film Thickness Dependence of Protein Adsorption from Blood Serum and Plasma onto Poly(Sulfobetaine)-Grafted Surfaces. Langmuir 2008, 24, 9211–9214. 10.1021/la801487f. [DOI] [PubMed] [Google Scholar]
- Kuzmyn A. R.; Nguyen A. T.; Zuilhof H.; Baggerman J. Bioactive Antifouling Surfaces by Visible-Light-Triggered Polymerization. Adv. Mater. Interfaces 2019, 6, 1900351 10.1002/admi.201900351. [DOI] [Google Scholar]
- Lísalová H.; Brynda E.; Houska M.; Víšová I.; Mrkvová K.; Song X. C.; Gedeonová E.; Surman F.; Riedel T.; Pop-Georgievski O.; Homola J. Ultralow-Fouling Behavior of Biorecognition Coatings Based on Carboxy-Functional Brushes of Zwitterionic Homo- and Copolymers in Blood Plasma: Functionalization Matters. Anal. Chem. 2017, 89, 3524–3531. 10.1021/acs.analchem.6b04731. [DOI] [PubMed] [Google Scholar]
- Schönemann E.; Laschewsky A.; Wischerhoff E.; Koc J.; Rosenhahn A. Surface Modification by Polyzwitterions of the Sulfabetaine-Type, and Their Resistance to Biofouling. Polymers 2019, 11, 1014 10.3390/polym11061014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Andel E.; Lange S. C.; Pujari S. P.; Tijhaar E. J.; Smulders M. M. J.; Savelkoul H. F. J.; Zuilhof H. Systematic Comparison of Zwitterionic and Non-Zwitterionic Antifouling Polymer Brushes on a Bead-Based Platform. Langmuir 2019, 35, 1181–1191. 10.1021/acs.langmuir.8b01832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobii M.; de los Santos Pereira A.; Pop-Georgievski O.; Kostina N. Y.; Rodriguez-Emmenegger C.; Percec V. Synthesis of Non-Fouling Poly[N-(2-Hydroxypropyl)Methacrylamide] Brushes by Photoinduced SET-LRP. Polym. Chem. 2015, 6, 4210–4220. 10.1039/C5PY00506J. [DOI] [Google Scholar]
- Rodriguez-Emmenegger C.; Brynda E.; Riedel T.; Houska M.; Šubr V.; Alles A. B.; Hasan E.; Gautrot J. E.; Huck W. T. S. Polymer Brushes Showing Non-Fouling in Blood Plasma Challenge the Currently Accepted Design of Protein Resistant Surfaces. Macromol. Rapid Commun. 2011, 32, 952–957. 10.1002/marc.201100189. [DOI] [PubMed] [Google Scholar]
- Surman F.; Riedel T.; Bruns M.; Kostina N. Y.; Sedláková Z.; Rodriguez-Emmenegger C. Polymer Brushes Interfacing Blood as a Route toward High Performance Blood Contacting Devices. Macromol. Biosci. 2015, 15, 636–646. 10.1002/mabi.201400470. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Cao Z. Ultralow-Fouling, Functionalizable, and Hydrolyzable Zwitterionic Materials and Their Derivatives for Biological Applications. Adv. Mater. 2010, 22, 920–932. 10.1002/adma.200901407. [DOI] [PubMed] [Google Scholar]
- Yang W.; Xue H.; Li W.; J Z.; Jiang S. Pursuing “Zero” Protein Adsorption of Poly(Carboxybetaine) from Undiluted Blood Serum and Plasma. Langmuir 2009, 25, 11911–11916. 10.1021/la9015788. [DOI] [PubMed] [Google Scholar]
- Blaszykowski C.; Sheikh S.; Thompson M. A Survey of State-of-the-Art Surface Chemistries to Minimize Fouling from Human and Animal Biofluids. Biomater. Sci. 2015, 3, 1335–1370. 10.1039/C5BM00085H. [DOI] [PubMed] [Google Scholar]
