

SUMMARY
Drosophila Dpr (21 paralogs) and DIP proteins (11 paralogs) are cell recognition molecules of the immunoglobulin superfamily (IgSF) that form a complex protein interaction network. DIP and Dpr proteins are expressed in a synaptic layer-specific fashion in the visual system. How interactions between these proteins regulate layer-specific synaptic circuitry is not known. Here we establish that DIP-α and its interacting partners Dpr6 and Dpr10 regulate multiple processes, including arborization within layers, synapse number, layer specificity, and cell survival. We demonstrate that heterophilic binding between Dpr6/10 and DIP-α and homophilic binding between DIP-α proteins promote interactions between processes in vivo. Knockin mutants disrupting the DIP/Dpr binding interface reveal a role for these proteins during normal development, while ectopic expression studies support an instructive role for interactions between DIPs and Dprs in circuit development. These studies support an important role for the DIP/Dpr protein interaction network in regulating cell-type-specific connectivity patterns.
In Brief
Xu et al. demonstrate that DIP-α and Dpr6/10 heterophilic and DIP-α homophilic interactions regulate multiple aspects of circuit assembly including layer-specific targeting, cell survival, and synapse number and distribution in the Drosophila visual system.
INTRODUCTION
The extraordinary diversity of cell surface proteins expressed in different neuron types during development and the complexity of synaptic connectivity suggest that molecular complexity at the cell surface contributes in important ways to synaptic specificity (Takemura et al., 2015; Tan et al., 2015; Zhang et al., 2016). One attractive notion is that different members of families of cell recognition molecules regulate interactions between related, but different, neurons in a similar way (Zipursky and Sanes, 2010). That is, diversity in their extracellular domains would allow for specific associations between different sets of neurons, but the output (e.g., synapse formation) might be similar. Here, we use genetic analysis guided by biochemical and developmental studies to assess the roles of one cognate pair of proteins within the DIP-Dpr interaction network in regulating circuit assembly in the Drosophila visual system.
In many regions of the brain, connections between neurons occur in discrete layers, with different neuron types forming connections in different layers. In the vertebrate and fly visual systems, the inner plexiform layer (IPL) in the retina and the medulla neuropil are arranged in an analogous fashion, with about a hundred different neuronal cell types forming layer-specific patterns of connectivity (Sanes and Zipursky, 2010). In the mouse IPL, classical cadherins and members of the Sema/Plexin family regulate the layer-specific organization of processes in the IPL (Duan et al., 2014; Matsuoka et al., 2011). In the chick IPL, different Ig superfamily (IgSF) proteins of the related sidekick (sdk), Dscam, and connectin families are expressed in different layers (Yamagata and Sanes, 2008, 2012; Yamagata et al., 2002). Gain- and loss-of-function experiments support their role in establishing layer-specific circuitry. The layer-specific expression of these proteins in the mouse is less pronounced and it remains unclear whether these proteins play the same role in the mouse IPL as in the chick. Genetic studies, however, have shown that Sdk2 regulates synaptic connectivity between one amacrine cell subtype and a specific retinal ganglion cell within a layer of the mouse IPL (Krishnaswamy et al., 2015). Thus, these data suggest that different related IgSF proteins may act together to determine different layer specificities through a common molecular strategy.
Cell recognition molecules regulating the layered organization of the Drosophila medulla neuropil have also been identified. These have been studied largely in the context of two photoreceptor neurons, R7 and R8, and five lamina interneurons, L1–L5. The axons of these neurons terminate in different layers, elaborate unique morphologies, and form specific patterns of connections with an array of different classes of medulla neurons. Cadherins, semaphorins, receptor tyrosine phosphatases, and netrin/DCC proteins, among others, have been shown to regulate layer-specific innervation in this system (Clandinin et al., 2001; Lee et al., 2001; Maurel-Zaffran et al., 2001; Nern et al., 2008; Pecot et al., 2013; Timofeev et al., 2012). Different recognition systems regulate targeting to different layers. Conversely, in some cases, different recognition molecules regulate targeting of different neurons to the same layer. Combinatorial mechanisms also contribute to layer-specific wiring. For instance, N-cadherin and the protein tyrosine phosphatases Lar and Ptp69D are required for targeting of R7 neurons to one layer (Clandinin et al., 2001; Hakeda-Suzuki et al., 2017; Lee et al., 2001; Newsome et al., 2000), whereas N-cadherin and Sema/Plexin signaling act in parallel to regulate targeting of L1 neurons to another (Pecot et al., 2013). Despite progress in identifying molecules controlling the patterning of connections in this system, there is little evidence for a common regulatory logic underlying the patterning of connectivity in different layers.
As a step toward uncovering mechanisms underlying layer-specific connectivity, we used RNA sequencing (RNA-seq) to profile mRNAs encoding cell surface and secreted proteins that are expressed in R7, R8, and L1–L5 neurons just prior to the onset of synapse formation (Tan et al., 2015). Each neuronal cell type expresses hundreds of genes encoding cell surface proteins, and striking differences in expression were observed between different cell types. This complexity mirrors the diversity of neuronal cell types that each neuronal process encounters within the developing neuropil and the specificity of synaptic connectivity within the mature neuropil revealed through serial EM reconstruction studies (Takemura et al., 2013, 2015). The data suggest that a complex choreography of cell surface protein interactions underlies the formation of neural circuits in the medulla, and also that considerable redundancy may complicate genetic analyses of their functions.
Among the many genes differentially expressed in these neurons, the dprs were particularly intriguing. The Dprs comprise a 21-member subfamily of IgSF cell surface proteins, with each paralog binding to one or more DIPs, a different, though closely related IgSF subfamily with eleven members (Carrillo et al., 2015; Özkan et al., 2013; Cosmanescu et al., 2018). DIPs are expressed in a striking layer-specific fashion, and many DIP/Dpr binding partners are expressed in neurons previously shown to be synaptic partners (Takemura et al., 2013, 2015; Carrillo et al., 2015; Tan et al., 2015; Cosmanescu et al., 2018; also see Figure S14). Previous genetic experiments indicate that DIP-γ and Dpr11 regulate circuitry in the M6 layer of the medulla neuropil, but how they function in this process remains unclear (Carrillo et al., 2015; see Results). This pair of binding partners also regulates the morphologies of synaptic terminals of motor neurons on larval body wall muscles (Carrillo et al., 2015).
Here we demonstrate that DIP-α and its interacting partners Dpr6 and Dpr10 play a crucial role in regulating the assembly of neural circuitry in the M3 layer of the medulla using analysis of loss-of-function null alleles, knockins selectively disrupting specific protein interaction sites, genetic mosaics, and gain-of-function perturbations. Together these data support a role for heterophilic interactions between DIP-α and Dpr6/10 in regulating arborization within layers, synapse number, layer specificity, and cell survival of amacrine-like Dm4 and Dm12 neurons. These studies also suggest that DIP-α/DIP-α homophilic interaction regulates circuit development. We propose that different DIP/Dpr binding partners are utilized in a layer- and context-specific way to regulate layer-specific circuitry in the medulla. Our work provides a framework for understanding the role of DIP/Dpr proteins in circuit assembly by combining genetics with biophysical data.
RESULTS
DIP-α, Dpr6, and Dpr10 Proteins Are Localized to Neuronal Processes in the Developing Medulla Neuropil
Using MiMIC technology, including protein (GFP) and transcription (GAL4) traps, we previously showed that DIP-α is expressed in the amacrine-like Dm4 and Dm12 medulla neurons in early pupae, after the onset of neuronal differentiation, and that expression continues into the adult (Figures 1A–1C) (Tan et al., 2015). Both neurons arborize in the M3 layer. Dm4 tiles (Figure 1B), whereas the arbors of Dm12 overlap (Figure 1C). EM reconstruction studies demonstrated that L3 neurons are presynaptic to Dm4 and are both pre- and postsynaptic to Dm12 neurons (Takemura et al., 2015; S. Takemura, I.A. Meinetzhagen, and L. Scheffer, personal communication). Consistent with RNA-seq studies, Dpr6 and Dpr10 MiMICs and their derivatives revealed expression in L3 neurons, prior to and during synapse formation (Tan et al., 2015).
Figure 1. DIP-α, Dpr6, and Dpr10 Proteins Are Localized to Neuronal Processes.

(A) Schematic of the adult Drosophila visual system. Cell types studied in this paper are shown. Adapted from Fischbach and Dittrich (1989).
(B and C) Wild-type Dm4 and Dm12 clones labeled by Multicolor Flip-out (MCFO) using Dm4- and Dm12-specific GAL4 lines. Dm4 neuron terminals tile; Dm12 terminals overlap.
(D) KD for interactions (see Cosmanescu et al., 2018).
(E–H) Developing medulla at 24 hr APF.
(E–E”) Antibody staining of wild-type pupa as indicated. Yellow dotted line, cell body region in the medulla; green dotted lines, medulla neuropil.
(F) Dm4 axons in the medulla neuropil. Image, maximum intensity projection from a 10 mm stack. Arrow, Dm4 target layer labeled by anti-chaoptin (red) and myrGFP-labeled Dm4 neurons (green).
(G) Dm4 dendrites contacting Dpr10-protein-rich layer. Green, myrGFP-labeled Dm4 neurons; red, Dpr10 antibody staining.
(H) L3 contacts Dm4 at 24 hr APF. Green, myrGFP labeled Dm4 neuron; red, a myr-tdTomato-labeled L3 growth cone.
See also Figures S1–S3.
DIP-α was originally shown to interact heterophilically with Dpr6 and Dpr10 (Özkan et al., 2013). Surface plasmon resonance (SPR) binding studies reported in the accompanying paper (Cosmanescu et al., 2018) have now demonstrated that DIP-α also displays homophilic interactions (Figure 1D). DIP-α binds strongly (KDs ≈ 1–2 μM) to Dpr6 and Dpr10D, the Dpr10 isoform predominantly expressed in lamina neurons (Figure S8). In a comprehensive biochemical study, the binding interactions of DIP-α with all other Dprs were either undetectable or weak, with KD > 200 μM (Cosmanescu et al., 2018). Dpr6 and Dpr10 interactions with DIPα were among the strongest observed for DIP/Dpr interactions. Analytical ultracentrifugation (AUC) analysis further revealed that DIP-α behaves as a homodimer, with KD = 24 μM (Figure 1D; Cosmanescu et al., 2018). In addition to the strong binding to DIP-α, Dpr6 and Dpr10D exhibited weaker binding to a few other DIPs, with KD binding affinities of 19.4 and 54.9 μM to DIP-β, respectively, and 28.4 and 88 μM to DIP-λ, respectively. Dpr6 also exhibited a weak affinity to DIP-ζ of 151 μM, while no measurable affinity was observed for Dpr10D. Thus, although there are no binding partners with substantial affinity for DIP-α other than Dpr6 and Dpr10, there is appreciable binding of other DIPs to both Dpr6/10. Given that each medulla neuron typically expresses a single or a small number of DIPs (see accompanying paper), but lamina neurons express many Dprs, we predicted that DIP-expressing medulla neurons were more likely to exhibit loss-of-function mutant phenotypes.
To assess the distribution of DIP-α, Dpr6, and Dpr10 proteins, we generated mouse antibodies to them and stained tissue at different developmental stages (Figure S1). At 24 hr after pupa formation (APF), staining with all three antibodies was observed within the nascent neuropil (Figures 1E–1E”). Staining was not observed in null mutants (insets in Figures 1E–1E”). By this stage in development, many Dm4 neurons have already extended their axons into the incipient M3 layer, where they encounter processes, including those of lamina L3 neurons, expressing Dpr6 and Dpr10 proteins (Figures 1F–1H, S1B, S1C”, and S2A– S2A”); Dm12 neurons project into the brain at about the same stage or slightly later than Dm4 (Figures S2B–S2B”). Dpr6 and Dpr10 are also expressed in other lamina and medulla neurons (Tan et al., 2015; Figure S3). DIP-α, Dpr6, and Dpr10 proteins were observed in a layer-specific fashion at 24 hr APF. Their expression patterns change during development (Figures S1A– S1C”). Specifically, expression within the incipient M3 layer was prominent at 24 and 48 hr. The expression of all three proteins decreased at later stages of development and was very weak in adults (Figures S1A–S1C”). At 24 hr both Dm4 and Dm12 neurons elaborate processes within the incipient layer, but their arbor patterns are immature (Figures S2B–S2B”). Thus, these data support the notion that interactions between DIP-α proteins or between DIP-α and Dpr6/10 may occur between neurites within target layers in the developing neuropil.
