

Abstract
Compared to the biological world’s rich chemistry for functionalizing carbon, enzymatic transformations of the heavier homologue silicon are rare. We report that a wild-type cytochrome P450 monooxygenase (P450BM3 from Bacillus megaterium, CYP102A1) has promiscuous activity for oxidation of hydrosilanes to make silanols. Directed evolution enhanced this non-native activity and created a highly efficient catalyst for selective silane oxidation under mild conditions with oxygen as terminal oxidant. The evolved enzyme leaves C–H bonds present in the silane substrates untouched, and this biotransformation does not lead to disiloxane formation, a common problem in silanol syntheses. Computational studies reveal that catalysis proceeds through hydrogen atom abstraction followed by radical rebound, as observed in the P450’s native C–H hydroxylation mechanism. Enzymatic silane oxidation now extends Nature’s impressive catalytic repertoire.
Keywords: biocatalysis, CYP102A1, monooxygenation, silanols, synthetic methods
Graphical Abstract
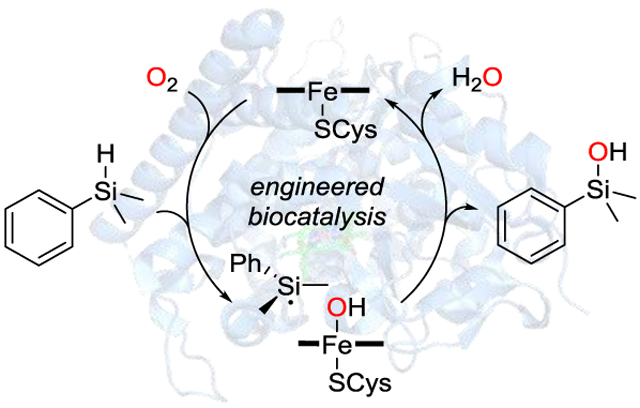
In rerum natura: Wild-type cytochrome P450BM3 catalyzes the oxidation of hydrosilanes to silanols both in vivo and in vitro. Directed evolution is used to generate an efficient and selective biocatalyst that delivers a broad range of aryl- and alkylsubstituted silanols. Computational studies reveal a sequence of H atom abstraction and OH rebound as the mechanism, in analogy to the native C–H hydroxylation activity.
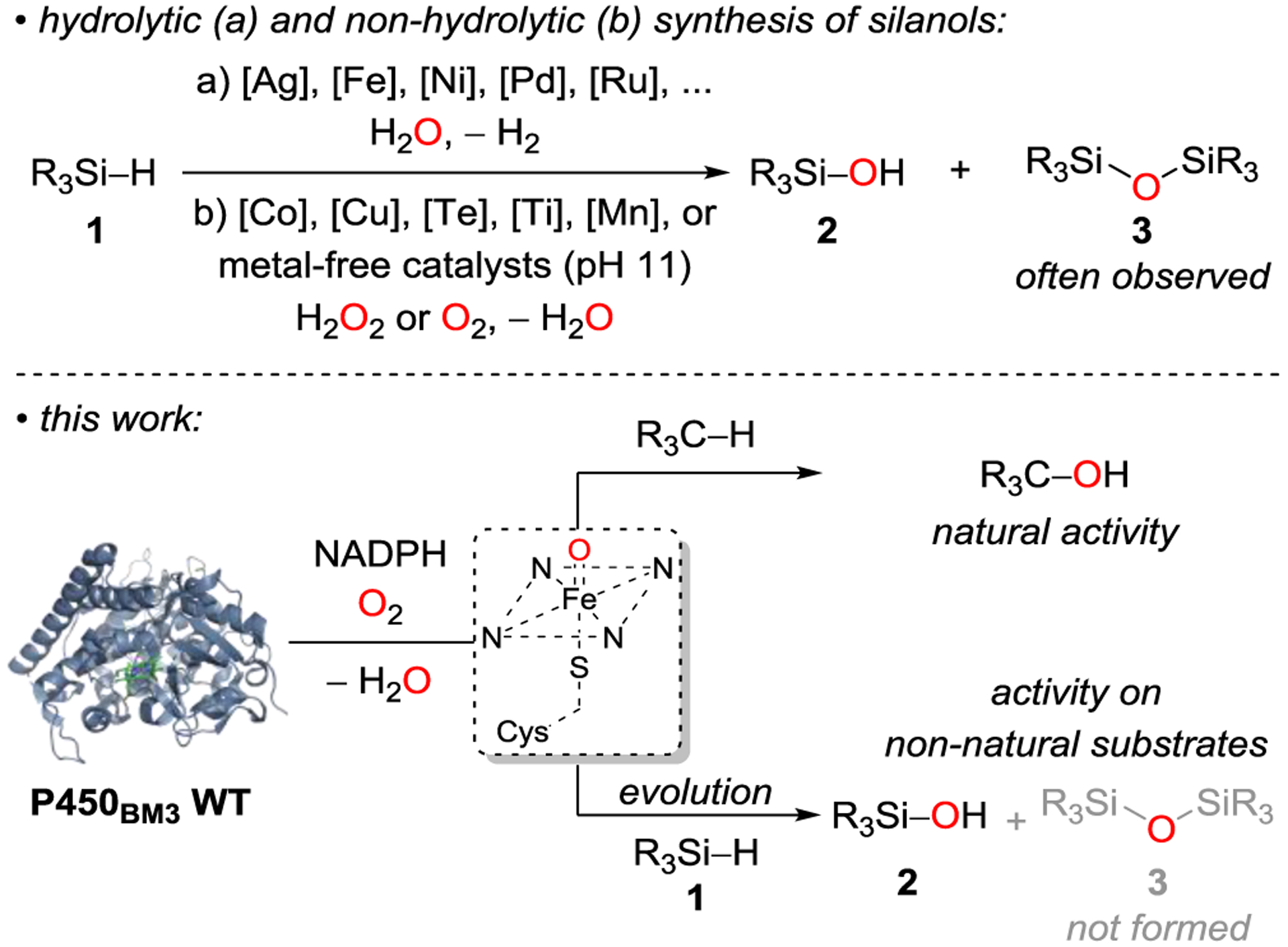
Silanols are widely used[1] to build silicone polymers[2], which are found in an enormous number of modern lifestyle products. Silanols are also synthetic intermediates in organic synthesis[3], catalysts,[4] and serve as functionally interesting isosteres for bioactive molecules.[5] The selective and mild synthesis of silanols has received substantial attention and remains of high interest.[1] Protocols that start from functionalized precursors such as chlorosilanes[6] or use strong oxidants[7] in combination with hydrosilanes often result in disiloxane formation, lead to other side reactions, or generate waste products in substantial amounts. Direct catalytic hydrosilane oxidation is an attractive alternative, and several methods have been developed, most of which rely on precious metals;[8] base-metal catalysts[9] or metal-free transformations[10] are rare.
