
Figure 7.
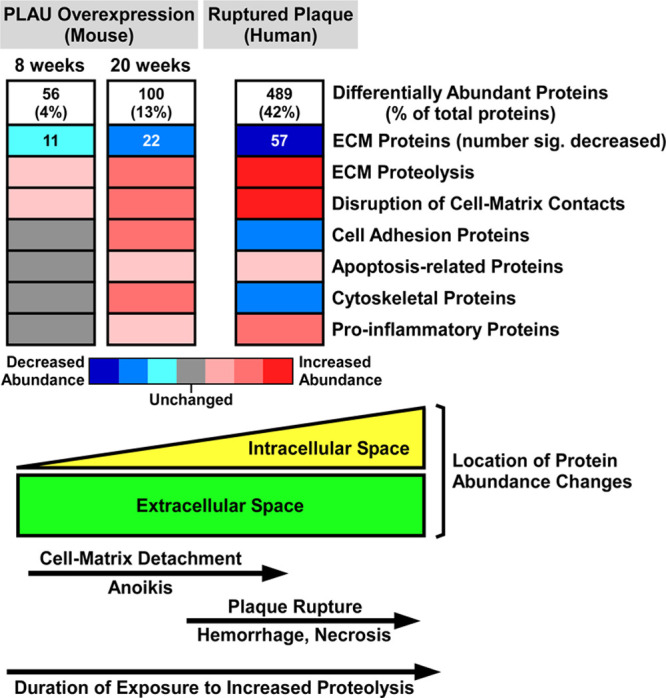
Shotgun proteomic analyses of mouse and human plaques suggest molecular and cellular mechanisms that connect extracellular proteolysis with plaque rupture.
Proteomic data from the mouse model of PLAU (urokinase-type plasminogen activator) overexpression (2 time points) and from human plaques show stepwise increases in the number of differentially abundant proteins (Data Sets II, IX, and XII in the Data Supplement) and corresponding decreases in ECM protein abundance (Data Sets IV, XI, and XIV in the Data Supplement). Decreased ECM (extracellular matrix) protein abundance is likely caused by increased proteolysis. Eight weeks exposure to elevated PLAU activity initiates disruption of cell-matrix contacts, which increases by 20 wk. Also by 20 wk, ECM protein loss stimulates homeostatic increases in cell adhesion and cytoskeletal proteins, inflammation, as well as programmed cell death (anoikis) triggered by loss of cell-matrix contacts. Advanced human plaques have more extensive proteolysis and disruption of cell-matrix contacts, causing anoikis, and cell necrosis with attendant loss of cell adhesion and cytoskeletal proteins. Plaque cell death weakens plaque structure, leading to rupture and an inflammatory response to tissue injury. The lower part of the figure illustrates the predominance of extracellular protein abundance changes at early stages of plaque proteolysis and the shift towards intracellular protein abundance changes at later stages.