DIP-α Promotes Interactions between Neurons within the Developing M3 Layer
We next sought to assess the role of DIP-α and Dpr6/10 regulating circuitry within the M3 layer. A previous study (Carrillo et al., 2015) reported subtle changes in the morphology of R7 terminals in both DIP-γ and dpr11 insertion mutants in the M6 layer. In this study, morphology defects were seen in R7 neurons in which terminals were visualized by overexpression of a short non-functional form of Brp fused to GFP. We did not observe defects in R7 terminal morphology or in the number of Brp puncta in dpr11null deletion mutant animals analyzed via STaR (Figure S4) (Chen et al., 2014). The reasons for the discrepancy between these results are not known. The background frequency of overshoots in heterozygote controls reported using Brp-short in Carrillo et al. (2015) is 3- to 4-fold higher than the frequency we obtained with STaR (Figure S4B). Overexpression of Brp-short, thus, appears to produce overshoot phenotypes on its own. Whether the increase in Brp-short overshoots over controls observed for dpr11MiMIC/deficiency transheterozygotes is due to loss of dpr11 or to a genetic background effect remains to be determined. Similarly, we did not observe targeting defects of L3 neurons to M3 in either DIP-α or dpr6,10 mutants. We, thus, turned our attention to the role of DIP/Dpr interactions in Dm4 and Dm12 development.
To assess whether DIP-α regulates the morphology of Dm4 and Dm12 neurons, we generated DIP-α null mutant neurons in an otherwise wild-type (i.e., heterozygous) background using mosaic analysis with a repressible cell marker (MARCM) (Lee and Luo, 1999). Single Dm4 mutant neurons target to M3 as in wild-type, but the branches of mutant neurons do not cover as many columns within the layer (WT, ~16 columns; DIP-αnull, ~11 columns) (Figures 2A, 2B, and 2K). By contrast, single Dm12 mutant neurons in a wild-type background exhibit robust defects in target layer specificity. Some 60% of mutant neurons (n = 150 neurons examined) extend an additional branch to M8, where they arborize within this layer (Figures 2E, 2F, and 2L). This mistargeting was first observed ~35 hr APF (Figures 2I and 2J), but not at 30 hr. The fraction of Dm12 neurons exhibiting mistargeting increased to that seen in the adult by about 50 hr APF. These data argue that DIP-α promotes interaction of Dm12 neurites with processes of other neurons that project to the M3 layer as early as 35 hr APF and continue to promote interactions to 50 hr APF.
Figure 2. DIP-α Is Required to Promote the Layer-Specific Patterning of Dm4 and Dm12 Axon Terminals.

(A–D) Single Dm4 neurons labeled using MARCM.
(E–H) Single Dm12 neurons labeled using MARCM. DIP-α het-homo: this mutation disrupts both heterophilic and homophilic binding. Isoleucine 83 was changed to aspartic acid (I83D). DIP-α homo: this mutant protein disrupts homophilic binding only. It is a double mutant with alanine 78 changed to lysine and asparagine 94 converted to aspartic acid (A78K+N94D) (see text and Cosmanescu et al., 2018).
(I and J) Single Dm12 neurons labeled using MARCM at 35 hr APF. Blue and orange dotted lines indicate two different medulla layers.
(K) Arbor coverage of Dm4 neurons within M3 (n: 11–16 neurons, except for DIP-α het-homo, which is 6. DIP-α mutant, p < 0.0001; DIP-α het-homo, p = 0.0005, unpaired t test).
(L) Dm12 targeting defects to M8 (brains analyzed, 7–13; number of labeled neurons analyzed, 40–166). Note that all mutant neurons are generated in an otherwise wild-type background (i.e., heterozygous background).
(K) and (L) are represented as mean ± SD. See also Figures S4–S6.
Dm1 neurons also express DIP-α and single mutant Dm1 neurons lacking DIP-α still target to M1 as they do in wild-type. These neurons exhibited a reduction in branch points, but no change in the number of boutons, or the number of active zones scored by the presence of Brp puncta (Figure S5). As the Dm1 phenotype is subtle compared with that of the other two Dm neurons, we focus here on defects in Dm4 and Dm12 arbors in the M3 layer.
We next sought to determine whether DIP-α function requires homophilic binding to DIP-α, or heterophilic binding to Dpr6/10. To address this question, we generated DIP-α mutant alleles that disrupt both heterophilic and homophilic binding (DIP-α homo-het) and homophilic binding only (DIP-α homo) (Cosmanescu et al., 2018). We were unable to design mutants that disrupted heterophilic interactions while leaving homophilic interactions intact. Mutant proteins were expressed at levels similar to wild-type, as assessed using antibodies to optic lobe tissue at 40 hr APF (Figures S6A–S6C’). We assessed the phenotype of single mutant neurons harboring knockin mutations using MARCM. Both Dm4 and Dm12 neurons homozygous for DIP-α homo were indistinguishable from wild-type (Figures 2D, 2H, 2K, and 2L). By contrast, the morphology and targeting of Dm4 and Dm12 neurons homozygous for DIP-α homo-het were indistinguishable from DIP-α null mutant neurons (Figures 2C, 2G, 2K, and 2L). The simplest interpretation of these data is that heterophilic, but not homophilic, binding is necessary for targeting. As we were unable to generate alleles that disrupted heterophilic interactions only, however, it remains possible that either heterophilic or homophilic binding is sufficient to promote interactions within M3.
Altering the Layer-Specific Expression of Dpr10 Leads to Dm4 and Dm12 Mistargeting
The analysis of MARCM clones described above suggested DIP-α binding to Dpr6/10 mediates interactions of Dm4 and Dm12 neurites with other neuronal processes within M3. But do Dpr6, Dpr10, or both Dprs in M3-projecting neurons specify targeting of Dm4 and Dm12 processes to this layer? If so, they must be partially redundant with other molecules, since processes of DIP-α mutant neurons still arborize in M3, although they have abnormal distributions within that layer (Dm4) or target to an additional layer (Dm12). To determine whether interactions of DIP-α with Dpr6/10 can direct Dm4 or Dm12 processes to a target layer, we used a gain-of-function approach. We expressed Dpr10 in the nascent inner medulla just underlying the incipient M3 layer using the 42F06-GAL4 driver in T4 neurons (T4-GAL4). The T4 dendrites arborize within this layer, the incipient M10 layer; subsequent growth of the medulla neuropil leads to substantial separation of the developing M3 and M10 layers in the mature medulla (Figures 3A’–3C’, 3A”–3C”, S7A, and S7A’). This driver is also expressed in T5, but as these neurons do not project into the medulla, this feature of the expression is unlikely to influence DIP-a expressing Dm4 or Dm12 neurons (Maisak et al., 2013).
Figure 3. Ectopic Expression of Dpr10 Promotes Mistargeting of Dm4 and Dm12 Terminals.
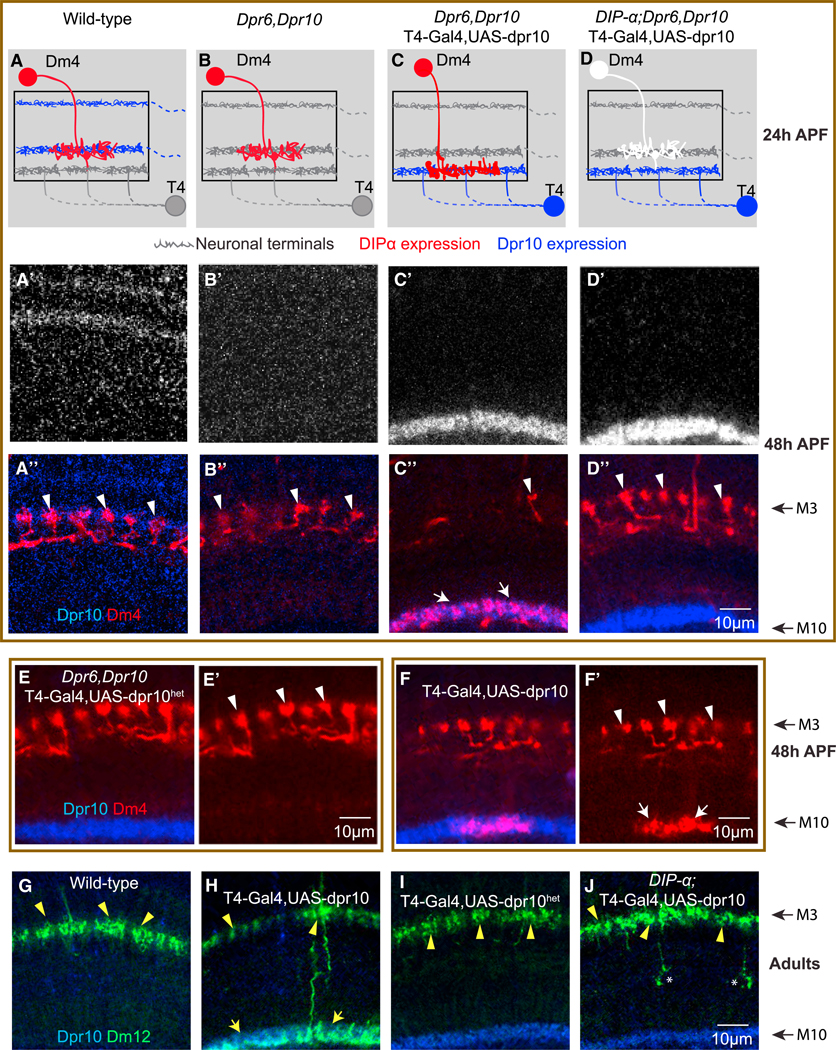
(A–D”) Wild-type (A–A”); dpr6, dpr10 double mutant animals (B–B”); T4-GAL4-driven expression of UAS-Dpr10 in the medulla M10 layer, in a dpr6, dpr10 double mutant animal (C–C”); and T4-GAL4-driven expression of UAS-Dpr10 in the M10 layer, in a DIP-α, dpr6, dpr10 triple mutant animal (D–D”).
(A–D) Schematic of targeting events at 24 hr APF. Red neuron, Dm4 expressing DIP-α; white neuron, Dm4 in DIP-αnull animal; blue neuron, expressing Dpr10; gray neuron, no expression of Dpr10.
(E and E’) T4-GAL4 driving expression of a Dpr10 mutant (Dpr10het) protein that does not bind to DIP-α does not promote Dm4 mistargeting.
(F and F’) T4-GAL4 drives expression of UAS-Dpr10 in the medulla M10 layer, in an otherwise wild-type background. Note phenotype is not as strong as in a dpr6,dpr10 double mutant background (see C”).
(A’–D’, A”–D”, E, E’, F, and F’) Images at 48 hr APF. Note layers at 48 hr are further apart than at 24 hr (schematically shown in A–D) due to intercalary growth of other neuronal processes. Red, myr-tdTomato-labeled Dm4; blue, anti-Dpr10 antibody; white arrowheads, Dm4 terminals in M3; white arrows, Dm4s mistargeted to M10.
(G–J) Wild-type Dm12 neurons target to the M3 layer in adults (G); T4-GAL4 driving expression of UAS-Dpr10 in the medulla M10 layer, in an otherwise wild-type animal, causes Dm12 to mistarget to the M10 layer. Note phenotype is not as strong as in a dpr6,dpr10 double mutant background (H); expressing a Dpr10 isoform (Dpr10het) that disrupts heterophilic binding does not cause Dm12 mistargeting (I); T4-GAL4 drives expression of UAS-Dpr10 in the M10 layer, in a DIP-α mutant animal; removing DIP-α suppresses mistargeting of Dm12 to the M10 layer induced by T4-Gal4 driven UAS-Dpr10 expression (J); green, myr-tdTomato- labeled Dm12; blue, anti-Dpr10 antibody; yellow arrowheads, Dm12 terminals at M3; yellow arrows, Dm12s mistargeted to M10.
See also Figures S7 and S8.
There are two protein isoforms of Dpr10 annotated in Flybase, Dpr10-PA and Dpr10-PD. Dpr10-PA contains an additional 46 amino acids between Ig1 and Ig2; these isoforms are otherwise identical. We performed RT-PCR experiments using mRNA from whole brain extracts from 3rd instar larvae and three stages of pupal development (i.e., 24, 42, and 72 hr APF) to assess expression of these isoforms. Only Dpr10-RD (renamed here as Dpr10D) was expressed (Figure S8). Accordingly, we expressed Dpr10D under the control of the T4-GAL4 driver. In early pupae Dpr10D protein accumulated in developing T4 dendrites located just beneath the incipient M3 layer in close proximity to nascent Dm4 and Dm12 processes (Figures 3A–3C, S7A, and S7A’).