- Honda T.; Nakao A.; Ishihara K.; Higaki Y.; Higaki K.; Takahara A.; Iwasaki Y.; Yusa S. I. Polymer Coating Glass to Improve the Protein Antifouling Effect. Polym. J. 2018, 50, 381–388. 10.1038/s41428-018-0026-x. [DOI] [Google Scholar]
- Michalek L.; Barner L.; Barner-Kowollik C. Polymer on Top: Current Limits and Future Perspectives of Quantitatively Evaluating Surface Grafting. Adv. Mater. 2018, 30, 1–18. 10.1002/adma.201706321. [DOI] [PubMed] [Google Scholar]
- Lange S. C.; Van Andel E.; Smulders M. M. J.; Zuilhof H. Efficient and Tunable Three-Dimensional Functionalization of Fully Zwitterionic Antifouling Surface Coatings. Langmuir 2016, 32, 10199–10205. 10.1021/acs.langmuir.6b02622. [DOI] [PubMed] [Google Scholar]
- Vorobii M.; De Los Santos Pereira A.; Pop-Georgievski O.; Kostina N. Y.; Rodriguez-Emmenegger C.; Percec V. Synthesis of Non-Fouling Poly[N-(2-Hydroxypropyl)Methacrylamide] Brushes by Photoinduced SET-LRP. Polym. Chem. 2015, 6, 4210–4220. 10.1039/C5PY00506J. [DOI] [Google Scholar]
- Sundaram H. S.; Han X.; Nowinski A. K.; Ella-Menye J. R.; Wimbish C.; Marek P.; Senecal K.; Jiang S. One-Step Dip Coating of Zwitterionic Sulfobetaine Polymers on Hydrophobic and Hydrophilic Surfaces. ACS Appl. Mater. Interfaces 2014, 6, 6664–6671. 10.1021/am500362k. [DOI] [PubMed] [Google Scholar]
- Xu L. Q.; Pranantyo D.; Neoh K. G.; Kang E. T.; Teo S. L. M.; Fu G. D. Synthesis of Catechol and Zwitterion-Bifunctionalized Poly(Ethylene Glycol) for the Construction of Antifouling Surfaces. Polym. Chem. 2016, 7, 493–501. 10.1039/C5PY01234A. [DOI] [Google Scholar]
- Kenausis G. L.; Vo J.; Elbert D. L.; Huang N.; Hofer R.; Ruiz-taylor L.; Textor M.; Hubbell J. A.; Spencer N. D. Poly (l-Lysine)-g-Poly (Ethylene Glycol) Layers on Metal Oxide Surfaces: Attachment Mechanism and Effects of Polymer Architecture on Resistance to Protein Adsorption. J. Phys. Chem. B 2000, 104, 3298–3309. 10.1021/jp993359m. [DOI] [Google Scholar]
- Pasche S.; De Paul S. M.; Vörös J.; Spencer N. D.; Textor M. Poly(l-Lysine)-Graft-Poly (Ethylene Glycol) Assembled Monolayers on Niobium Oxide Surfaces: A Quantitative Study of the Influence of Polymer Interfacial Architecture on Resistance to Protein Adsorption by ToF-SIMS and in Situ OWLS. Langmuir 2003, 19, 9216–9225. 10.1021/la034111y. [DOI] [Google Scholar]
- Morgese G.; Verbraeken B.; Ramakrishna S. N.; Gombert Y.; Cavalli E.; Rosenboom J. G.; Zenobi-Wong M.; Spencer N. D.; Hoogenboom R.; Benetti E. M. Chemical Design of Non-Ionic Polymer Brushes as Biointerfaces: Poly(2-Oxazine)s Outperform Both Poly(2-Oxazoline)s and PEG. Angew. Chem., Int. Ed. 2018, 57, 11667–11672. 10.1002/anie.201805620. [DOI] [PubMed] [Google Scholar]
- Perry S. S.; Yan X.; Limpoco F. T.; Lee S.; Müller M.; Spencer N. D. Tribological Properties of Poly(l-lysine)-graft-poly(ethylene glycol) Films: Influence of Polymer Architecture and Adsorbed Conformation. ACS Appl. Mater. Interfaces 2009, 1, 1224–1230. 10.1021/am900101m. [DOI] [PubMed] [Google Scholar]