In most cases, these are hydrolytic processes, i.e. water is the oxidant, and stoichiometric amounts of dihydrogen are released (Scheme 1, top, a). This process is also exothermic, which presents a safety issue when dihydrogen is evolved. Furthermore, contamination with disiloxane is often unavoidable, as the silanol product can react with the activated hydrosilane instead of water. Some catalysts can also catalyze silanol self-condensation to yield disiloxanes. A limited number of protocols use other oxidants like oxygen or hydrogen peroxide together with base metals[9d–g] or even metal-free, though very basic, conditions (Scheme 1, top, b).[10] Few reported selective syntheses of silanols employ environmentally friendly catalysts, proceed under mild reaction conditions, and avoid competing side reactions.[1] We hypothesized that a biocatalytic protocol employing nature’s versatile oxidation catalysts could be a valuable complement to existing synthetic methods.
Scheme 1.
Synthesis of silanols (top) and native vs. evolved activity of P450s (bottom).
Even though silicon is the second most abundant element in the Earth’s crust, the silicon chemistry of the biological world is very limited. Diatoms, for example, can incorporate ortho-silicic acid as stabilizing units in skeletons and materials; this process is usually based on the hydrolysis or alcoholysis of Si–O bonds.[15,16] Also, biocatalytic approaches for the formation of silicon polymers and small molecules are limited to the formation of Si–OR bonds through condensation reactions.[16] Although enzymatic manipulations of functional groups in the neighborhood of or remote to the silicon in various organosilicon compounds have been reported,[17] the direct biocatalytic functionalization of silicon centers remained unknown until this group engineered a cytochrome c in 2016 to catalyze carbene insertion into Si–H bonds in vitro and in vivo.[18,19] Biocatalytic oxidation of silanes to silanols is unknown.
The ubiquitous cytochrome P450 enzymes catalyze a range of oxidative reactions using atmospheric oxygen as terminal oxidant. [11,12] These transformations typically proceed via a reactive, high-valent iron-oxene intermediate (Compound I, Scheme 1, bottom),[13] which undergoes, for example, hydroxylations or epoxidations with C–H or C=C bonds.[14] We envisioned that we could use the natural promiscuity of P450s and repurpose a native enzyme for the efficient and selective oxidation of hydrosilanes using directed evolution.