Both wild-type Dm4 and Dm12 axons were efficiently re-targeted to T4 dendrites in response to ectopic Dpr10D (Figure 3). Dm4 targeting was virtually complete in a dpr6,10 double mutant background in which Dpr 10D was misexpressed in T4 (Figures 3C–3C”). Only a few small branches remained in M3 (Figure 3C’, arrowhead), suggesting that all Dm4 neurons mistargeted. This phenotype was suppressed by removing DIP-α (Figures 3D–3D”) or in animals carrying the DIP-αhet-homo allele (Figures S7A–S7A”), but not in animals carrying the DIP-α homo allele (Figures S7B–S7B”). Mistargeting was not seen in reponse to misexpression of Dpr10Dhet, a mutant form of Dpr10D that does not bind to DIP-α (Figures 3E, 3E’, S7D, and S7D’; Cosmanescu et al., 2018). The effect of competition for targeting between the two layers was highlighted in mistargeting experiments carried out in an otherwise wild-type background. Here the mistargeting of Dm4 to M10, though substantial, is attenuated (Figures 3F and 3F’). The substantial mistargeting in this background presumably reflects the higher level of expression from T4-GAL4 than endogenous expression. Similar mistargeting was seen for Dm12 neurons in response to T4-driven Dpr10D (Figures 3G–3J), as well as for both Dm4 and Dm12 in response to Dpr6 misexpression (Figures S7C–S7C”). These data indicate that DIP-α/Dpr10 (or Dpr6) binding is sufficient to promote interactions between processes in a layer-specific fashion.
Changes in the Number and Distribution of Presynaptic Sites in DIP-α Mutant Dm4 and Dm12 Neurons
We next sought to assess whether DIP-α was required in Dm4 and Dm12 neurons to control synaptogenesis. This was done by combining MARCM with the STaR technique (Figure 4A). This facilitates the generation of single mutant DIP-α neurons in which the presynaptic active zones in these neurons, and only in these mutant neurons, are selectively labeled with a unique epitope tag inserted into Brp, a presynaptic protein expressed in a modified BAC under the control of its endogenous regulatory mechanisms (Figure 4A). These Brp puncta are easily counted; in control experiments the number of these puncta corresponds well to synapse counts determined by electron microscopy (Chen et al., 2014). In mutant Dm4 and Dm12 there was a 37% and 35% loss of Brp puncta, respectively (Figures 4B, 4C, 4E, 4F, and 4H).
Figure 4. DIP-α Regulates the Number and Distribution of Presynaptic Sites.

(A) MARCM-STaR to assess the distribution of Brp puncta (active zone marker) in identified neuron types. Schematic diagram for MARCM-STaR (for genotype, see below). Note that in (C), (D), (F), and (G), single mutant neurons in an otherwise wild-type background are visualized.
(B–D) Dm4: wild-type (B), DIP-α mutant (C), and DIP-αhomo (D) Dm4 neurons labeled with MARCM-STaR.
(E–G) Dm12: wild-type (E), DIP-α mutant (F), and DIP-αhomo (G) Dm12 neurons with presynaptic sites labeled with MARCM-STaR.
(B–G) Red, myr-tdTomato; green, Brp puncta. Note in (F) that Dm12 neurons with processes in a deeper layer (M8) also accumulate Brp puncta consistent with elaborating synapses in an inappropriate layer.
(H) Quantification of the total number of presynaptic sites per neuron at the M3 layer (Dm4: number of labeled neurons, 5–11; Dm12: number of labeled neurons, 8–12; ***p < 0.001; **p < 0.01).
(I) Quantification of the density of presynaptic sites in the M3 layer (Dm4 group: number of terminals [N.B. one terminal/column], 16–36 [exception, 108 terminals were analyzed in DIP-αhomo], p < 0.0001; Dm12 group: number of labeled neurons, 6–10, p = 0.001; unpaired t test).
(H) and (I) are represented as mean ± SD. The genotype for MARCM-STaR is DIP-α, FRT9–2/Act-GAL80, FRT9–2; hs-Flp:PEST/UAS-R, LexAOP-myrTdTom; Dm4, or Dm12-GAL4/Brp-RSR-smGFP-V5–2A-LexA. See also Figure S6.
For Dm4, this decrease was commensurate with the reduction in the number of columns covered; the density of synapses in each column remained the same as in wild-type (Figures 4B, 4C, 4H, and 4I).
By contrast, the decrease in the number of Brp puncta in Dm12 branches within M3 was not matched by a decrease in the number of columns covered within the layer; a slight decrease in column coverage in mutant Dm12 neurons (19.8 ± 2.1 to 17.4 ± 2.3 with a p value of 0.02) was observed, but this decrease was modest compared to the decrease in synapse number. Indeed, here the density of synapses was decreased by ~30% (Figures 4E, 4F, 4H, and 4I). This decrease in M3 synapses was seen both in Dm12 mutant neurons that arborize in M3 only and in Dm12 mutant neurons arborizing in both the M3 and M8 layers (Brp puncta in M3: wild-type Dm12, 80.1 ± 12.3; DIP-α mutant Dm12 arborizing in M3 only, 54.3 ± 13.7; DIP-α mutant Dm12 arborizing in both layers, 51.6 ± 13.4). In addition, Dm12 neurons that arborized within M8 also elaborated Brp puncta within this layer (19.1 ± 14.6), consistent with these mistargeted branches forming synapses in an inappropriate layer (Figure 4F). The total number of Brp puncta in mutant neurons that target to both M3 and M8, however, was not significantly different from the number of Brp puncta in M3 observed in wild-type Dm12 neurons. DIP/Dpr interactions may play a direct role in regulating synapse formation or maintenance in M3, or alternatively may act only indirectly to regulate this process by promoting interactions between appropriate synaptic partners.
Synaptic defects in DIP-αhomo and DIP-αhomo-het were also assessed. For Dm12, a similar decrease in the density of puncta per column was seen in DIP-αhomo-het as seen in DIP-αnull, whereas synapse density in DIP-αhomo was similar to wild-type. Although DIP-αhomo-het did not exhibit differences in synapse density in Dm4, consistent with the DIP-αnull result, there was about a 50% increase in Brp puncta per column in DIP-αhomo mosaics (Figures 4D and 4G–4I; see Regulation of Cell Interactions through DIP-α Homophilic Binding for further discussion of DIP/DIP interactions in vivo). Together, these findings support a role for DIP/DIP and DIP/Dpr interactions in regulating synapse number
In the course of generating single DIP-α mutant Dm4 neurons via MARCM, we observed a marked decrease in the frequency of generating null mutant neurons compared to wild-type controls. The decrease in frequency was also observed for DIP-αhomo-het, whereas the frequency of DIP-αhomo neurons was similar to wild-type. By contrast, generating single neurons expressing higher levels of DIP-α than wild-type neighbors increased their representation over wild-type. (Figure S9A). These observations suggested that DIP-α levels may also regulate cell number through survival, competition, or proliferation. This is consistent with previous studies from the Zinn group demonstrating that loss of DIP-γ led to a reduction in Dm8 cell number (Carrillo et al., 2015).
DIP-α and Dpr6/10 Interactions Regulate Cell Number during Development by Antagonizing Apoptosis
Cell Loss in DIP-α and Dpr6/10 Mutants
Both DIP-α and Dpr6/10 double homozygous mutant animals were viable and fertile. A modest decrease in cell number was observed in DIP-α and Dpr6/10 double mutant animals (~20%) for Dm12 (Figure S9B), and a stronger phenotype of Dm4 cell loss was seen in both mutant backgrounds (~40%–50%) (Figure 5A). The penetrance of the phenotype caused by removing both Dpr6 and Dpr10 is similar to that seen for DIP-α mutants. Single dpr6 and dpr10 mutants had less severe phenotypes. This supports the idea that they have partially overlapping functions, which is consistent with their biophysically determined DIP-a-binding properties (Figure 5A). Removing all three genes led to a cell loss phenotype similar to DIP-α alone or the dpr6/dpr10 double mutants, consistent with these genes acting in the same pathway (Figure 5A).
Figure 5. DIP-α Antagonizes Dm4 Apoptosis in Early Pupal Development.

(A) Dm4 cell number in adult whole-animal wild-type or null mutants as indicated (n = 8–18 optic lobes; ***p < 0.0001, **p = 0.0003, unpaired t test).
(B) Dm4 cell number at different developmental stages (n: 4–20 optic lobes).
(C) Schematic of apoliner transgene. TM, trans-membrane domain; nls, nuclear localization signal; dotted line, caspase-sensitive cleavage site.
(D) Dm4 neurons (%) in all animals analyzed that are positive for active caspase (nuclear eGFP) (n = 14, 13 optic lobes for DIP-α and wild-type).
(E) Apoliner expression pattern in Dm4 neurons at 24 hr APF. Upon caspase activation, the apoliner sensor is cleaved and eGFP translocates to the nucleus, leaving mRFP at membranes. Arrow indicates a Dm4 cell body with nuclear GFP and cytoplasmic RFP, while arrowheads point to Dm4 cell bodies with membrane-tethered eGFP and mRFP.
(F) Dm4 neurons with (arrow) or without (arrowhead) hid-GFP expression in wild-type or DIP-α null animals as indicated.
(G) Percentage of Dm4 neurons that are positive for the expression of hid-GFP or grimMiMIC in wild-type or DIP-α null animals (n: for the hid-GFP group, 26 and 28 optic lobes; for grimMiMIC group, 8 and 6 optic lobes; ***p = 0.0007, unpaired t test).
(H) Dm4 cell number in adult wild-type and DIP-α mutants is increased in animals heterozygous for hid or H99. The H99 deficiency removes four pro- apoptotic genes (hid, rpr, grim, and skl) (n: 5–16 optic lobes, **p = 0.0023, 0.0002, 0.0001 from left to right; ***p < 0.0001; unpaired t test).
(A), (B), (D), (G), and (H) are represented as mean ± SD.
See also Figures S9–S11.
We next sought to determine when cell loss occurred in mutants. As Dm4 cell loss is more dramatic than Dm12 loss, and as suitable cell-type-specific markers are available as early as 24 hr APF for Dm4, but not for Dm12, we focused on the kinetics of Dm4 cell loss (Figure 5B). Using several different GAL4 lines expressed in Dm4 during development, we assessed cell number from 24 hr APF into the adult. At 24 hr APF, there were, on average, 20 Dm4 neurons in wild-type and this increased to the adult number of ~40 neurons by 40 hr APF. By contrast, in DIP-αnull animals there were ~14 neurons at 24 hr and the adult number of ~20 was achieved at 40 hr.
The Reduction in Cell Number Results from Apoptosis of Differentiating Neurons
In principle, DIP/Dpr interactions could regulate cell number via cell proliferation, differentiation, or survival of postmitotic neurons. As DIP-α is not expressed in dividing cells (Figures S9D, S9D’, S9E, and S9E’), a role in survival seemed more likely. To assess whether the reduction in cell number in DIP-α is a consequence of apoptosis, we tested whether expression of caspase inhibitors can rescue the cell loss phenotype. Targeted expression of either the caspase inhibitor baculovirus p35 protein or the Drosophila death-associated inhibitor of apoptosis 1 (Diap1) protein in DIPα-expressing cells prevented the loss of Dm4 neurons (Figures 5B and S9C). Interestingly, the number of Dm4 neurons in animals in which p35 was driven in DIP-α-ex-pressing neurons, in either wild-type or DIP-α mutant animals, exceeded the number in wild-type, suggesting that about 10–15 Dm4 neurons are lost through apoptosis during normal development (Figure S9C). The finding that the number of Dm4 neurons generated at different stages of development was increased at 24 hr in animals expressing P35 supports this interpretation (Figure 5B). This is consistent with an increase in the fraction of Dm4 neurons with increased caspase activity from 2% in wild-type to 20% in DIP-α mutants at this stage in development (as assessed using Apoliner, a genetically encoded caspase sensor; Figures 5C–5E) (Bardet et al., 2008).
Importantly, all Dm4 neurons expressing DIP-α at 24 hr APF had extended axons into the medulla neuropil. Most, if not all, of these axons had already reached the nascent M3 layer, where DIP-α overlaps with the expression of both Dpr6 and Dpr10 proteins (Figures 1F–1H). Cell death appears to occur after Dm4 processes have extended into the Dpr6/10-rich developing M3 layer, but prior to synapse formation. Together, these data indicate that the number of Dm4 neurons is controlled by apoptosis during normal development and that survival is influenced, at least in part, via DIP/Dpr signaling.