- Huang W. M.; Gibson S. J.; Facer P.; Gu J.; Polak J. M. Improved Section Adhesion for Immunocytochemistry Using High Molecular Weight Polymers of l-Lysine as a Slide Coating. Histochemistry 1983, 77, 275–279. 10.1007/BF00506570. [DOI] [PubMed] [Google Scholar]
- Yan X.; Perry S. S.; Spencer N. D.; Pasche S.; De Paul S. M.; Textor M.; Lim M. S. Reduction of Friction at Oxide Interfaces upon Polymer Adsorption from Aqueous Solutions. Langmuir 2004, 20, 423–428. 10.1021/la035785b. [DOI] [PubMed] [Google Scholar]
- Parks G. A. The Isoelectric Points of Solid Oxides, Solid Hydroxides, and Aqueous Hydroxo Complex Systems. Chem. Rev. 1965, 65, 177–198. 10.1021/cr60234a002. [DOI] [Google Scholar]
- Weber D.; Torger B.; Richter K.; Nessling M.; Momburg F.; Woltmann B.; Müller M.; Schwartz-Albiez R. Interaction of Poly(l-Lysine)/Polysaccharide Complex Nanoparticles with Human Vascular Endothelial Cells. Nanomaterials 2018, 8, 358 10.3390/nano8060358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold D. A.; Keil K.; Bruns D. E. Oxidation of Polyethylene Glycols by Alcohol Dehydrogenase. Biochem. Pharmacol. 1989, 38, 73–76. 10.1016/0006-2952(89)90151-2. [DOI] [PubMed] [Google Scholar]
- Lubich C.; Allacher P.; de la Rosa M.; Bauer A.; Prenninger T.; Horling F. M.; Siekmann J.; Oldenburg J.; Scheiflinger F.; Reipert B. M. The Mystery of Antibodies Against Polyethylene Glycol (PEG) - What Do We Know?. Pharm. Res. 2016, 33, 2239–2249. 10.1007/s11095-016-1961-x. [DOI] [PubMed] [Google Scholar]
- Garay R. P.; El-Gewely R.; Armstrong J. K.; Garratty G.; Richette P. Antibodies against Polyethylene Glycol in Healthy Subjects and in Patients Treated with PEG-Conjugated Agents. Expert Opin. Drug Delivery 2012, 9, 1319–1323. 10.1517/17425247.2012.720969. [DOI] [PubMed] [Google Scholar]
- Armstrong J. K.; Hempel G.; Koling S.; Chan L. S.; Fisher T.; Meiselman H. J.; Garratty G. Antibody against Poly(Ethylene Glycol) Adversely Affects PEG-Asparaginase Therapy in Acute Lymphoblastic Leukemia Patients. Cancer 2007, 110, 103–111. 10.1002/cncr.22739. [DOI] [PubMed] [Google Scholar]
- Branden R.; Matthew G.; Anirudha S.; Janis T.; Cecilia F.; Melissa M.; Jennifer E. PEG Hydrogel Degradation and the Role of the Surrounding Tissue Environment. J. Tissue Eng. Regener. Med. 2015, 9, 315–318. 10.1002/term.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. N.; Chu D. S. H.; Shi J.; SChellinger J. G.; Carlson P. M.; Pun S. H. HPMA-Oligolysine Copolymers for Gene Delivery: Optimization of Peptide Length and Polymer Molecular Weight Russell. J. Controlled Release 2011, 155, 303–311. 10.1016/j.jconrel.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tappertzhofen K.; Weiser F.; Montermann E.; Reske-Kunz A.; Bros M.; Zentel R. Poly-l-lysine-poly[HPMA] Block Copolymers Obtained by RAFT Polymerization as Polyplex-Transfection Reagents with Minimal Toxicity. Macromol. Biosci. 2015, 15, 1159–1173. 10.1002/mabi.201500022. [DOI] [PubMed] [Google Scholar]