We chose cytochrome P450BM3 (CYP102A1) from the soil bacterium Bacillus megaterium[12] to investigate Si–H oxidation activity. This self-sufficient monooxygenase is a 118-kDa enzyme with its NADPH-dependent reductase domain fused to the C-terminus of the heme domain; it has served as a robust scaffold for directed evolution of hydroxylation activity in previous work.[20] We tested the ability of wild-type P450BM3 to oxidize dimethylphenylsilane (1a) in Escherichia coli (E. coli) cells under aerobic conditions (Figure 1A). The enzyme delivered product 2a with 210 TTN in 2.1% yield at 1.0 μM protein concentration. A negligible amount of 2a was observed in control reactions under anaerobic conditions and otherwise identical setup, indicating that oxygen serves as the oxidant (see Supporting Information). We then used directed evolution by sequential rounds of saturation mutagenesis at selected amino acid residues to increase the enzyme’s activity for hydrosilane oxidation. Mutations at a number of amino acid residues in P450BM3 are known to affect oxidation activity.[20] We chose F87 as a first target for directed evolution due to its close proximity to the heme cofactor (Figure 1B).[21] A single site-saturation mutagenesis (NNK) library was screened in whole E. coli cells, from which we identified improved variant P450SiOx1 (containing mutation F87G) with a 1.5-fold improvement in both yield and TTN of product 2a (Figure 1A). Subsequently, we targeted residues T327 and A328 with double site-saturation mutagenesis using the 22-codon trick.[23] These residues are close to the iron cofactor in the distal heme-binding pocket, and mutations at these positions have influenced performance and selectivity in previous engineering studies.[24] A double site-saturation mutagenesis strategy was chosen in an attempt to discover potential beneficial epistatic interactions between these sites. Screening identified only a single beneficial mutation, A328L, which further increased the yield of silanol 2a to 8.5% (850 TTN) using 2nd-generation variant P450SiOx2. A further round of double site-saturation mutagenesis at residues L181 and A184[25] led to the discovery of mutations L181D and A184H in P450SiOx3, which improved the TTN to 1,200.
Figure 1.
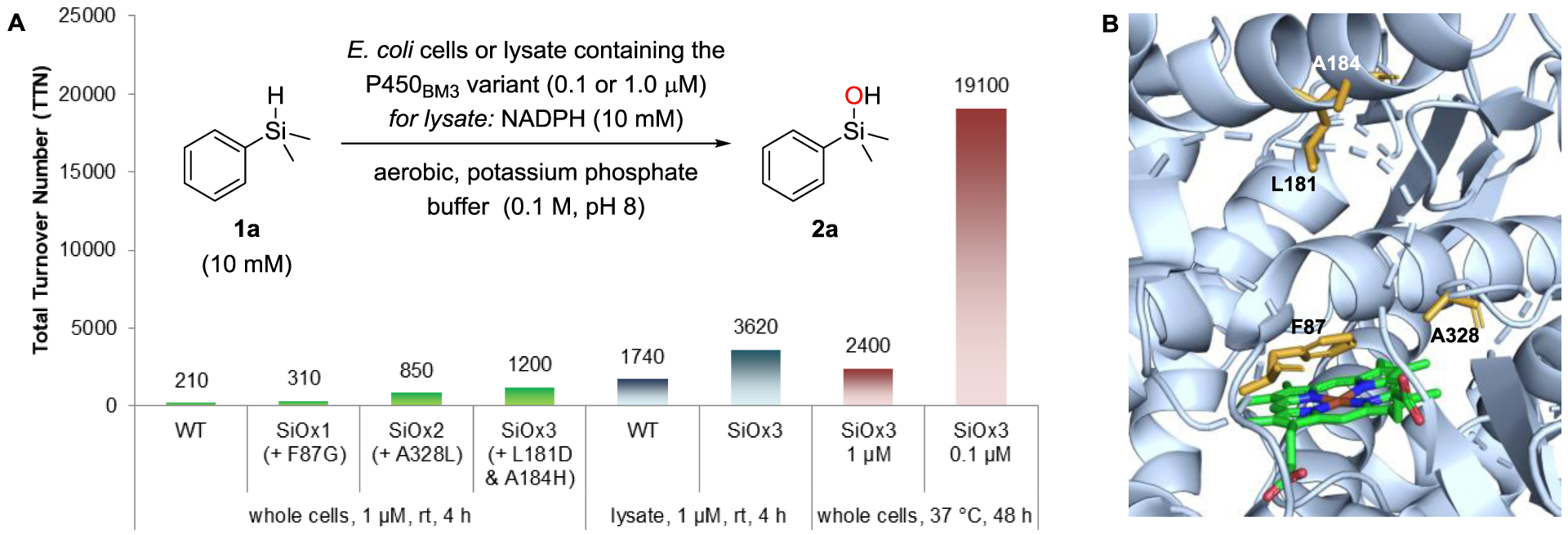
Activity of P450BM3 variants in whole cells (green) or lysate (blue) over the course of directed evolution and under optimized conditions (red), given as total turnover number for silanol production. See the Supporting Information for experimental details. B) Structure of wild-type cytochrome P450BM3 (PDB:1JPZ)[22] showing bound heme cofactor. Amino acid residues mutated during evolution are highlighted in orange (A = alanine, F = phenylalanine, L = leucine; NADPH = nicotinamide adenine dinucleotide phosphate).
