
Figure 2.
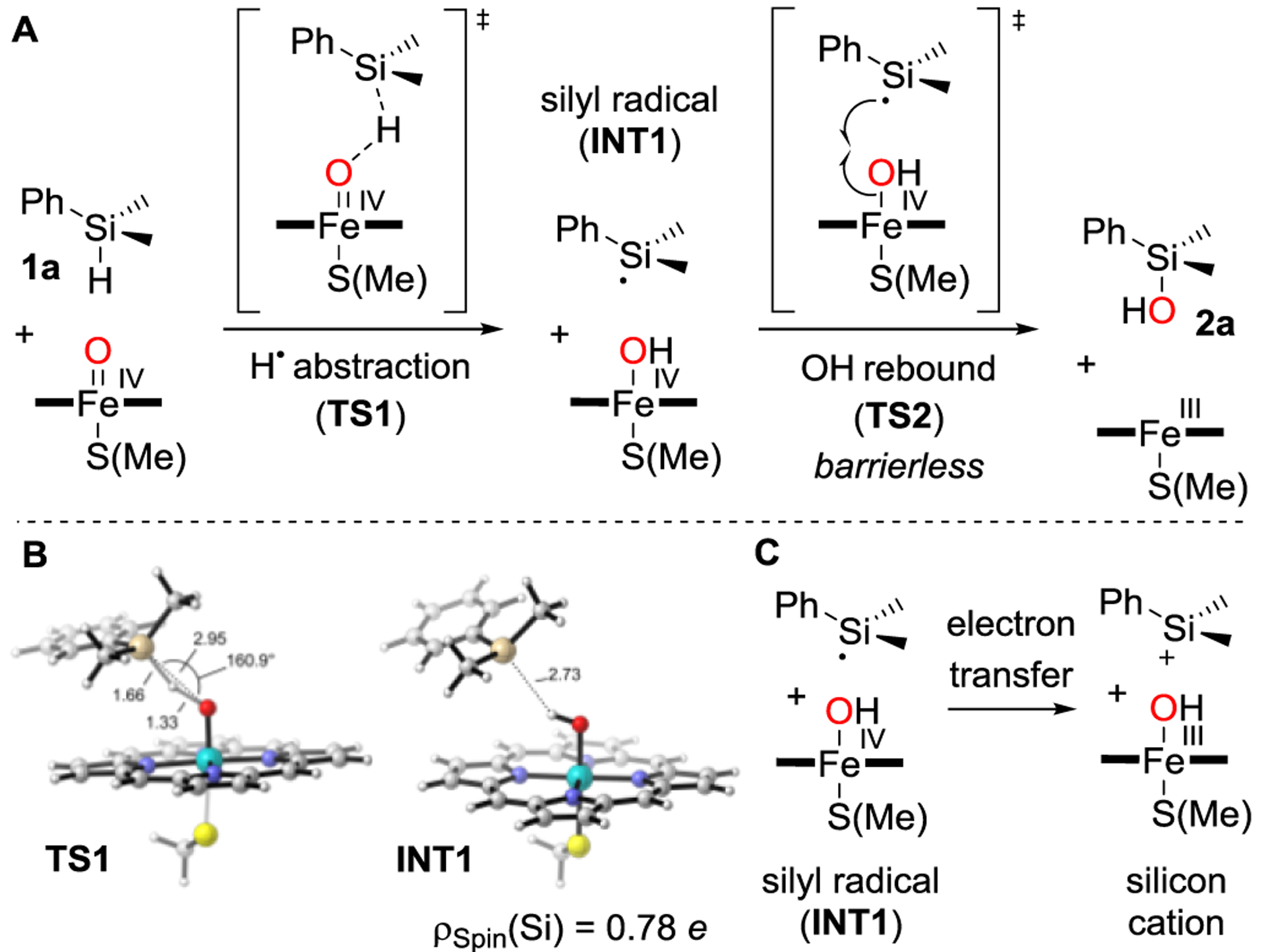
A) Computed mechanism for FeIV–oxene-catalyzed oxidation of 1a to 2a. B) DFT-optimized, lowest energy and rate-determining H atom abstraction, transition state TS1 and radical intermediate INT1 (quartet electronic state). The spin density localized at the Si atom (ρspin(Si)) in INT1 is shown. C) The endergonic (ΔG = 10.2 kcal·mol–1) electron transfer for the silyl cation formation pathway reinforces the conclusion that the radical oxidation pathway is the most plausible. Key distances are given in Å and angles in degrees.