DIP-a and Dpr6,10 Proteins Suppress Dm4 Cell Death
In Drosophila, cell death signals typically activate transcription of pro-apoptotic proteins, including Reaper, Hid, and Grim (Vernooy et al., 2000). These proteins bind to and antagonize inhibitor of apoptosis proteins (IAPs), thereby activating caspases and cell death. We assessed the expression of reaper, hid, and grim using transcriptional reporters (rpr-11-lacZ [Nordstrom et al., 1996], hid-GFP [Tanaka-Matakatsu et al., 2009], and a MiMIC for grim [Venken et al., 2011]) in wild-type and DIP-α mutant animals. At 24 hr APF, hid-GFP and grim MiMIC, but not rpr-LacZ, were detected in both wild-type and DIP-α mutant Dm4 cells (Figures 5F, 5F’, 5G, S9F, and S9G). The number of Dm4 cells positive for grim was the same in wild-type and DIP-α mutant pupae. By contrast, there was an increase in the number of Dm4 cells positive for hid-GFP signals in DIP-α mutants compared to wild-type (Figure 5G). This is consistent with DIP-α antagonizing a hid-mediated apoptotic pathway. Indeed, removal of one copy of hid, but not of a single copy of grim, led to an increase in cell number in both DIP-α null and DIP-α homo-het mutants (Figure 5H). Removing one copy of rpr also did not change Dm4 cell number (Figures S9H and S9I). These data support a model in which DIP-α acts upstream of hid to dampen its expression and thereby promotes Dm4 survival.
The Reduced Number of Dm4 Neurons Leads to an Increase in Column Coverage in DIP-α and dpr6,dpr10 Mutants
Like wild-type Dm4 neurons, Dm4 neurons in a DIP-a homozygous or dpr6,dpr10 double mutant animal tile. In these mutants, however, there is a marked decrease in cell density. As a consequence, the number of columns covered by Dm4 arbors increases considerably (from 18.6 columns to 26.1 columns) (Figure S10A). By contrast, in animals in which cell death is suppressed either in a wild-type background (i.e., excess Dm4 neurons are generated during normal development) or in a DIP-α homozygous mutant, there is an increase in cell number and a commensurate decrease in the number of columns covered (11.3 columns) (Figure S10A). In summary, there is a reciprocal relationship between the number of Dm4 neurons and the number of columns covered (Figure S10A).
Paradoxically, DIP-α mutant neurons in an otherwise wild-type background cover fewer columns than their wild-type counter-parts, whereas the column coverage of individual Dm4 mutant neurons in a homozygous animal is increased. Thus, in homozygous animals Dm4 neurons compete equally for territory, but as there is a reduction in the number of Dm4 neurons there is an increase in the number of columns covered. By contrast, in mosaic animals a homozygous mutant cell competes poorly with wild-type neighbors for space and, hence, one sees a reduction in the number of columns covered by the mutant cells.
Summary
Blocking apoptosis increased Dm4 cell number beyond wild-type, both in wild-type animals and in mutant backgrounds. Thus, DIP/Dpr interactions during normal development play a role in regulating cell number. As the processes of Dm4 neurons tile, in wild-type, mutant, and diap1-rescued mutants (i.e., with increase Dm4 cell number), cell number correlates with the size of the receptive fields, and thus modifies their function. The selection against single neurons lacking DIP-α and for single neurons expressing an increased level of DIP-α suggests that competitive interactions between neurons mediated by DIP/Dpr interactions regulate cell number.
DIP/Dpr Interactions Regulate Cell Survival in Other Dm Neurons
DIP-α is also expressed in Dm1 neurons, which arborize within the M1 layer, where they co-localize with processes expressing Dpr6 and Dpr10. A 25% decrease in these cells was also seen in both DIP-α mutants and animals lacking both Dpr6 and Dpr10 (Figure S11A). In a previous study, Zinn and colleagues reported a reduction in Dm8 neurons, which arborize in the M6 layer, in an insertion allele of DIP-γ. By contrast, loss of cells was not seen for insertions in Dpr11, a high-affinity ligand of DIP-γ expressed in R7, a synaptic partner of Dm8 (Carrillo et al., 2015). We generated protein null alleles of both DIP-γ and dpr11. We observed a marked decrease in the number of Dm8 neurons in both mutants, consistent with our findings on DIP-α and its Dpr ligands (Figure S11B). Here DIP-γ and dpr11 also suppress cell death (data not shown), and this occurs through a competitive mechanism (K. Menon, M. Courgeon, C. Desplan, and K. Zinn, personal communication). Thus, DIP/Dpr interactions may regulate Dm cell number in different layers through a common molecular strategy.
DIP/Dpr Binding Is Sufficient to Promote Cell Survival
We next sought to assess whether interactions between DIP-α and Dpr6/10 were necessary for cell survival. DIP-αhomo-het showed a cell loss phenotype similar in severity to DIP-αnull mutants, whereas Dm4 cell number in DIPαhomo knockins was indistinguishable from wild-type (Figure 6A). Furthermore, a knockin mutation into dpr10, which prevents binding to DIP-α (Figures S6D and S6D’), in an otherwise dpr6 mutant background, also resulted in a cell loss phenotype indistinguishable from either DIP-αnull or dpr6,10 double null mutant animals (Figure 6B). Thus, these findings support the notion that DIP-α/Dpr6,10 interactions are necessary for cell survival.
Figure 6. Ligand Binding to DIP-α Regulates Cell Survival.

(A) Dm4 cell number in DIP-α mutant alleles defective for heterophilic and homophilic binding (DIP-α het+homo) or homophilic binding only (DIP- α homo) (n: 6–10 optic lobes; ***p < 0.0001, unpaired t test).
(B) Total Dm4 cell number in dpr10 mutant allele defective for heterophilic binding. Note with the exception of wild-type, all other animals were null for dpr6 (n: 6–13 optic lobes; ***p < 0.0001, unpaired t test).
(C) Total Dm4 cell number in wild-type animals and dpr6,dpr10 double mutant animals with and without expression of Dpr6 or Dpr10 transgenes in different neuronal populations, as indicated (n: 6–18 optic lobes; ***p < 0.0001, unpaired t test).
(D) Domain structure and binding affinity of DIP- α Nectin1 and Dpr10Nectin3 chimeras. Homodimerization KDs were determined by analytical ultracentrifugation and the KD for heterophilic binding was determined by SPR.
(E) Total Dm4 cell number in DIP-αNectin1 knockin flies, and in knockin flies expressing Dpr10Nectin3 driven by the combined activity of dpr6-GAL4 and dpr10-GAL4 (n: 10–24 optic lobes; ***p < 0.0001, unpaired t test).
(F) Total Dm4 cell number in rescue experiments using UAS-DIP-α driven by various GAL4 drivers in dpr6,dpr10 double mutants. Note that although loss of DIP-α homophilic binding does not lead to a reduction in cell number (see A), ectopic expression of DIP-α can rescue cell survival in animals lacking Dpr6 and Dpr10 through homophilic binding (n = 8–15 optic lobes; ***p < 0.0001, unpaired t test).
Cell numbers for (A) and (B) were quantified as adults and (C), (E), and (F) in pupae at 48 hr APF. (A)–(C), (E), and (F) are represented as mean ± SD. See also Figures S6, S12, and S13.
To determine which cells are capable of presenting Dpr6 and Dpr10 to Dm4 neurons to promote survival of DIP-α-expressing neurons, we assessed the ability of expression of Dpr6 or Dpr10 in different neurons to rescue the loss of Dm4 neurons in dpr6/10 double mutants (Figure 6C). Dpr10D transgene expression in L3 rescued cell death in dpr6/10 double mutants, as did expression of Dpr10D in all lamina neurons. Interestingly, expression of Dpr10D in developing T4 neurons, which leads to mistargeting of Dm4 neurons to M10 (see above), also suppressed the mutant phenotype (Figure 6C). These data support the notion that it is not the nature of the cell itself expressing Dpr6 or Dpr10 that is crucial for cell survival, but rather it is the binding of Dpr6 or Dpr10 to DIP-α that is sufficient to transduce a survival signal.
To provide additional support for the importance of binding between DIPs and Dprs to regulate cell number, we generated chimeric proteins comprising interacting domains of a different receptor ligand complex not found in Drosophila, which selectively bind to each other, and tested their ability to rescue the Dm4 cell number phenotype. Özkan and co-workers employed a similar approach to explore the role of interactions between the D1 domains of Syg1 and Syg2 in C. elegans through chimeras with members of the same family from vertebrates Özkan et al., 2014). Previous biochemical studies demonstrated that heterophilic binding between Nectin1 and Nectin3 was mediated by their N-terminal Ig domains with an interface similar to that seen for DIP/Dpr interactions (Carrillo et al., 2015; Harrison et al., 2012) (Figure S12A). Like DIP-α, Nectin1 also promotes homophilic interactions. We generated chimeras between Nectins and DIP/Dprs, such that the DIP/Dpr heterophilic interaction was replaced by interacting domains of Nectin1 and Nectin3 (Figure 6D). These were then tested to assess whether Nectin 1/Nectin 3 interactions were sufficient to rescue the DIP-α mutant phenotype (Figure 6E).
The DIP chimera comprised the N-terminal Ig domain of Nectin1 in place of the N-terminal domain of DIP-α (DIP-αNectin1), and the Dpr chimera contained the N-terminal domain of Nectin3 in place of the N-terminal Ig domain of Dpr10 (Dpr10Nectin3) (Figure 6D). AUC experiments showed that the DIP-αNectin1 chimera dimerized with a KD of ~2 mM, whereas SPR experiments showed heterophilic binding between the two chimeras was of similar strength, with a KD of less than 5 mM (Figure S12B). Thus, the chimeric proteins showed heterophilic binding properties of similar affinities to that of DIP-α/Dpr10, but, by contrast, DIP-αNectin1 homophilic binding was stronger than DIP-α.
To assess whether the nectin chimeras were sufficient to promote cell survival, we assessed whether a homozygous DIP-αNectin1 knockin, with a strong loss-of-function phenotype (the chimeric protein is expressed similarly to wild-type protein; Figures S12C–S12F), could be rescued by expressing a cDNA encoding the Dpr10Nectin3 chimera under the control of two GAL4s representing the composite pattern of expression of both Dpr6 and Dpr10. Substantial rescue was observed, further supporting the view that interaction between wild-type DIP-α and Dpr6/10 is sufficient to provide trophic support (Figures 6E). By contrast to Dpr10D, however, expression of the Dpr10Nectin3 chimera in the nascent M10 layer in this genetic background was not sufficient to promote mistargeting. This may reflect lower levels of expression of either of the nectin chimeras or both of them, an important role for structural determinants outside the D1 domains for this function, or competition from the substantially stronger homophilic binding of the DIP-αNectin1 chimera (2 μM) retaining Dm4 within M3 compared to DIP-α homophilic binding (24 mM) in wild-type, which may not be sufficient to do so.
Together, these experiments indicate that DIP-α/Dpr10 binding promotes trophic interactions between neurons. Dpr6/Dpr10 binding to DIP-α may directly activate a signaling pathway suppressing apoptosis, or alternatively this binding may promote interactions between processes of neurons destined to be synaptic partners within the layer, which in turn, allow other anti-apoptotic ligands and receptors to bind to each other to promote cell survival.
Regulation of Cell Interactions through DIP-α Homophilic Binding
As shown in Cosmanescu et al., several DIP paralogs including DIP-α exhibit homophilic binding in addition to heterophilic interactions with Dprs. In the previous sections, we presented evidence that DIP-α/Dpr6,10 heterophilic binding regulates patterning of visual system circuits. These studies make a strong case for heterophilic interactions in regulating cell survival, but it remains unclear to what extent DIP-α homophilic binding contributes to patterning. That DIP-α homophilic interaction functions during normal development was indicated by the 50% increase in presynaptic sites per column in DIP-αhomo mutant Dm4 neurons in an otherwise wild-type background (Figures 4F and 4G). This increase in Brp puncta in DIP-αhomo mutant neurons may reflect an inhibitory role of homophilic binding (i.e., antagonizing a synaptogenic Dpr6/10 heterophilic interaction), a decrease in KD for heterophilic interactions also seen in these mutants (i.e KDs for Dpr6 and Dpr10 binding to wild-type DIP-α range from 1.7 to 2.4 mM, whereas their binding is considerably stronger to DIP-αhomo protein (i.e., KD = 0.4 μM) (see Cosmanescu et al., 2018), or both.
To directly test whether interactions can occur between DIP-α proteins in the absence of Dpr6 and Dpr10, we assessed whether homophilic binding could substitute for heterophilic interactions in transgene rescue and gain-of-function experiments. Expression of DIP-α in the outer medulla effectively rescued the Dm4 cell loss phenotype in a dpr6,dpr10 double mutant background (Figure 6F). By contrast, unlike misexpression of Dpr6 or Dpr 10, DIP-α misexpression under the control of the T4 driver did not lead to massive mistargeting of Dm4 or Dm12 neurons to M10, and no significant rescue of Dm4 cell number was observed (Figure 6F). Interestingly, however, some 50% of the samples analyzed showed a small number of Dm4 neurons, expressing DIP-α at endogenous levels, projecting through the medulla and into the lobula (Figure S13).