We also compared the performance of wild-type P450BM3 and the final variant P450SiOx3 in E. coli lysate, which requires the addition of the cofactor NADPH in stoichiometric amounts (Figure 1A, blue bars). Turnover numbers were generally higher in lysate than in whole cells, which we ascribe to the absence of a diffusion barrier for the substrate and product through the cell wall and the limited availability of NADPH in whole cells. The final variant P450SiOx3 proved superior in lysate as well, with 3,620 TTN (36% yield of 2a) compared to 1740 (17% yield) for wild-type P450BM3. We next investigated the effect of changing various conditions on the production of 2a (Figure 1A, red bars; see Supporting Information). Running the reaction for 48 h at 37 °C substantially increased the yield, now delivering silanol 2a with 2,400 TTN and also indicating that the protein retains activity over this time. Furthermore, the yield of 2a was almost as high at a lower catalyst loading (0.1 μM protein concentration), resulting in remarkably high TTNs of 19,100. Even though these conditions show the potential of variant P450SiOx3, the yield of 2a obtained in this setup is not synthetically useful. Increasing the protein concentration to 8.1–9.0 μM and lowering the substrate concentration to 5.0 mM, we were able to achieve the transformation of 1a to 2a in quantitative yield as determined by GC analysis (76% isolated yield after 72 h at 37 °C; Scheme 2). Similarly, ethyl-substituted silanol 2b was formed in high yield. The conversion of vinylsilane 1c to silanol 2c was lower, but chemoselectivity was good, and epoxidation of the double bond was not observed.[26] Both 2b and 2c are silicon-stereogenic, but as we expect configurational lability on the silicon center of acyclic silanols under aqueous, slightly basic conditions, we did not attempt to monitor enantioselectivity.[27]
Scheme 2.
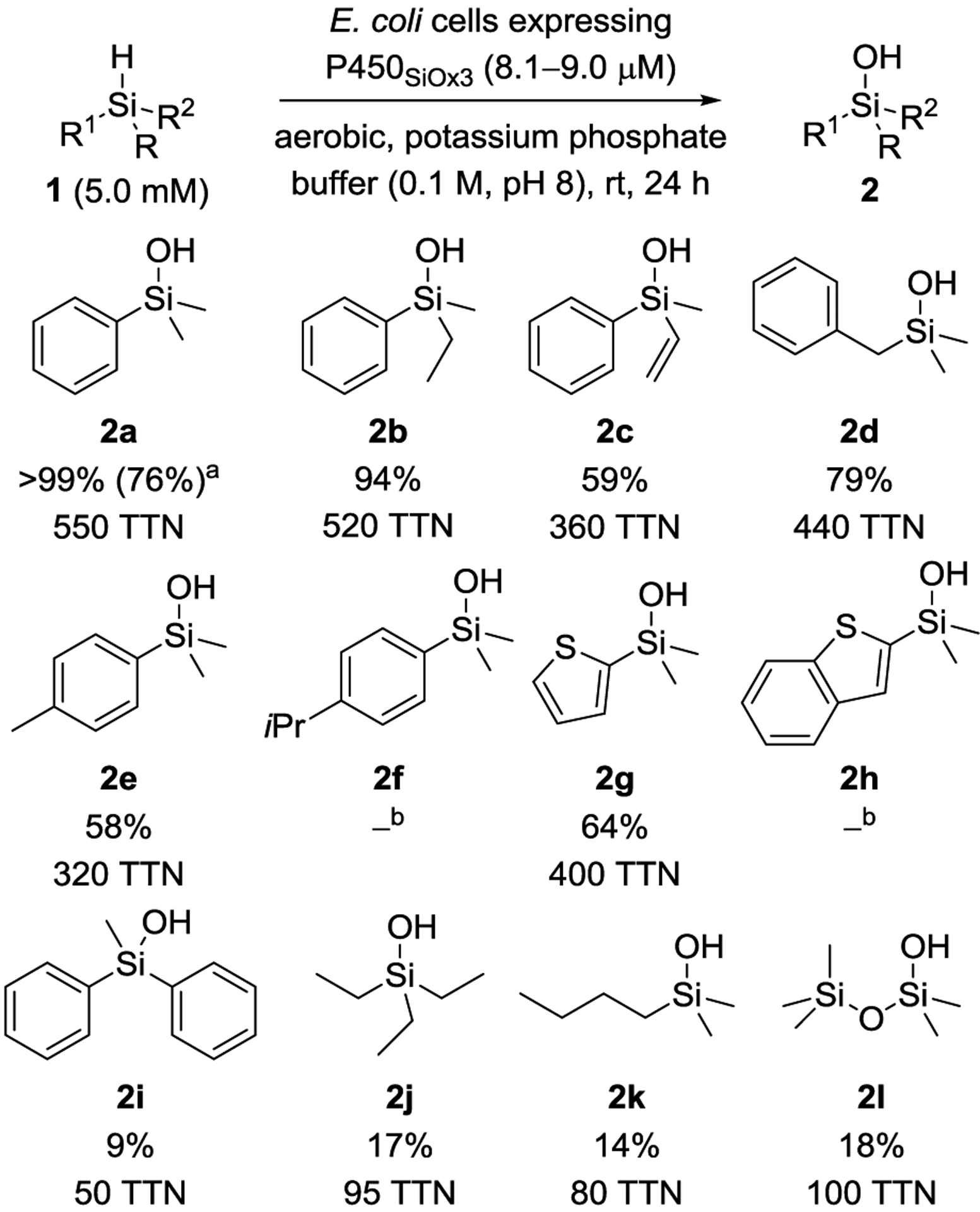
Substrate scope of P450SiOx3. GC yields are given as average of triplicate runs (see the Supporting Information for further details). a Isolated yield after 72 h at 37 °C in parentheses. b No conversion to the silanol observed.