- Thomson D.; Zilkie A.; Bowers J. E.; Komljenovic T.; Reed G. T.; Vivien L.; Marris-Morini D.; Cassan E.; Virot L.; Fédéli J. M.; Hartmann J. M.; Schmid J. H.; Xu D. X.; Boeuf F.; O’Brien P.; Mashanovich G. Z.; Nedeljkovic M. Roadmap on Silicon Photonics. J. Opt. 2016, 18, 1–20. 10.1088/2040-8978/18/7/073003. [DOI] [Google Scholar]
- Qi Z. B.; Xu L.; Xu Y.; Zhong J.; Abedini A.; Cheng X.; Sinton D. Disposable Silicon-Glass Microfluidic Devices: Precise, Robust and Cheap. Lab Chip 2018, 18, 3872–3880. 10.1039/C8LC01109E. [DOI] [PubMed] [Google Scholar]
- Niu J.; Lunn D. J.; Pusuluri A.; Yoo J. I.; O’Malley M. A.; Mitragotri S.; Soh H. T.; Hawker C. J. Engineering Live Cell Surfaces with Functional Polymers via Cytocompatible Controlled Radical Polymerization. Nat. Chem. 2017, 9, 537–545. 10.1038/nchem.2713. [DOI] [PubMed] [Google Scholar]
- Lueckerath T.; Strauch T.; Koynov K.; Barner-Kowollik C.; Ng D. Y. W.; Weil T. DNA-Polymer Conjugates by Photoinduced RAFT Polymerization. Biomacromolecules 2019, 20, 212–221. 10.1021/acs.biomac.8b01328. [DOI] [PubMed] [Google Scholar]
- Kuzmyn A. R.; Nguyen A. T.; Teunissen L. W.; Zuilhof H.; Baggerman J. Antifouling Polymer Brushes via Oxygen-Tolerant Surface-Initiated PET-RAFT. Langmuir 2020, 36, 4439–4446. 10.1021/acs.langmuir.9b03536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallart X.; Henry de Villeneuve C.; Allongue P. Truly Quantitative XPS Characterization of Organic Monolayers on Silicon: Study of Alkyl and Alkoxy Monolayers on H-Si(111). J. Am. Chem. Soc. 2005, 127, 7871–7878. 10.1021/ja0430797. [DOI] [PubMed] [Google Scholar]
- Scheres L.; Giesbers M.; Zuilhof H. Organic Monolayers onto Oxide-Free Silicon with Improved Surface Coverage: Alkynes versus Alkenes. Langmuir 2010, 26, 4790–4795. 10.1021/la9035503. [DOI] [PubMed] [Google Scholar]
- Morga M.; Adamczyk Z.; Gödrich S.; Oćwieja M.; Papastavrou G. Monolayers of Poly-l-lysine on Mica - Electrokinetic Characteristics. J. Colloid Interface Sci. 2015, 456, 116–124. 10.1016/j.jcis.2015.05.044. [DOI] [PubMed] [Google Scholar]
- Elbert D. L.; Herbert C. B.; Hubbell J. A. Thin Polymer Layers Formed by Polyelectrolyte Multilayer Techniques on Biological Surfaces. Langmuir 1999, 15, 5355–5362. 10.1021/la9815749. [DOI] [Google Scholar]
- Xiao A.; Dhand C.; Leung C. M.; Beuerman R. W.; Ramakrishna S.; Lakshminarayanan R. Strategies to Design Antimicrobial Contact Lenses and Contact Lens Cases. J. Mater. Chem. B 2018, 6, 2171–2186. 10.1039/C7TB03136J. [DOI] [PubMed] [Google Scholar]
- Poręba R.; de los Santos Pereira A.; Pola R.; Jiang S.; Pop-Georgievski O.; Sedláková Z.; Schönherr H. “Clickable” and Antifouling Block Copolymer Brushes as a Versatile Platform for Peptide-Specific Cell Attachment. Macromol. Biosci. 2020, 20, 1–10. 10.1002/mabi.201900354. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy M.; Hakobyan S.; Ramstedt M.; Gautrot J. E. Surface-Initiated Polymer Brushes in the Biomedical Field: Applications in Membrane Science, Biosensing, Cell Culture, Regenerative Medicine and Antibacterial Coatings. Chem. Rev. 2014, 114, 10976–11026. 10.1021/cr500252u. [DOI] [PubMed] [Google Scholar]