In summary, gain-of-function and transgene rescue studies support the notion that DIP-α/DIPα binding also can regulate interactions between cells. These interactions are sufficient to promote cell survival, but only weakly promote motility when compared to DIP/Dpr interactions. That loss of DIP-α homophilic binding in single Dm4 neurons increases synapse number appreciably raises the intriguing possibility that homophilic DIP interactions may regulate cellular interactions during normal development through inhibiting heterophilic DIP/Dpr binding.
DISCUSSION
The DIP/Dpr protein families exhibit complex biochemical interactions. Some DIP and Dpr proteins bind homophilically and all paralogs bind heterophilically, albeit with different affinities and degrees of specificities. Furthermore, these proteins are expressed in cell-type-specific patterns and high-affinity interactors are frequently expressed on synaptic partners. These findings, the cellular complexity of the visual system, and the specificity of synaptic connectivity led us to propose that DIP/ Dpr proteins contribute to the establishment of layer-specific neural circuitry. As a step toward critically addressing this possibility, we report here that DIP-α and its high-affinity binding partners Dpr6 and Dpr10 regulate interactions between processes within the M3 layer.
As the phenotypes in DIP-a homozygous mutants and DIP-α homozygous mutant neurons in a wild-type background are different from each other, for clarity we summarize them here before discussing the results in more detail. In homozygous animals lacking either DIP-α or both Dpr6 and Dpr10, there is a reduction in the number of Dm4 and Dm12 neurons. Layer-specific targeting of these cell types is unaffected. There is no obvious change in the morphology Dm12 neurons in DIP-α or dpr6/10 homozygous animals. There is, however, an increase in the number of columns covered by each Dm4 neuron within the M3 layer. As both wild-type and mutant Dm4 neurons tile, the increase in the number of columns covered may reflect the decrease in cell number and argues that column coverage is governed by homotypic interactions (i.e., Dm4/Dm4 interactions independent of either DIP-α or Dpr6/10).
We explored the role of DIP-α and Dpr6,10 in controlling cell number in depth in the context of Dm4. We demonstrated that DIP-α and Dpr6,10 heterophilic interactions promote cell survival by antagonizing a hid-activated cell death pathway. Developmental studies, antibody staining, and knockin mutant and chimeric rescue experiments support the notion that the interactions between this DIP/Dpr pair occur between axonal processes as they first encounter one another within the incipient M3 layer. The simplest interpretation of these data is that Dm4 neurons are generated in excess during normal development and interactions between them and L3 afferents (and perhaps other Dpr6- and Dpr10-expressing processes in M3) act as a source of limited trophic support, thereby determining the number of Dm neurons surviving into the adult. As Dm4 neurons tile, the number of Dm4 neurons indirectly sets the number of columns covered. This is consistent with the decrease in the extent of Dm4 arborization in animals with more Dm4 neurons as a consequence of Diap1 expression.
That interactions between DIP-α and Dpr6/10 regulate other aspects of Dm4 and Dm12 development was seen in genetically mosaic animals, in which DIP-α was selectively removed from single Dm4 or Dm12 neurons in an otherwise wild-type background. Different phenotypes in DIP-α mutant Dm4 and Dm12 neurons were observed: (1) There was a 30% decrease in the number of columns covered by mutant Dm4 neurons in mosaic animals. This is different from the number of columns covered by mutant neurons in a mutant background; presumably extension of processes within the layer is promoted by these DIP/Dpr interactions, such that mutant neurons compete less effectively for territory within the layer with their wild-type counterparts. There was no defect in layer-specific targeting. (2) By contrast to Dm4, in mosaic animals single DIP-α null mutant Dm12 neurons exhibited a robust mistargeting to another layer. Although all mutant Dm12 neurons targeted to M3, 60% of these sent additional processes to M8, where they arborized within this layer. A modest (~10%) reduction in column coverage in M3 was observed. (3) Removal of DIP-α from Dm12 led to a 30% reduction in the density of presynaptic sites. By contrast, the removal of DIP-α did not lead to a change in the density of presynaptic sites in Dm4. (4) DIP-αhet-homo mutant neurons, in which DIP-α heterophilic and homophilic interactions were disrupted, led to phenotypes in Dm4 and Dm12 indistinguishable from those seen in DIP-α null mutant neurons. (5) DIP-αhomo mutant neurons in a wild-type background led to an increase in the number of presynaptic sites in Dm4, but not in Dm12. Together, these data support a role for interactions between DIP-α on the surface of Dm4 and Dm12 neurons on mediating interactions with the processes of other neurons, notably L3, and perhaps other neurons within the developing M3 layer that are important for establishing neural circuitry.
Gain-of-function studies provide additional strong support for this conclusion. Misexpression of Dpr10 (or Dpr6) in a different layer from the expression in wild-type animals led to a nearly complete re-specification of targeting to this layer of both Dm4 and Dm12 axonal processes. This finding and the dependence of mistargeting upon DIP-α provide compelling evidence that binding observed in vitro occurs in vivo and contributes to the establishment of layer-specific circuitry. These observations are also consistent with the overlapping expression patterns of Dpr6/10 and DIP-α proteins in the developing neuropil. Thus, gain- and loss-of-function mutations leading to defects in arborization, layer-specific targeting, synapse number, and cell survival provide compelling evidence that interactions between DIP-α and Dpr6/10 on the surface of neurons in the developing M3 layer are necessary for normal circuit development. The mechanisms by which these interactions regulate these specific developmental processes, however, remain poorly understood.
It remains plausible that these different phenotypes result from a range of effects on cell viability, from death to compromised cell function. But as P35 expression in single mutant Dm12 neurons does not rescue the targeting defects (Figure S10B), we favor the view that these wiring phenotypes are independent of cell survival. Other ligand/receptor pairs regulating both wiring and cell survival have been described, including the classical neurotrophins (Lykissas et al., 2007), and clustered protocadherins (Ing-Esteves et al., 2018; Kostadinov and Sanes, 2015; Lefebvre et al., 2012; Wang et al., 2002). Whether the interactions between DIP/Dpr proteins directly control survival, targeting, or synapse number, or whether they facilitate interactions between other cell surface proteins that, in turn, directly regulate specific effector functions, is not known.
Striking differences between the Dm12 mutant phenotypes in different genetic contexts were observed. Mistargeting of Dm12 was seen in sparsely distributed DIP-α mutant neurons in a wild-type background, whereas the targeting of mutant Dm12 neurons in a whole-animal DIP-αnull mutant was unaffected. Whether this difference reflects the activation of compensatory mechanisms in homozygous animals or whether the juxtaposition of single null neurons with wild-type neighbors artificially uncovers redundancy by creating neighboring neurons with different “competitive” fitness is not known. In addition to the aforementioned targeting differences, mutant Dm12 neurons nestled within an otherwise wild-type background showed a reduced density of presynaptic sites compared to wild-type or mutant neurons in an all mutant background. Indeed, there was no difference in the density of synapses seen in wild-type Dm12 neurons compared to mutant Dm12 neurons in an all mutant background (data not shown). As each L3 neuron receives input from three different Dm12 neurons (S. Takemura and L. Scheffer, personal communication), these data are consistent with DIP-α mutant Dm12 neurons in mosaics being at a competitive disadvantage relative to wild-type Dm12 neurons synapsing on the same L3. That is, we would anticipate a compensatory increase in the number of synapses in the two remaining wild-type Dm12 partners.
The discrepancy in phenotypes between mutant neurons in an all mutant background and mutant neurons with wild-type neighbors is similar to recent observations on the effects of neurexin knockouts in climbing fiber synapses on Purkinje neuron dendrites (Chen et al., 2017). Here, severe synaptic phenotypes were observed in sparsely labeled triple mutant neurons in a largely wild-type background, compared to only very weak phenotypes observed in sparsely labeled triple mutant neurons with many triple mutant neighbors. Similar observations were made on the dendritic targeting behavior of Dscam4 mutant lamina L4 neurons. Phenotypes were seen in homozygous Dscam4 mutant neurons with wild-type neighbors, but not in homozygous neurons in a homozygous mutant background (Tadros et al., 2016; W. Tadros and S.L.Z., unpublished data). These studies suggest that genetic mosaic analyses may establish artificial competitive interactions between neurons, which, in turn, uncovers gene function.
Correlating the expression patterns and binding specificities of different DIPs and Dprs revealed that many cognate DIP/Dpr pairs are expressed on synaptic partners throughout the visual system (Figure S14). That matched expression patterns reflect function is supported by the finding that two DIPs (DIP-α and DIP-γ) and their high-affinity ligands (Dpr6/10 and Dpr11, respectively) regulate layer-specific circuit assembly. The role of the DIP and Dpr families in regulating specificity more broadly is likely to be complex as binding affinities between different DIPs and Dprs vary over two orders of magnitude and some Dprs and DIPs also exhibit homophilic binding.
The increase in synapses in Dm4 neurons seen in DIP-αhomo mutants raises the interesting possibility that homophilic interactions may inhibit and thereby modify heterophilic interactions.
In principle, homophilic interactions regulating synapse number could occur between DIP-α proteins acting in cis (i.e., in Dm4), or alternatively in trans with other cells with arbors within the layer (e.g., in Dm12) to antagonize a synaptogenic signal generated by heterophilic interactions between synaptic partners. In other contexts, however, such as cell survival, homophilic interactions in trans may also promote similar responses to heterophilic binding, as we observed targeted expression of DIP-α substituting for a cell survival phenotype seen in dpr6/ dpr10 mutants. The inconsistencies of these results, the anti-synaptogenic signal and pro-cell survival signals, could also reflect differences in expression levels. In the former case, the homophilic binding-deficient form was expressed from the endogenous locus, acted cell autonomously at normal levels, and with the same spatiotemporal pattern as wild-type. By contrast, overexpression of DIP-α under the control of the GAL4/UAS system was sufficient to promote survival in a cell-non-autonomous fashion. Together these data raise the exciting possibility that circuit organization, in part, reflects different types of interactions between various DIPs and Dprs on neuronal processes leading to a variety of functional outputs.
Although there are many Dprs and DIPs, they represent only a small number of the vast array of cell surface proteins expressed in neurons in the developing visual system. There are over 100 neuronal cell types contributing processes, axons, and dendrites to the medulla neuropil, and each neuron type makes a characteristic pattern of connections (Takemura et al., 2013, 2015). Different types of neurons express many cell surface proteins in common (e.g., hundreds) and they also express others in a cell-type-enriched fashion (Tan et al., 2015). Many of these proteins exhibit homophilic or heterophilic binding or both, and thus may interact with proteins expressed on the surface of other neurons in the developing neuropil. We propose that DIPs and Dprs act with other specificity molecules in a combinatorial and partially redundant fashion to allow axons and dendrites to discriminate between the diverse neuronal cell surfaces they encounter during visual circuit assembly. As DIPs and Dprs are expressed in a cell-type-specific fashion throughout the developing CNS (Carrillo et al., 2015; Özkan et al., 2013), it seems likely that these proteins will act in different combinations to contribute to wiring specificity beyond the developing visual system.