Silanol 2d was obtained in good yield as well, again showing that a certain degree of steric flexibility close to the silicon center is accepted. Conversely, steric congestion of the aromatic ring limited reactivity: whereas a methyl group at the para-position of the aryl substituent in 2e was still tolerated, increasing the steric bulk thwarted the reaction so that isopropyl-substituted 2f was not formed. C–H oxidation in the benzylic positions did not occur for 1d and 1e.[28] The performance of the catalyst on electronically similar hydrosilanes 1g and 1h again indicates a certain size limitation. The smaller thiophenyl substituent was accepted and 2g was obtained in 64% yield; we did not observe the formation of 2h. Methyldiphenylsilane (1i) as well as aliphatic hydrosilanes 1j and 1k reacted selectively to the corresponding silanols 2i–k, albeit with lower yields. Notably, siloxane 1l was oxidized to 2l, even though uncatalyzed background hydrolysis occurred in significant amounts (see Supporting Information). It is worth noting that, if moderate or low yields of the silanols were obtained, they are a consequence of incomplete conversion of the starting hydrosilane. Products of competing C–H or C=C oxidation were not observed, nor was the formation of disiloxanes.
To explain this selectivity, we investigated the mechanism of the P450-catalyzed Si–H oxidation in further detail using density functional theory (DFT) calculations of a truncated computational model (Figure 2A, see Supporting Information for computational details). This model consists of an iron–porphyrin pyrrole core and methanethiol as axial ligand, mimicking the heme cofactor bond to a cysteine in the enzyme active site. The calculations indicate that the oxidation of dimethylphenylsilane (1a) proceeds through a transition state (TS1, Figure 2A–B) in which hydrogen atom transfer (HAT) from the hydrosilane to an FeIV-oxene intermediate generates a silyl radical (INT1, Figures 2A–B). This intermediate subsequently undergoes a barrierless process (TS2) where hydroxyl rebound occurs to yield the final silanol product 2a. The hydrogen atom abstraction corresponds to the rate-limiting step of the overall reaction (ΔG‡ = 18.1 kcal·mol–1). We also explored the possibility of silyl cation formation, which might be formed through electron transfer (ET) from the silyl radical to the porphyrin–Fe–OH species (Figure 2C). This step was found to be thermodynamically unfavorable (ΔG = 10.2 kcal·mol–1 for 1a); in contrast, the OH rebound step is barrierless (see Figure S1), hence silyl cation formation is unlikely. We found similar energy barriers for the rate-limiting hydrogen atom abstraction when using benzyldimethylsilane (1d, ΔG‡ = 19.6 kcal·mol–1) or a simple siloxane (HMe2Si–O–SiMe2H, ΔG‡ = 17.7 kcal·mol–1) as substrate. This is expected, since C–Si bonds are longer than C–C bonds, and orbital overlap and resonance stabilization by phenyl of the radical intermediate at the silicon center is less effective (see Figure S5).[29] Thus, Si–H-bond strengths are less prone to vary substantially with the substitution pattern.[28] We concluded that the limitations we observe in the substrate scope are related to potential issues with binding the substrate in a catalytically competent pose and steric effects, which the truncated DFT model does not account for, and not to intrinsic Si–H-bond strengths or electronic effects.
