

Abstract
Inherited retinal degeneration is a major cause of incurable blindness characterized by loss of retinal photoreceptor cells. Inherited retinal degeneration is characterized by high genetic and phenotypic heterogeneity with several genes mutated in patients affected by these genetic diseases. The high genetic heterogeneity of these diseases hampers the development of effective therapeutic interventions for the cure of a large cohort of patients. Common cell demise mechanisms can be envisioned as targets to treat patients regardless the specific mutation. One of these targets is the increase of intracellular calcium ions, that has been detected in several murine models of inherited retinal degeneration. Recently, neurotrophic factors that favor the efflux of calcium ions to concentrations below toxic levels have been identified as promising molecules that should be evaluated as new treatments for retinal degeneration. Here, we discuss therapeutic options for inherited retinal degeneration and we will focus on neuroprotective approaches, such as the neuroprotective activity of the Pigment epithelium-derived factor. The characterization of specific targets for neuroprotection opens new perspectives together with many questions that require deep analyses to take advantage of this knowledge and develop new therapeutic approaches. We believe that minimizing cell demise by neuroprotection may represent a promising treatment strategy for retinal degeneration.
Keywords: achromatopsia, calcium, calpains, calpastatin, congenital stationary night blindness, Leber's congenital amaurosis, retinitis pigmentosa, stargardt disease
Introduction
Inherited retinal degeneration (IRD) is a group of diseases characterized by progressive loss of photoreceptor cells which affects vision and ultimately leads to complete blindness (Broadgate et al., 2017). The complexity of genetic variations in this group of diseases demands for gene-independent therapeutic strategies and requires a strong collaboration among ophthalmologists, geneticists and biotechnologists (Wubben et al., 2019). The pathophysiological events occurring at the subcellular and molecular levels in the degenerating photoreceptors have been partially characterized and represent possible targets for neuroprotective therapeutic strategies. Specifically, Ca2+ overloads have been recently identified as harmful events at early stages of photoreceptor degeneration. Interestingly, increased intracellular Ca2+ was detected in animal models caused by mutations in different genes, identifying high intracellular Ca2+ as a common mechanism during the degeneration process (Power et al., 2019). A rationale for developing neuroprotective approaches can be the treatment with either calcium pump blockers or molecules able to boost calcium pumps favoring the extrusion of the ion from the photoreceptor cell (Frasson et al., 1999; Comitato et al., 2018). The topic of this review is the discussion of recent characterizations of the molecular events activated during photoreceptor degeneration and how these molecules can be taargeted by neuroprotective approaches.
Search Strategy and Selection Criteria
The databases used to select the most relevant papers included in this article were: https://www.ncbi.nlm.nih.gov/pubmed and https://sph.uth.edu/retnet/. Keywords for searching (selection criteria): retinal degeneration, photoreceptors, rods, neuroprotection, gene therapy, cell replacement, optogenetics, retinal prosthesis, calcium, calpains, PEDF. We set dates of searching: 2000–2019.
Inherited Retinal Degeneration
IRD is a group of diseases that can lead to vision loss and eventually to blindness due, primarily, to photoreceptor cell death (Broadgate et al., 2017). The incidence of IRD is estimated 1:2000, thus being the most common cause for visual loss in the working population of the industrialized world (Cremers et al., 2018). The term IRD groups several diseases in which photoreceptors are affected and can be stationary, as for congenital stationary night blindness and achromatopsia, or progressive as in retinitis pigmentosa (RP), Leber’s congenital amaurosis (LCA) and Stargardt disease (Verbakel et al., 2018). IRD are genetically and clinically heterogeneous retinopathies. More than 100 different genes have been linked to the disease and each of them can bear different mutations (https://sph.uth.edu/retnet/). There is a great functional diversity in the types of genes that have been implicated in IRD and they can be eye specific (e.g., components of the visual transduction cascade or of the retinoid cycle, involved in outer segment renewal, photoreceptor specific structural proteins, transcription factors) or ubiquitously expressed (e.g., splicing factors, contributing to nucleotide metabolism) (Daiger et al., 2013). The genetic and functional multiplicity of the involved proteins might cause the activation of distinctive molecular mechanisms for the different forms of RP. However, the most recent findings suggest that some common mechanisms are associated to photoreceptor demise in the animal models for IRD analyzed so far (Marigo, 2007).
While the mutations behind many of the disease types are known, only one treatment based on gene therapy for the RPE65 gene is available for Leber’s congenital amaurosis, and some other gene therapy treatments are developing for mutations in specific genes (Trapani and Auricchio, 2019). Nevertheless, development of gene therapy strategies for each mutated gene or, even, for specific mutations in one gene is not feasible and requires such highly personalized therapy approaches, that only few patients may benefit from every single new treatment that will be generated. Otherwise, recent studies characterized cell death mechanisms in several models of IRD, either caused by dominant or recessive mutations, and found some events that play key roles in all the analysed retinas, such as increases of cyclic guanosine monophosphate (cGMP) and high levels of intracellular calcium ions (Power et al., 2019). Based on this evidence, development of new treatments targeting the characterized common molecular mechanisms leading to photoreceptor cell death may benefit a larger cohort of patients (Marigo, 2007).