Hassan and Hiesinger have recently proposed that wiring can be understood through simple cellular rules rather than through molecular dissection of the pathways regulating these processes (Hassan and Hiesinger, 2015). While we too share the wish that circuit assembly relies upon simple cellular rules, we believe it is only through molecular and genetic studies that rules, simple or not, will be established. One possibility is that the vast diversity of neuronal morphologies and patterns of connectivity will rely, in part, on the duplication and divergence of binding specificities of different classes of cell recognition molecules (e.g., whether homophilic or heterophilic) and the precise patterns of expression of these proteins in discrete neuronal sub-classes. These proteins must act in various combinations with other broadly expressed proteins, such as N-cadherin, different levels of proteins (e.g., Ephs and Ephrins) expressed in a graded fashion, and a core set of evolutionarily conserved guidance molecules (e.g., netrins, Slits, and semaphorins) to regulate the interactions between developing neurons as they assemble into circuits. Dramatic advances in technology—from CRISPR-based mutagenesis, to single-cell sequencing, microscopy, and optogenetics—provide unprecedented opportunities to un-cover the molecular solutions that have evolved to create neural circuits, and the developmental principles upon which circuit assembly rests.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Larry Zipursky (lzipursky@mednet.ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Flies were reared at 25°C on standard medium. For developmental analysis and sorting experiments white pre-pupae were collected and incubated for the indicated number of hours. Fly lines used in this study are listed in the Key Resources Table. Geotypes and sex of flies used for each experiment are provided in Table S1. FreeStyle 293F cells were cultured in suspension in Freestyle 293 Expression medium at 37°C and 10% CO2. For monoclonal antibody production, mice were handled following protocols approved by the Chancellor’s Animal Research Committee (ARC) at UCLA.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| chicken-anti-GFP | Abcam | Cat#ab13970, RRID: AB_300798 |
| rabbit-anti-DsRed | Clontech | Cat#632496, RRID: AB_10013483 |
| mouse-anti-24B10 | DSHB | Cat#24B10, RRID: AB_528161 |
| mouse-anti-Brp | DSHB | Cat#Nc82, RRID: AB_2314866 |
| rabbit-anti-HA | Cell Signaling Technologies | Cat#3724S, RRID: AB_1549585 |
| rat-anti-FLAG | Novus Biologicals | Cat#NBP1–06712, RRID: AB_1625981 |
| chicken-anti-V5 | Abcam | Cat#9113, RRID: AB_307022 |
| Nectin1 monoclonal antibody CK8 | Thermo Fisher | Cat#37–5900, RRID: AB_2533329 |
| mouse-anti-DyLight549-conjugated V5 | AbD Serotec | Cat#MCA1360D549, RRID: AB_915420 |
| mouse-anti-DIP-α | This paper | N/A |
| mouse-anti-Dpr6 | This paper | N/A |
| mouse-anti-Dpr10 | This paper | N/A |
| Alexa Fluor 488 donkey-anti-chicken | Jackson Immuno Research Lab | Cat#703–545-155, RRID: AB_2340375 |
| Alexa Fluor 488 donkey-anti-mouse | Jackson Immuno Research Lab | Cat#715–545-151, RRID: AB_2341099 |
| Alexa Fluor 594 donkey-anti-rabbit | Jackson Immuno Research Lab | Cat#711–585-152, RRID: AB_2340621 |
| Alexa Fluor 647 donkey-anti-rat | Jackson Immuno Research Lab | Cat#712–605-153, RRID: AB_2340694 |
| Alexa Fluor 647 donkey-anti-mouse | Jackson Immuno Research Lab | Cat#715–605-151, RRID: AB_2340863 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Everbrite Mounting Reagent | Biotium | Cat#23001 |
| Para-formaldehyde | Electron Microscope Science | Cat#15710 |
| LE Agarose | Genemate | Cat#E-3210–500 |
| Ethidium Bromide | amresco | Cat#E406–15ml |
| Phalloidin | Thermo-fisher | Cat#A22287 |
| DIP-α, Dpr6, Dpr10 proteins | Cosmanescu et al., 2018 | N/A |
| Tris Base | Fisher Scientific | Cat# BP152–5 |
| Sodium Chloride | Fisher Scientific | Cat# S271–10 |
| Calcium Chloride Dihydrate | JT Baker | Cat# 1336–01 |
| Imidazole | ACROS | Cat# 301870025 |
| Polyethylenimine | Polysciences | Cat# 24765–2 |
| N-Hydroxysuccinimide | Thermo Fisher Scientific | Cat# 24500 |
| 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide | Thermo Fisher Scientific | Cat# 22980 |
| Sodium Acetate | Sigma | Cat# S7545 |
| Ethanolamine | Sigma | Cat# 398136 |
| Tween-20 | Sigma | Cat# P7949 |
| BSA | Sigma | Cat# A7906 |
| BIS-Tris | Sigma | Cat# B9754 |
| HEPES | Sigma | Cat# H3375 |
| EDTA | ACROS | Cat# 409930010 |
| Ammonium Citrate Tribasic | Sigma | Cat# A1332 |
| EX-CELL 420 Serum-Free Medium | Sigma | Cat# 24420C |
| Freestyle 293 Expression Media | Thermo Fisher Scientific | Cat# 12338–018 |
| Opti-MEM Reduced Serum Media | Thermo Fisher Scientific | Cat# 31985–070 |
| IMAC Sepharose 6 Fast Flow | GE Healthcare | Cat# 17092109 |
| Series S CM4 chip | GE Healthcare | Cat# BR100539 |
| Critical Commercial Assays | ||
| 2x Gibson Assembly Master Mix | New England Biolabs | Cat#E2611L |
| Dreamtaq Green PCR Master Mix | Thermo Scientific | Cat#K1081 |
| cDNA Synthesis with SuperScript III RT | Thermo Fisher Scientific | Cat#18080093 |
| NotI restriction enzyme | New England Biolabs | Cat#R0189S |
| XbaI restriction enzyme | New England Biolabs | Cat#R0145S |
| Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| Hispeed Plasmid Maxi Kit | QIAGEN | Cat# 12663 |
| Experimental Models: Cell Lines | ||
| Human: FreeStyle 293-F cells | Thermo Fisher Scientific | Cat# R79007 |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: 24F10-GAL4 | Bloomington Drosophila Stock Center | BDSC 49090, RRID: BDSC_49090 |
| D. melanogaster: 75F06-GAL4 | Bloomington Drosophila Stock Center | BDSC 39901, RRID: BDSC_39901 |
| D. melanogaster: 23G11-LexA | Bloomington Drosophila Stock Center | BDSC 54775, RRID: BDSC_54775 |
| D. melanogaster: 24F10-LexA | Bloomington Drosophila Stock Center | BDSC 52696, RRID: BDSC_52696 |
| D. melanogaster: 75F06-LexA | Bloomington Drosophila Stock Center | BDSC 54100, RRID: BDSC_54100 |
| D. melanogaster: 47G08-GAL4 | Bloomington Drosophila Stock Center | BDSC 50328, RRID: BDSC_50328 |
| D. melanogaster: 87B02-GAL4 | Bloomington Drosophila Stock Center | BDSC 41316, RRID: BDSC_41316 |
| D. melanogaster: 9B08-GAL4 | Bloomington Drosophila Stock Center | BDSC 41369, RRID: BDSC_41369 |
| D. melanogaster: 42F06-GAL4 | Bloomington Drosophila Stock Center | BDSC 41253, RRID: BDSC_41253 |
| D. melanogaster: 9D03-GAL4 | Bloomington Drosophila Stock Center | BDSC 47364, RRID: BDSC_47364 |
| D. melanogaster: 9–9-GAL4 | (Nern et al., 2008) | FlyBase FBti0141173 |
| D. melanogaster: Rh4-GAL4 | Bloomington Drosophila Stock Center | BDSC 8627, RRID: BDSC_8627 |
| D. melanogaster: 13xLexAop-CD4-tdTom (attp2) | This lab | N/A |
| D. melanogaster: 10xUAS-myr::GFP (attP2; attP40) | This lab | N/A |
| D. melanogaster: LexAop-myr::GFP | This lab | N/A |
| D. melanogaster: LexAopmyrtdTomato | This lab | N/A |
| D. melanogaster: UAS-FSF-myrGFP | This lab | N/A |
| D. melanogaster: hs-Flp:PEST | Bloomington Drosophila Stock Center | BDSC 77141, RRID: BDSC_77141 |
| D. melanogaster: FRT 9–2 | Bloomington Drosophila Stock Center | BDSC 5763, RRID: BDSC_5763 |
| D. melanogaster: Gal80 on X chromosome | Gift from Gerald Rubin | This lab |
| D. melanogaster: 27G05Flp on X chromosome | Gift from Gerald Rubin | This lab |
| D. melanogaster: MCFO-1 (pBPhsFlp2::PEST;+; HA_V5_FLAG) | Bloomington Drosophila Stock Center | BDSC 64085, RRID: BDSC_64085 |
| D. melanogaster: STaR constructs | (Chen et al., 2014) | This lab |
| D. melanogaster: hid-GFP | Bloomington Drosophila Stock Center | BDSC 50751, RRID: BDSC_50751 |
| D. melanogaster: grimMiMIC | Bloomington Drosophila Stock Center | BDSC 36978, RRID: BDSC_36978 |
| D. melanogaster: rpr-lacZ | Bloomington Drosophila Stock Center | BDSC 58793, RRID: BDSC_58793 |
| D. melanogaster: H99 | Bloomington Drosophila Stock Center | BDSC 1576, RRID: BDSC_1576 |
| D. melanogaster: hidML66 | gift from Hermann Steller | Flybase FBal0045286 |
| D. melanogaster: rpr[87] | gift from Kristin White | Flybase FBal0260732 |
| D. melanogaster: Df(3L)416 | Bloomington Drosophila Stock Center | BDSC 24920, RRID: BDSC_24920 |
| D. melanogaster: Df(3L)grimA6C | Bloomington Drosophila Stock Center | BDSC 32061, RRID: BDSC_32061 |
| D. melanogaster: Df(3L)6113 | Bloomington Drosophila Stock Center | BDSC 7612, RRID: BDSC_7612 |
| D. melanogaster: UAS-P35 | (Pecot et al., 2014) | This lab |
| D. melanogaster: UAS-diap1.myc | (Pecot et al., 2014) | Flybase FBtp0019712 |
| D. melanogaster: UAS-FLP,y,w | Bloomington Drosophila Stock Center | BDSC 8208, RRID: BDSC_8208 |
| D. melanogaster: UAS-Apoliner | Bloomington Drosophila Stock Center | BDSC 32122, RRID: BDSC_32122 |
| D. melanogaster: DIP-αGAL4 (MI02031) | This paper | N/A |
| D. melanogaster: DIP-γGAL4(MI03222) | This paper | N/A |
| D. melanogaster: Dpr6GAL4 (MI01358) | This paper | N/A |
| D. melanogaster: Dpr10GAL4 (MI03557) | This paper | N/A |
| D. melanogaster: DIP-αnull1 | This paper | N/A |
| D. melanogaster: DIP-αnull2 | This paper | N/A |
| D. melanogaster: DIP-αhet-homo | This paper | N/A |
| D. melanogaster: DIP-αhomo | This paper | N/A |
| D. melanogaster: dpr6null | This paper | N/A |
| D. melanogaster: dpr10null | This paper | N/A |
| D. melanogaster: dpr6–10L | This paper | N/A |
| D. melanogaster: dpr6–10S | This paper | N/A |
| D. melanogaster: dpr10het | This paper | N/A |
| D. melanogaster: DIP-αNectin1 Y172K L116E | This paper | N/A |
| D. melanogaster: DIP-γnull | This paper | N/A |
| D. melanogaster: dpr11null | This paper | N/A |
| D. melanogaster: UAS-Dpr10D.NV5 | This paper | N/A |
| D. melanogaster: UAS-Dpr6F.NV5 | This paper | N/A |
| D. melanogaster: UAS-DIP-α−2A-tdTomato | This paper | N/A |
| D. melanogaster: UAS-Dpr10DNectin3-T2A-EGFP | This paper | N/A |
| D. melanogaster: UAS-Dpr11-T2A-EGFP | This paper | N/A |
| Oligonucleotides | ||
| Please refer to Table S2. | This paper | N/A |
| Recombinant DNA | ||
| VRC-8400 | Vaccine Research Center (NIH), Gary Nabel | N/A |
| Software and Algorithms | ||
| Imaris 8.2.0 | Bitplane | http://www.bitplane.com/imaris |
| Fiji | Schindelin et al., 2012 | http://fiji.sc/ |
| Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Scrubber 2.0 | BioLogic Software | http://www.biologic.com.au |
| SednTerp | Dr. Thomas Laue | http://bitcwiki.sr.unh.edu/index.php/Main_Page |
| HeteroAnalysis | (Cole and Lary) | https://core.uconn.edu/auf |
| Other | ||
| Genotypes of all animals used in this study are provided in Table S1. | This paper | N/A |
METHOD DETAILS
Generation of null alleles using CRISPR
We generally chose two protospacer sequences that are 50–400 bp away from each other, in the upstream exons, to create a short deletion leading to a nonsense mutation upstream of the protein sequence. High score protospacer sequence was chosen on http://crispr.dfci.harvard.edu/SSC/. We cloned each protospacer into pU6–2-sgRNA-short (Addgene 41700) plasmid and coinjected two plasmids into vas-Cas9 line (BDSC 51323 or 51324, depending on which chromosome the gene is at) in Bestgene. Injected larvae were crossed with balancer lines, and screened in F1for single flies carrying the mutation. A mutant stock was established from this single F1. sgRNA sequences are listed in Table S2. Detailed protocols are available upon request.
DIP-αnull1 deleted sequence: CGCCGGACTTCATCAGCGAGGACACCTCATCCGATGTGATTGTGCCGGAG
DIP-αnull2 deleted sequence: CGTCGGACGCGATGCAACCTTCACGTGTCACGTCCGACACTTGGGCGGCTATCGGGTAAGT CATCAGTTAACGCCAGTGCACTGGAAATCCAGCATGCATCGAACTAAGTATTTTCCAAGTGTTTGTCATGTCGAATTTAACATAC TTTCCAATAAGGTGGGCTGGCTCAAGGCCGACACCAAGGCCATTCAAGCCATCCACGAGAACGTAATCACGCACAATCCTCGC
dpr6null deleted sequence: CCCATAGAGGGCTACAATTCGCTGGATGACCTGCTGACAACCACGCCCACACCCGGCCAGGCG GCCCTCCTCCTGCCCACCGCGCCCACCGCCGCCTACACGCATCCCAAGTGGAT
dpr10null deleted sequence: GTGCTACCAGCGGCAATCTGTAAGCAACAACAATCACAATAACGCCGAGGCGAAGCCCACCCAT GCGCCACCCTCGC
dpr6–10L deleted sequence: large deletion was made spanning dpr6 and 10 loci.