Figure 2.
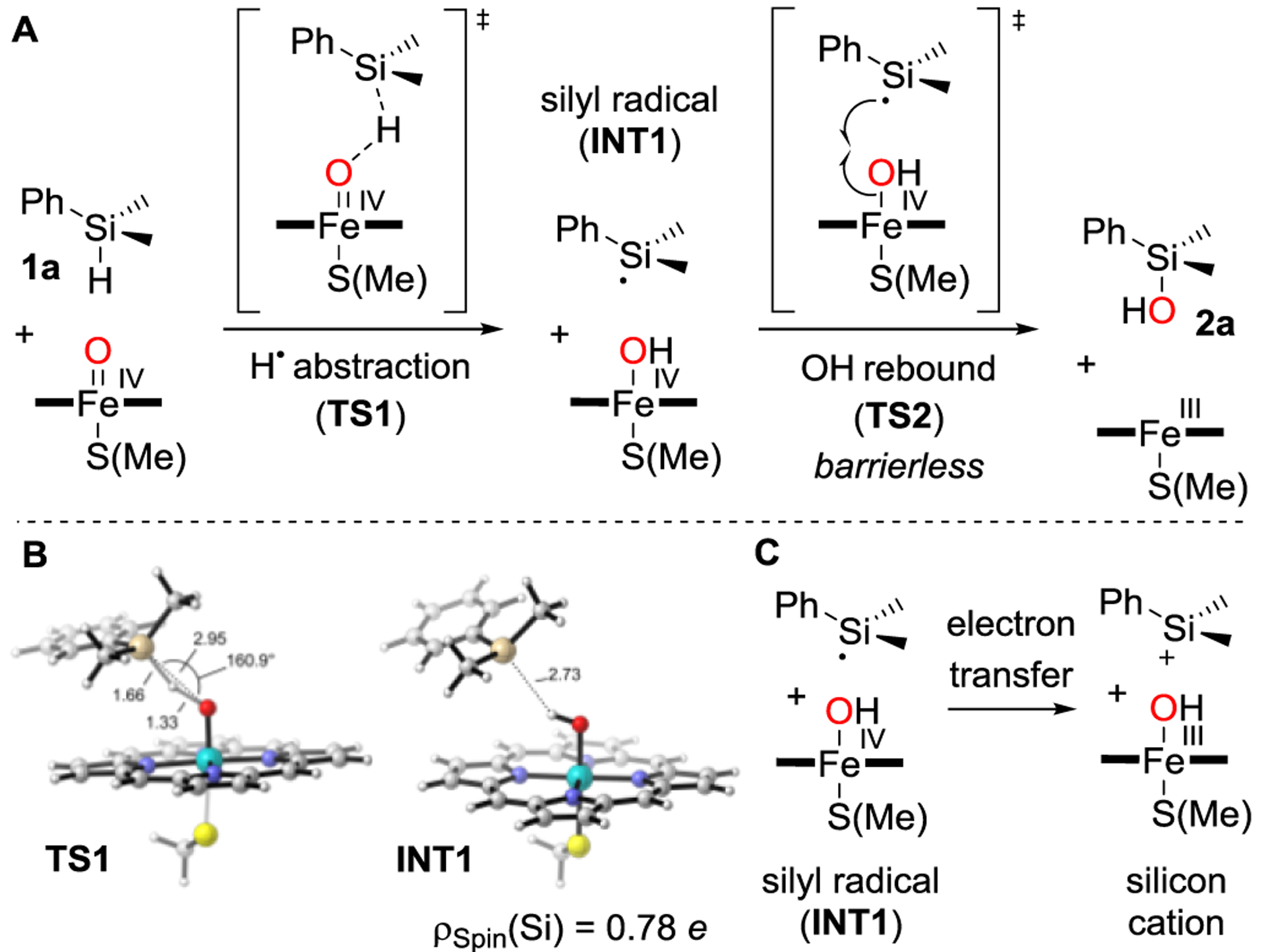
A) Computed mechanism for FeIV–oxene-catalyzed oxidation of 1a to 2a. B) DFT-optimized, lowest energy and rate-determining H atom abstraction, transition state TS1 and radical intermediate INT1 (quartet electronic state). The spin density localized at the Si atom (ρspin(Si)) in INT1 is shown. C) The endergonic (ΔG = 10.2 kcal·mol–1) electron transfer for the silyl cation formation pathway reinforces the conclusion that the radical oxidation pathway is the most plausible. Key distances are given in Å and angles in degrees.
Overall, the Si–H-oxidation mechanism described here is similar to the mechanism for native P450-catalyzed C–H oxidation,[12] and it involves the same FeIV-oxene catalytic intermediate and sequence of H-atom abstraction and rebound. The high chemoselectivity observed for Si–H oxidations over C–H can be directly attributed to the lower bond dissociation energy (BDE) of the Si–H bond as compared to the sterically accessible C–H bonds from the Si–Me groups (see Figure S5). This DFT study confirms that the P450 enzyme can adapt its native mechanism to oxidize hydrosilanes, and it also explains the absence of disiloxane side products which would require the activation of the hydrosilane to invite attack of the silanol.
In summary, we have engineered a fully genetically encoded catalyst capable of functionalizing silicon-containing compounds inside and outside cells, expanding our ability to manipulate this element in living systems. Three rounds of directed evolution created a highly active variant with four mutations from wild-type P450BM3. This work demonstrates that P450s are easily evolvable for unnatural hydrosilanes, although yields are currently limited for sterically hindered and trialkyl silanols. Furthermore, this renewable, iron-based biocatalyst shows high selectivity for forming silanols instead of disiloxanes; it also favors Si–H over the native C–H and C=C oxidation. This ability to enzymatically oxidize hydrosilanes in concert with previously identified biocatalysts for C–Si bond formation[18] could offer access to a range of useful organosilicon compounds in engineered microbial systems and open more sustainable paths to producing the silicon-containing molecules that have become ubiquitous in our modern world.
Supplementary Material
Acknowledgements
This work was supported by the Dow University Partnership Initiative (227027AO), the Rothenberg Innovation Initiative (RI2) Program (F.H.A.), the NIH National Institute for General Medical Sciences GM-124480 (K.N.H.), the Deutsche Forschungsgemeinschaft (DFG) Postdoctoral Fellowship (BA 6604/1-1 to S.B.), and the Spanish MINECO (JdC-I contract IJCI-2017-33411 to M.G.-B.). We thank N. W. Goldberg, Dr. S. B. J. Kan, and A. M. Knight for productive discussions, and Dr. Zhijun Jia for experimental assistance.