- Yang W.; Xue H.; Li W.; J Z.; Jiang S. Pursuing “Zero” Protein Adsorption of Poly(Carboxybetaine) from Undiluted Blood Serum and Plasma. Langmuir 2009, 25, 11911–11916. 10.1021/la9015788. [DOI] [PubMed] [Google Scholar]
- Vaisocherová-Lísalová H.; Surman F.; Víšová I.; Vala M.; Špringer T.; Ermini M. L.; Šípová H.; Šedivák P.; Houska M.; Riedel T.; Pop-Georgievski O.; Brynda E.; Homola J. Copolymer Brush-Based Ultralow-Fouling Biorecognition Surface Platform for Food Safety. Anal. Chem. 2016, 88, 10533–10539. 10.1021/acs.analchem.6b02617. [DOI] [PubMed] [Google Scholar]
- Wang Y. S.; Yau S.; Chau L. K.; Mohamed A.; Huang C. J. Functional Biointerfaces Based on Mixed Zwitterionic Self-Assembled Monolayers for Biosensing Applications. Langmuir 2019, 35, 1652–1661. 10.1021/acs.langmuir.8b01779. [DOI] [PubMed] [Google Scholar]
- Lin X.; Jain P.; Wu K.; Hong D.; Hung H. C.; O’Kelly M. B.; Li B.; Zhang P.; Yuan Z.; Jiang S. Ultralow Fouling and Functionalizable Surface Chemistry Based on Zwitterionic Carboxybetaine Random Copolymers. Langmuir 2019, 35, 1544–1551. 10.1021/acs.langmuir.8b02540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr L. R.; Xue H.; Jiang S. Functionalizable and Nonfouling Zwitterionic Carboxybetaine Hydrogels with a Carboxybetaine Dimethacrylate Crosslinker. Biomaterials 2011, 32, 961–968. 10.1016/j.biomaterials.2010.09.067. [DOI] [PubMed] [Google Scholar]
- Kowalczyńska H. M.; Nowak-Wyrzykowska M.; Szczepankiewicz A.; Dobkowski J.; Dyda M.; Kamiński J.; Kołos R. Albumin Adsorption on Unmodified and Sulfonated Polystyrene Surfaces, in Relation to Cell-Substratum Adhesion. Colloids Surf., B 2011, 84, 536–544. 10.1016/j.colsurfb.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Price W. S.; Tsuchiya F.; Arata Y. Lysozyme Aggregation and Solution Properties Studied Using PGSE NMR Diffusion Measurements. J. Am. Chem. Soc. 1999, 121, 11503–11512. 10.1021/ja992265n. [DOI] [Google Scholar]
- Zhu L. J.; Zhu L. P.; Jiang J. H.; Yi Z.; Zhao Y. F.; Zhu B. K.; Xu Y. Y. Hydrophilic and Anti-Fouling Polyethersulfone Ultrafiltration Membranes with Poly(2-Hydroxyethyl Methacrylate) Grafted Silica Nanoparticles as Additive. J. Membr. Sci. 2014, 451, 157–168. 10.1016/j.memsci.2013.09.053. [DOI] [Google Scholar]
- Alswieleh A. M.; Cheng N.; Canton I.; Ustbas B.; Xue X.; Ladmiral V.; Xia S.; Ducker R. E.; Zubir O.; El; Cartron M. L.; Hunter C. N.; Leggett G. J.; Armes S. P. Zwitterionic Poly(Amino Acid Methacrylate) Brushes. J. Am. Chem. Soc. 2014, 136, 9404–9413. 10.1021/ja503400r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai C.; Lee J.; Park J.; Takhistov P. Antibody Immobilization on a Nanoporous Aluminum Surface for Immunosensor Development. Appl. Surf. Sci. 2012, 263, 195–201. 10.1016/j.apsusc.2012.09.027. [DOI] [Google Scholar]
- Jain P.; Dai J.; Grajales S.; Saha S.; Baker G. L.; Bruening M. L. Completely Aqueous Procedure for the Growth of Polymer Brushes on Polymeric Substrates. Langmuir 2007, 23, 11360–11365. 10.1021/la701735q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.