The sequence deleted in this mutant.is 120kb, between dpr6 exon 2 to dpr10 exon 2.
dpr6–10S deleted sequence: short deletions were made in both dpr6 and dpr10 loci.
Sequence deleted in dpr6 region (same as dpr6null): CCCATAGAGGGCTACAATTCGCTGGATGACCTGCTGACAACCACGCCCA CACCCGGCCAGGCGGCCCTCCTCCTGCCCACCGCGCCCACCGCCGCCTACACGCATCCCAAGTGGAT
Sequence deleted in dpr10 region: GTGTGCTACCAGCGGCAATCTGTAAGCAACAACAATCACAATAACGCCGAGGCGAAGCC CACCCATGCGCCACC
DIP-γnull deleted sequence: GGCAGGACGCGAGGCCATCCTGGCCTGCTCGGTGCGCAATCTCGGCAAGAATAAGGTGAGCTA GAATGATTTACCTTGCATTGCAATATATATAATATGATATATAATCCCCTGATAATAGGTTGGTTGGCTGAGAGCCTCCGATCAGA CCGTTTTAGCTCTCCAAGGTCGCGTTGTCACCCATAATGCGAGA
Insertion right upstream of the deleted sequence: ATGCCGGCACAT
dpr11null deleted sequence: CGACATCGGGACTCGTGGAGCCCTATCTGGATGGCTACGCCACTTCCAATGTGACCACTCAG
Insertion right upstream of the deleted sequence: ATATCT
Generation of heterophilic and homophilic interaction-defective flies
DIP-αhet-homo and DIP-αhomo: The genomic sequence of DIP-a exon2-exon4 was first replaced with sequence of attP-3XP3-DsRed-attP using a CRISPR-based knock-in strategy (DIP-α NM flies). DIP-α exon2-exon4 carrying I83D or A78K+N94D point mutants, which disrupt hetero-homophilic interaction or homophilic interaction only, were cloned into the pBluscript KS(+) vector. Product constructs were injected into DIP-α NM flies generated above and mutations were introduced into the genome through ΦC31 recombinase-mediated cassette exchange (Bestgene).
Dpr10het: Dpr10 exon3 carrying Y103D mutant, which disrupts heterophilic interaction, was cloned into pHD-DsRed-attP vector (with the attp site removed), and this plasmid was injected into Vas-Cas9(X);+/+;dpr6null flies at Bestgene to generate the mutants using a CRISPR-based knock-in strategy.
sgRNA sequences are listed in Table S2. Detailed protocols are available upon request.
Generation of UAS-transgenic flies
cDNA encoding Dpr6F, Dpr10D, Dpr11 and DIP-α were cloned into the pJFRC28 vector (Pfeiffer et al., 2012) using standard molecular biology methods. V5 sequence was inserted after signal peptide and before Ig1 for DIP-α and Dpr6F, Dpr10D. Transgenes were inserted into specific landing sites by injection of fertilized embryos (Bestgene). UAS-Dpr6, UAS-Dpr10 used attP-3B site, UAS-Dpr-11 used the attP40 site, and UAS-DIP-α used attP9A site. Plasmid and primer design were carried out using the software Snapgene. Plasmids and detailed sequences are available upon request.
Nectin-Chimera protein cloning, expression and purification
Proteins were produced in human embryonic kidney cells (HEK293F) with complementary DNA sequences encoding the extracellular regions of human nectin-1 (Gln31-Met143) followed by DIP-α isoform A (Ile142-Pro341) and human nectin-3 (Gly58-Leu167) followed by Dpr10 isform D (Val155-Glu255) amplified and inserted into the mammalian expression vector VRC-8400 (Barouch et al., 2005) between the NotI and BamHI sites. Nectin-1 L116E and DIP-α Y172K mutations were introduced to enhance DIP-αNectin1 expression. Point mutations were introduced using the QuickChange method (Agilent). All sequences were preceded by the signal sequence of human binding immunoglobulin protein BiP (MKLSLVAAMLLLLSAARA), and the Kozak sequence (GCCACC). Protein sequences were followed by a C-terminal hexa-histidine tag. Dpr6, Dpr10, and DIP-α proteins were produced as in Cosmanescu et al. (2018).
HEK293 cells were transiently transfected with expression constructs by the Polyetheleneimmine method (Baldi et al., 2012). Conditioned media was equilibrated to 500mM NaCl, 10mM Tris-HCl pH 8.0, 3mM CaCl2, 5mM Imidazole pH 8.0 and incubated with Ni2+ charged IMAC Sepharose 6 Fast Flow resin (GE Healthcare) for 1 hour at 25°C. Resin was washed with at least 20 column volumes of 10mM Imidazole pH 8.0 before proteins were eluted with 90mM Imidazole pH 8.0. Gel electrophoresis with NuPage 4%–12% Bis-TRIS gels (Life Technologies) was used to detect which elutions contained desired protein.
Further purification by size-exclusion chromatography was performed using a Superdex 200 column (GE Healthcare) on an AKTA pure fast protein liquid chromatography system (GE Healthcare). DIP-αNectin1 was stored in 150mM NaCl, 10mM Bis-TRIS pH 6.0 and Dpr10Nectin3 was stored in 150mM NaCl, 10mM Bis-TRIS pH 6.6. UV absorbance at 280nm was used to determine protein concentration and verification of purity was determined by gel electrophoresis. Accurate molecular weights were determined through MALDI-TOF mass spectrometry at the Proteomics Shared Resource facility at Columbia University.
Generation of DIP-αNectin1 knock-in fly via CRISPR
In designing the DIP-αNectin1 chimeric protein, to allow for a more favorable interaction between chimeric proteins and protein expression, we introduced two point mutations in the DIP-αNectin1 chimera, L116E (in Nectin1 Ig1) and Y172K (in DIP-a Ig2). To generate the knock-in flies, we used homologous recombination after CRISPR-Cas9 mediated DNA cleavage. We chose two protospacer sequences in DIP-α intronic region in the genome and generated two protospacer-gRNA plasmids. We also generated a donor plasmid by cloning the DIP-a genomic region spanning Ig1 and Ig2, with Ig1 replaced by Nectin1 Ig1, and two point mutations, L116E and Y172K, into pHD-DsRed-attP vector. We co-injected three plasmids into vas-Cas9 line (BDSC 51324) in Bestgene, and screened the fly carrying the knock-in allele by two rounds of screening. First by the Dsred marker in the larval body and adult eye, then by genomic screen verifying our knock-in sequence. The knock-in stock was then established.
Generation of antibody
DIP-α and Dpr6 monoclonal antibodies: Phe40-Pro341 of DIP-α-PA and Asp24-Glu275 of Dpr6-PC were purified and injected into mice (Caltech Monoclonal Antibody Facility). Clones were screened for immune-reactivity to the purified DIP-α or Dpr6 protein fragments by ELISA and verified by western blotting to the purified protein fragments and immunohistochemistry of 40hrs Drosophila pupal optic lobes. One positive clone 4G11 was identified and saved for DIP-α. Two positive clones 1F10 and 4G6 were identified and saved for Dpr6.
Dpr10 mouse polyclonal antibody: Tyr19-Glu255 of Dpr10-PD protein was purified and injected into three mice. Six bleeds from each mouse were collected and tested by immunohistochemistry of 40hrs Drosophila pupal optic lobes. One bleed from one animal was tested to be specific to Dpr10 and saved.
Immunohistochemistry
Fly brains were dissected in Schneider’s Drosophila Media and fixed in PBL (4% paraformaldehide, 75mM lysine, and 37mM sodium phosphate buffer, pH 7.4) for 25 min at room temperature (RT). After several rinses with PBS (137mM NaCl, 2.7mM KCl, 10mM Na2HPO4, 1.8mM KH2PO4) at RT, samples were incubated in PBT (PBS 0.5% Triton-X10) containing 10% normal goat serum (blocking solution) for at least 1hr at RT. Brains were incubated overnight at 4°C in primary and secondary antibodies for at least one day each with multiple PBT rinses at RT in between and afterward. Brains were mounted in EverBrite mounting medium (Biotium).
The following primary antibodies were used in this study: chicken-anti-GFP (1:1000, Abcam ab13970); rabbit-anti-DsRed (1:200, Clontech 632496); mouse-anti-24B10 (Zipursky et al., 1984) (1:20, DSHB); mouse-anti-Brp (nc82) (1:20, DSHB); rabbit-anti-HA (1:300 Cell Signaling Technologies 3724S); rat-anti-FLAG (1:200, Novus Biologicals, NBP1–06712); chicken-anti-V5 (1:500, Abcam 9113); Nectin1 monoclonal antibody (CK8) (1:200, Thermo-fisher 37–5900); mouse-anti-DyLight549-conjugated V5 (1:300, AbD Serotec MCA1360D549); mouse-anti-DIP-α (4G11), mouse-anti-Dpr6 (1F10 and 4G6), and mouse-anti-Dpr10(polyclonal) were developed in the lab (see ‘Generation of monoclonal antibodies’) and used at 1:10, undiluted and 1:1000 respectively.
Secondary antibodies were used as 1:500 dilution. From Jackson Immuno Research Lab: Alexa Fluor 488 donkey-anti-chicken (703–545-155); Alexa Fluor 488 donkey-anti-mouse (715–545-151); Alexa Fluor 594 donkey-anti-rabbit (711–585-152); Alexa Fluor 647 donkey-anti-rat (712–605-153); From Life Technologies: Alexa Fluor 647 donkey-anti-mouse (A-21236). Phalloidin (1:100, Thermo-fisher A22287)
MCFO Immunohistochemistry
We crossed the MCFO-1 line with each GAL4 line. Flies with GAL4 and MCFO transgenes were raised at 25°C and receive heat-shock at 37°C for 10–20 min at mid-pupal stage, then they were dissected within two days after eclosion and the brains were stained following MCFO immunohistochemistry protocol as described before (Nern et al., 2015).
Microscopy and Image Analysis
Confocal images were acquired on a Zeiss LSM780 confocal microscope. The staining patterns were reproducible between samples. However, some variation on the overall fluorescence signal and noise levels existed between sections and samples. Thus, proper adjustments of laser power, detector gain, and black level settings were made to obtain similar overall fluorescence signals. Single plane or maximum intensity projection confocal images were exported into TIFF files using ImageJ software (Schindelin et al., 2012).
Numbers of cell soma and presynaptic sites are counted semi-automatically or manually using Imaris (Bitplane) spheres function. 3D images were taken by snapshot function in Imaris 3D mode.
Sedimentation equilibrium by analytical ultracentrifugation
Experiments were performed in a Beckman XL-A/I analytical ultracentrifuge (Beckman-Coulter, Palo Alto CA, USA), utilizing six-cell centerpieces with straight walls, 12 mm path length and sapphire windows. Protein samples were dialyzed over-night in 150mM NaCl, 10mM Bis-TRIS pH 6.6. The samples were diluted to an absorbance at 10 mm and 280 nm of 0.65, 0.43 and 0.23 in channels A, B and C, respectively. Dilution buffer were used as blank. The samples were run at four speeds: 12,000, 16,000, 20,000 and 24,000 rpm. The lowest speed was held for 20hr then four scans were taken with 1hr interval, the second lowest held for 10hr then four scans with 1hr interval, and the third lowest and highest speed measured as the second lowest. Measurements were done at 25°C, and detection was by UV at 280 nm. Solvent density and protein v-bar were determined using the program SednTerp. (Alliance Protein Laboratories). For calculation of dimeric KD and apparent molecular weight, all useful data were used in a global fit, using the program HeteroAnalysis, obtained from University of Connecticut. (https://core.uconn.edu/auf).
Surface Plasmon Resonance (SPR) binding experiments
SPR binding assays were performed using a Biacore T100 biosensor equipped with a Series S CM4 sensor chip. DIP-α, DIP-αNectin1 and Dpr10Nectin3 were immobilized over independent flow cells using amine-coupling chemistry in HBS-P (10mM HEPES, 150mM NaCl, 0.005% (v/v) tween-20) pH 7.4 buffer as described in Cosmanescu et al. (2018), to yield immobilization levels of 625–700 RU. A BSA-immobilized surface was used as a reference surface. All binding experiments were performed at 25°C in a running buffer of 10mM Tris-HCl, pH 7.2, 150mM NaCl, 1mM EDTA, 1 mg/mL BSA and 0.01% (v/v) Tween-20. Dpr10 protein was tested for binding at 27 to 0.004 μM and Dpr6, DIP-α, DIP-αNectin1 and Dpr10Nectin3 were tested at 9 to 0.004 μM using a similar protocol as described in Cosmanescu et al. (2018). The responses between 35 and 39 s were plotted against the Dpr concentration and fit to an 1:1 interaction model to calculate the KD. The data was processed and analyzed using Scrubber 2.0 (BioLogic Software).