References
- [1].a) Jeon M, Han J, Park J, ACS Catal. 2012, 2, 1539–1549; [Google Scholar]; b) Chandrasekhar V, Boomishankar R, Nagendran S, Chem. Rev 2004, 104, 5847–5910. [DOI] [PubMed] [Google Scholar]
- [2].Colas A, Silicones: Preparation, Properties and Performance; Dow Corning, Life Sciences, 2005. [Google Scholar]
- [3].a) Denmark SE, Regens CS, Acc. Chem. Res 2009, 41, 1486–1499; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mewald M, Schiffner JA, Oestreich M, Angew. Chem 2012, 124, 1797–1799; Angew. Chem. Int. Ed. 2012, 51, 1763–1765; [DOI] [PubMed] [Google Scholar]; c) Denmark SE, Ambrosi A, Org. Process Res. Dev 2015, 19, 982–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Especially silanediols in catalysis:Diemoz KM, Hein JE, Wilson SO, Fettinger JC, Franz AK, J. Org. Chem 2017, 82, 6738–6747 and references cited therein.Schafer AG, Wieting JM, Fisher TJ, Mattson AE, Angew. Chem 2013, 125, 11531–11534; Angew. Chem. Int. Ed. 2013, 52, 11321–11324.
- [5].a) Franz AK, Wilson SO, J. Med. Chem 2013, 56, 388–405; [DOI] [PubMed] [Google Scholar]; b) Ramesh R, Reddy DS, J. Med. Chem 2018, 61, 3779–3798. [DOI] [PubMed] [Google Scholar]
- [6].Cella JA, Carpenter JC, J. Organomet. Chem 1994, 480, 23–36. [Google Scholar]
- [7].Selected examples:Lickiss PD, Lucas R, J. Organomet. Chem 1995, 521, 229–234 (KMnO4);Valliant-Saunders K, Gunn E, Shelton GR, Hrovat DA, Borden WT, Mayer JM, Inorg. Chem 2007, 46, 5212–5219 (OsO4);Adam W, Mello R, Curci R, Angew. Chem 1990, 102, 916–917; Angew. Chem. Int. Ed. Engl. 1990, 29, 890–891 (dioxirans); Sommer LH, Ulland LA, Parker GA, J. Am. Chem. Soc 1972, 94, 3469–3471 (peracids). See also Ref [1].
- [8].The use of Pt, Pd, Au, Ag, Ru and Ni as heterogeneous catalysts is reported. Leading reference:Barnes GH Jr., Daughenbaugh NEJ, J. Org. Chem 1966, 31, 885–887. See also Ref [1]. Hydrolytic, homogeneous catalysis:Lee M, Ko S, S. Chang, J. Am. Chem. Soc 2000, 122, 12011–12012 (Ru);Shi M, Nicholas KM, J. Chem. Res. (S), 1997, 400–401;Yu M, Jing H, Liu X, Fu X, Organometallics 2015, 34, 5754–5758 (both Rh);Kikukawa Y, Kuroda Y, Yamaguchi K, Mizuno, Angew. Chem 2012, 124, 2484–2487; Angew. Chem. Int. Ed. 2012, 51, 2434–2437 (silver);Lee Y, Seomoon D, Kim S, Han H, Chang S, Lee PH, J. Org. Chem 2004, 69, 1741–1743 (Ir); Ison EA, Corbin RA, Abu-Omar MM, J. Am. Chem. Soc 2005, 127, 11938–11939 (Re).
- [9]. Base metals in hydrolytic processes:Matarasso-Tchiroukhine E, J. Chem. Soc., Chem. Commun 1990, 681–682 (Cr, no selectivity for the silanol);Schubert U, Lorenz C, Inorg. Chem 1997, 36, 1258–1259 (Cu, low selectivity for the silanol);Liang Teo AK, Fan WY, Chem. Commun 2014, 50, 7191–7194 (Fe). A Te complex with O2: Okada Y, Oba, Arai A, Tanaka K, Nishiyama K, Ando W, Inorg. Chem 2010, 49, 383–385. A Ti zeolite with H2O2:Adam W, Garcia H, Mitchell CM, Saha-Möller CR, Weichold O, Chem. Commun 1998, 2609–2610. A Mn complex with H2O2:Wang K, Zhou J, Jiang Y, Zhang M, Wang C, Xue D, Tang W, Sun H, Xiao J, Li C, Angew. Chem 2019, 131, 6446–6450; Angew. Chem. Int. Ed. 2019, 58, 6380–6384. Cu- or Co-catalysis with O2:Arzumanyan AV, Goncharova IK, Novikov RA, Milenin SA, Boldyrev KL, Solyev PN, Tkachev YV, Volodin AD, Smol’yakov AF, Korlyukov AA, Muzafarov AM, Green Chem. 2018, 20, 1467–1471. For heterogeneous, hydrolytic Ni-catalysis, see ref. [8a].
- [10].Metal-free protocols, both at pH 11:; a) Limnios D, Kokotos CG, ACS Catal. 2013, 3, 2239–2243; [Google Scholar]; b) Kelly AT, Franz AK, ACS Omega 2019, 4, 6295–6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Manikandan P, Nagini S, Curr. Drug. Targets 2018, 19, 38–54. [DOI] [PubMed] [Google Scholar]
- [12].Whitehouse CJC, Bell SG, Wong L-L, Chem. Soc. Rev 2012, 41, 1218–1260. [DOI] [PubMed] [Google Scholar]
- [13].Rittle J, Green MT, Science 2014, 330, 933–938. [DOI] [PubMed] [Google Scholar]
- [14].a) Isin EM, Guengerich FP, Biochim. Biophys. Acta 2007, 3, 314–329; [DOI] [PubMed] [Google Scholar]; b) Ortiz de Montellano PR, Chem Rev. 2010, 110, 932–948; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W, Chem. Rev 2010, 210, 949–1017. [DOI] [PubMed] [Google Scholar]
- [15].Treguer P, Nelson DM, Bennekom AJV, DeMaster DJ, Leynaert A, Quéguiner B, Science 1995, 268, 375–379. [DOI] [PubMed] [Google Scholar]
- [16].Frampton MB, Zelisko PM, Chem. Asian J 2017, 12, 1153–1167. [DOI] [PubMed] [Google Scholar]
- [17].Frampton MB, Zelisko PM, Silicon 2009, 1, 147–163. [Google Scholar]
- [18].Kan SBJ, Lewis RD, Chen K, Arnold FH, Science 2016, 354, 1048–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].The oxidation of hydrosilanes in rats was reported almost fifty years ago, but an enzyme responsible for this transformation has never been identified:; Fessenden RJ, Hartman RA, J. Med. Chem 1970, 13, 52–54. [DOI] [PubMed] [Google Scholar]
- [20].Jung ST, Lauchli R, Arnold FH, Curr. Op. Biotech 2011, 22, 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li H, Poulos TL, Biochim. Biophys. Acta 1999, 1441, 141–149. Whereas F87, A328 and L181 are important for substrate recognition, A184 plays a role in the thermostability of P450BM3 variants. See Ref. [20].
- [22].Haines DC, Tomchick DR, Machius M, Peterson JA, Biochemistry 2001, 40, 13456–13465. [DOI] [PubMed] [Google Scholar]
- [23].Kille S, Acevedo-Rocha CG, Parra LP, Zhang ZG, Opperman DJ, Reetz MT, Acevedo JP, ACS Synth. Biol 2013, 2, 83–92. [DOI] [PubMed] [Google Scholar]
- [24]. Examples for the mutation of site A328:Roiban G, Reetz MT, Chem. Comm 2015, 51, 2208–2224 and cited references. For a recent example, see:Zhou H, Wang B, Wang F, Yu X, Ma L, Li A, Reetz M, Angew. Chem 2019, 131, 774–778; Angew. Chem. Int. Ed. 2019, 58, 764–768. See also Ref. [20].
- [25].For the importance of sites 181 and 184, see for example:; Zhang K, Shafer BM, Denars MD II, Stern HA, Fasan R, J. Am. Chem. Soc 2012, 134, 18695–19704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26]. Double-bond epoxidation was observed for example in a manganese-catalyzed protocol with H2O2 as oxidant, see Ref. [9f].
- [27].a) Corriu RJP, Guerin C, J. Organomet. Chem 1980, 198, 231–320; [Google Scholar]; b) Chuit C, Corriu RJP, Reye C, Young JC, Chem. Rev 1993, 93, 1371–1448. [Google Scholar]
- [28].For a comparison of Si–H and C–H bond strengths, see; a) Walsh R, Acc. Chem. Res 1981, 14, 246–252; [Google Scholar]; b) Blanksby SJ, Ellison GB, Acc. Chem. Res 2003, 36, 255–263 (BDE of a benzylic C–H bonds: 89.8 kcal/mol). Phenyl-substitution weakens the Si–H bond strength by ca. 2 kcal/mol: [DOI] [PubMed] [Google Scholar]; c) Chatgilialoglu C, Acc. Chem. Res 1992, 25, 188–194 (BDE of Ph3Si–H: 84 kcal/mol, Et3Si–H: 91 kcal/mol). For reviews, see also: [Google Scholar]; c) Tumanskii B, Karni M, Apeloig Y in Organosilicon Compounds Theory and Experiment (Synthesis) (Ed.: Lee VY), Elsevier, Academic Press, 2017, pp. 231–294; [Google Scholar]; d) Tumanskii B, Karni M, Apeloig Y in Encyclopedia of Radicals in Chemistry, Biology and Materials, Vol. 4 (Eds.: Chatgilialoglu C, Studer A), J. Wiley, Chichester, 2012, pp. 2117–2146. [Google Scholar]
- [29].a) Sheldrick WS in The Chemistry of Organic Silicon Compounds, Vol. 1 (Eds.: Patai S, Rapport Z), Wiley, Chichester, 1989, pp. 227‒303; [Google Scholar]; b) Alabugin IV in Stereoelectronic Effects: A Bridge Between Structure and Reactivity, Vol.1, John Wiley & Sons, Hoboken, 2016, 1–392. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.