QUANTIFICATION AND STATISTICAL ANALYSIS
Images were analyzed with FIJI (https://fiji.sc/) (Schindelin et al., 2012) and Imaris (http://www.bitplane.com/imaris). Statistial analysis was done using Prism software. All data are shown as mean ± standard deviation (SD). Statistical test: unpaired t-test.
DATA AND SOFTWARE AVAILABILITY
Please refer to Key Resources Table.
Supplementary Material
Highlights.
DIP-α binding to Dpr6/Dpr10 regulates layer-specific circuit assembly
DIP-α and Dpr6/10 function in multiple aspects of circuit development
DIP-α and Dpr6/10 proteins are expressed on neuronal processes
DIP/Dpr and DIP/DIP binding promote interactions between neuronal processes in vivo
ACKNOWLEDGMENTS
We thank Kai Zinn, Kaushiki Menon, and members of the Zipursky Lab for critical reading of the manuscript and helpful discussion during the course of these studies. Dorian Gunning, Western Kramer, and Jeanette Hernandez helped with generation of antibody to DIP-α, Dpr6, and Dpr10. We are grateful for unpublished fly lines and advice from Aljoscha Nern and Gerald Rubin. This work was supported by the Training Program in Neural Microcircuits from the National Institute of Neurological Disease and Stroke (T32NS058280) (S.X.), NIH grant R01GM067858 (H.J.B.), the G. Harold and Leila Y. Mathers Foundation (S.L.Z.), and US National Science Foundation grant MCB-1412472 (B.H.). The Microscopy Core at Baylor College of Medicine is supported by IDDRC (U54 HD083092) from National Institute of Child Health and Human Development. H.J.B., B.H., and S.L.Z. are Investigators of the Howard Hughes Medical Institute.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes fourteen figures and two tables and can be found with this article online at https://doi.org/10.1016/j.neuron.2018.11.001.
REFERENCES
- Baldi L, Hacker DL, Meerschman C, and Wurm FM (2012). Large-scale transfection of mammalian cells. Methods Mol. Biol. 801, 13–26. [DOI] [PubMed] [Google Scholar]
- Bardet PL, Kolahgar G, Mynett A, Miguel-Aliaga I, Briscoe J, Meier P, and Vincent JP (2008). A fluorescent reporter of caspase activity for live imaging. Proc. Natl. Acad. Sci. USA 105, 13901–13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barouch DH, Yang ZY, Kong WP, Korioth-Schmitz B, Sumida SM, Truitt DM, Kishko MG, Arthur JC, Miura A, Mascola JR, et al. (2005). A human T-cell leukemia virus type 1 regulatory element enhances the immunogenicity of human immunodeficiency virus type 1 DNA vaccines in mice and nonhuman primates. J. Virol. 79, 8828–8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo RA,Özkan E, Menon KP, Nagarkar-Jaiswal S, Lee PT, Jeon M, Birnbaum ME, Bellen HJ, Garcia KC, and Zinn K. (2015). Control of synaptic connectivity by a network of Drosophila IgSF cell surface proteins. Cell 163, 1770–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Akin O, Nern A, Tsui CYK, Pecot MY, and Zipursky SL (2014). Cell-type-specific labeling of synapses in vivo through synaptic tagging with recombination. Neuron 81, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Jiang M, Zhang B, Gokce O, and Südhof TC (2017). Conditional deletion of all neurexins defines diversity of essential synaptic organizer functions for neurexins. Neuron 94, 611–625.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clandinin TR, Lee CH, Herman T, Lee RC, Yang AY, Ovasapyan S, and Zipursky SL (2001). Drosophila LAR regulates R1-R6 and R7 target specificity in the visual system. Neuron 32, 237–248. [DOI] [PubMed] [Google Scholar]
- Cosmanescu F, Katsamba PS, Sergeeva AP, Ahlsen G, Patel SD, Brewer JJ, Tan L, Xu S, Xiao Q, Nagarkar-Jaiswal S, et al. (2018). Neuron subtype-specific expression, interaction affinities, and specificity determinants of DIP/Dpr cell recognition proteins. Neuron. Published online November 19, 2018. 10.1016/j.neuron.2018.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Krishnaswamy A, De la Huerta I, and Sanes JR (2014). Type II cadherins guide assembly of a direction-selective retinal circuit. Cell 158, 793–807. [DOI] [PubMed] [Google Scholar]
- Fischbach K-F, and Dittrich APM (1989). The optic lobe of Drosophila melanogaster. I: A. Golgi analysis of wild-type structure. Cell Tissue Res. 258, 441–475. [Google Scholar]
- Hakeda-Suzuki S, Takechi H, Kawamura H, and Suzuki T. (2017). Two receptor tyrosine phosphatases dictate the depth of axonal stabilizing layer in the visual system. eLife 6, 10.7554/eLife.31812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison OJ, Vendome J, Brasch J, Jin X, Hong S, Katsamba PS, Ahlsen G, Troyanovsky RB, Troyanovsky SM, Honig B, and Shapiro L. (2012). Nectin ectodomain structures reveal a canonical adhesive interface. Nat. Struct. Mol. Biol. 19, 906–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan BA, and Hiesinger PR (2015). Beyond molecular codes: simple rules to wire complex brains. Cell 163, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ing-Esteves S, Kostadinov D, Marocha J, Sing AD, Joseph KS, Laboulaye MA, Sanes JR, and Lefebvre JL (2018). Combinatorial effects of alpha- and gamma-protocadherins on neuronal survival and dendritic self-avoidance. J. Neurosci. 38, 2713–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostadinov D, and Sanes JR (2015). Protocadherin-dependent dendritic self-avoidance regulates neural connectivity and circuit function. eLife 4, 10.7554/eLife.08964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswamy A, Yamagata M, Duan X, Hong YK, and Sanes JR (2015). Sidekick 2 directs formation of a retinal circuit that detects differential motion. Nature 524, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, and Luo L. (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451–461. [DOI] [PubMed] [Google Scholar]
- Lee CH, Herman T, Clandinin TR, Lee R, and Zipursky SL (2001). N-cadherin regulates target specificity in the Drosophila visual system. Neuron 30, 437–450. [DOI] [PubMed] [Google Scholar]
- Lefebvre JL, Kostadinov D, Chen WV, Maniatis T, and Sanes JR (2012). Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature 488, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykissas MG, Batistatou AK, Charalabopoulos KA, and Beris AE (2007). The role of neurotrophins in axonal growth, guidance, and regeneration. Curr. Neurovasc. Res. 4, 143–151. [DOI] [PubMed] [Google Scholar]
- Maisak MS, Haag J, Ammer G, Serbe E, Meier M, Leonhardt A, Schilling T, Bahl A, Rubin GM, Nern A, et al. (2013). A directional tuning map of Drosophila elementary motion detectors. Nature 500, 212–216. [DOI] [PubMed] [Google Scholar]
- Matsuoka RL, Nguyen-Ba-Charvet KT, Parray A, Badea TC, Ché dotal, A., and Kolodkin, A.L. (2011). Transmembrane semaphorin signalling controls laminar stratification in the mammalian retina. Nature 470, 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel-Zaffran C, Suzuki T, Gahmon G, Treisman JE, and Dickson BJ (2001). Cell-autonomous and -nonautonomous functions of LAR in R7 photoreceptor axon targeting. Neuron 32, 225–235. [DOI] [PubMed] [Google Scholar]
- Nern A, Zhu Y, and Zipursky SL (2008). Local N-cadherin interactions mediate distinct steps in the targeting of lamina neurons. Neuron 58, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nern A, Pfeiffer BD, and Rubin GM (2015). Optimized tools for multicolor stochastic labeling reveal diverse stereotyped cell arrangements in the fly visual system. Proc. Natl. Acad. Sci. USA 112, E2967–E2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsome TP, Asling B, and Dickson BJ (2000). Analysisof Drosophilaphotoreceptor axon guidance in eye-specific mosaics. Development 127, 851–860. [DOI] [PubMed] [Google Scholar]
- Nordstrom W, Chen P, Steller H, and Abrams JM (1996). Activation of the reaper gene during ectopic cell killing in Drosophila. Dev. Biol. 180, 213–226. [DOI] [PubMed] [Google Scholar]
- Özkan E, Carrillo RA, Eastman CL, Weiszmann R, Waghray D, Johnson KG, Zinn K, Celniker SE, and Garcia KC (2013). An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell 154, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özkan E, Chia PH, Wang RR, Goriatcheva N, Borek D, Otwinowski Z, Walz T, Shen K, and Garcia KC (2014). Extracellular architecture of the SYG-1/SYG-2 adhesion complex instructs synaptogenesis. Cell 156, 482–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot MY, Tadros W, Nern A, Bader M, Chen Y, and Zipursky SL (2013). Multiple interactions control synaptic layer specificity in the Drosophila visual system. Neuron 77, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot MY, Chen Y, Akin O, Chen Z, Tsui CYK, and Zipursky SL (2014). Sequential axon-derived signals couple target survival and layer specificity in the Drosophila visual system. Neuron 82, 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BD, Truman JW, and Rubin GM (2012). Using translational enhancers to increase transgene expression in Drosophila. Proc. Natl. Acad. Sci. USA 109, 6626–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, and Zipursky SL (2010). Design principles of insect and vertebrate visual systems. Neuron 66, 15–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadros W, Xu S, Akin O, Yi CH, Shin GJ, Millard SS, and Zipursky SL (2016). Dscam proteins direct dendritic targeting through adhesion. Neuron 89, 480–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura SY, Bharioke A, Lu Z, Nern A, Vitaladevuni S, Rivlin PK, Katz WT, Olbris DJ, Plaza SM, Winston P, et al. (2013). A visual motion detection circuit suggested by Drosophila connectomics. Nature 500, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura SY, Xu CS, Lu Z, Rivlin PK, Parag T, Olbris DJ, Plaza S, Zhao T, Katz WT, Umayam L, et al. (2015). Synaptic circuits and their variations within different columns in the visual system of Drosophila. Proc. Natl. Acad. Sci. USA 112, 13711–13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Zhang KX, Pecot MY, Nagarkar-Jaiswal S, Lee PT, Takemura SY, McEwen JM, Nern A, Xu S, Tadros W, et al. (2015). Ig superfamily ligand and receptor pairs expressed in synaptic partners in Drosophila. Cell 163, 1756–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka-Matakatsu M, Xu J, Cheng L, and Du W. (2009). Regulation of apoptosis of rbf mutant cells during Drosophila development. Dev. Biol. 326, 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev K, Joly W, Hadjieconomou D, and Salecker I. (2012). Localized netrins act as positional cues to control layer-specific targeting of photoreceptor axons in Drosophila. Neuron 75, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Schulze KL, Haelterman NA, Pan H, He Y, Evans-Holm M, Carlson JW, Levis RW, Spradling AC, Hoskins RA, and Bellen HJ (2011). MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat. Methods 8, 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernooy SY, Copeland J, Ghaboosi N, Griffin EE, Yoo SJ, and Hay BA (2000). Cell death regulation in Drosophila: conservation of mechanism and unique insights. J. Cell Biol. 150, F69–F76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Weiner JA, Levi S, Craig AM, Bradley A, and Sanes JR (2002). Gamma protocadherins are required for survival of spinal interneurons. Neuron 36, 843–854. [DOI] [PubMed] [Google Scholar]
- Yamagata M, and Sanes JR (2008). Dscam and Sidekick proteins direct lamina-specific synaptic connections in vertebrate retina. Nature 451, 465–469. [DOI] [PubMed] [Google Scholar]
- Yamagata M, and Sanes JR (2012). Expanding the Ig superfamily code for laminar specificity in retina: expression and role of contactins. J. Neurosci. 32, 14402–14414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata M, Weiner JA, and Sanes JR (2002). Sidekicks: synaptic adhesion molecules that promote lamina-specific connectivity in the retina. Cell 110, 649–660. [DOI] [PubMed] [Google Scholar]
- Zhang KX, Tan L, Pellegrini M, Zipursky SL, and McEwen JM (2016). Rapid changes in the translatome during the conversion of growth cones to synaptic terminals. Cell Rep. 14, 1258–1271. [DOI] [PubMed] [Google Scholar]
- Zipursky SL, and Sanes JR (2010). Chemoaffinity revisited: dscams, protocadherins, and neural circuit assembly. Cell 143, 343–353. [DOI] [PubMed] [Google Scholar]
- Zipursky SL, Venkatesh TR, Teplow DB, and Benzer S. (1984). Neuronal development in the Drosophila retina: monoclonal antibodies as molecular probes. Cell 36, 15–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Please refer to Key Resources Table.