In IRD, the causative mutations frequently affect rods but degeneration of diseased rods leads to a secondary loss of cones, even if cones are genetically unaffected (Campochiaro and Mir, 2018). There are several evidences that preservation of rods, although not functional, can save sight, because vision in humans is mainly mediated by cones (Sahel and Léveillard, 2018; Vighi et al., 2018). The purpose of neuroprotection is, indeed, based on preservation of rod photoreceptor cells and consequently cones to save vision.
Therapeutic Prospects for Retinal Degeneration
The slow progression of the disease allows a wide time window for treatments but different stages of the disease may be more appropriate to be targeted by different therapeutic approaches (Figure 1). At early stages of the disease, when the retinal structure and histology are still not fully degenerated and photoreceptor cells are present, gene therapy and neuroprotection are the most appropriate approaches (Pardue and Allen, 2018). In fact, a successful gene therapy based on AAV2 delivery was recently approved for patients bearing mutations in the RPE65 gene and was applied to patients that, based on optical coherent tomography analysis, showed a preserved photoreceptor cell layer (Trapani and Auricchio, 2019). When most of photoreceptors are lost, cell replacement is an option. Transplantation of photoreceptors in patients is not in clinical trials yet, but several studies evaluated photoreceptor transplantation in wild type or IRD mutant mice (Jayakody et al., 2015). Seminal studies showed that post-mitotic photoreceptor precursor cells or mature photoreceptors can integrate in the degenerating retina of murine models of IRD, express photoreceptor markers, are light sensitive and improve function (MacLaren et al., 2006; Lakowski et al., 2010; Gust and Reh, 2011). The challenges of these studies are to obtain long-term survival of transplanted cells and sufficient integrated cells for improved functionality. Endogenous sources of photoreceptors are the ciliary epithelium and Müller glia cells (Tropepe et al., 2000; Giannelli et al., 2011), and recent studies provided evidences that the regenerative ability of Müller glia cell may represent a new therapeutic approach for retinal degeneration (Langhe and Pearson, 2019). While several protocols have been developed to differentiate rod-like cells from embryonic stem cells (Osakada et al., 2008; Lamba et al., 2009), induced pluripotent stem cells (Osakada et al., 2009; Lamba et al., 2010; Tucker et al., 2014) and adult retinal stem cells (Coles et al., 2004; Giordano et al., 2007; Demontis et al., 2012), none of these differentiation protocols can, at the moment, provide cells in number and integration capacity appropriate for an efficient transplantation of photoreceptors (Marigo and Casarosa, 2014; Gasparini et al., 2019). Differently, transplantation of in vitro differentiated retinal pigment epithelium is at a much more advanced stage and in clinical trials, as differentiated human embryonic stem cells-derived retinal pigment epithelium cells have been transplanted in patients with age-related macular degeneration and Stargardt disease (Bertolotti et al., 2014; Schwartz et al., 2015). An autologous transplant of retinal pigment epithelium derived from induced pluripotent stem cells was performed in a patient with age-related macular degeneration (Mandai et al., 2017). One year after surgery, the transplant demonstrated to be safe but with no improved visual acuity. Perception of light can also be restored by optogenetic approaches in advanced stages of degeneration (Fortuny and Flannery, 2018). Optogenetics is a biotechnological approach to allow light perception by a light-sensitive protein ectopically expressed in retinal cells that are not photoreceptors. The idea behind optogenetics is that, provided the complete loss of the light-sensitive photoreceptors in IRD, new light-sensitive cells can be generated by misexpression of proteins, that can be membrane integral ion channels, i.e., channelrhodopsin and halorhodopsin, or retinal G-protein coupled receptor, like opsins, that can change membrane potential upon light stimuli (Ostrovsky and Kirpichnikov, 2019). The low light sensitivity of channelrhodopsin and halorhodopsin was recently overcome by the finding that cone opsin can activate a G-protein-coupled inward-rectifier potassium channel and transduce the signal (Berry et al., 2019). The delivery of the genes encoding for optogenetic tools was based on viral delivery, like in gene therapy, in animal models of IRD. The targets of the optogenetic gene therapy are second-order neurons, such as bipolar cells, or retinal ganglion cells, third-order neurons, because IRD leads to loss of photoreceptors but the rest of the retina can be preserved for long time (Stefanov et al., 2019).
Figure 1.

Stages of photoreceptor degeneration and applicable therapies.
Degeneration is represented in a graph showing the reduction in photoreceptors cells during degeneration. Below we report the time windows of different therapeutic options. Gene therapy is appropriate for early stages of photoreceptor degeneration and neuroprotective strategies can treat ongoing photoreceptor cell degeneration. Both these treatments act on endogenous photoreceptors. Cell replacement, optogenetics and retinal prosthesis are strategies to treat patients at advanced/late stages of degeneration.
In case degeneration reaches a terminal stage with complete photoreceptor loss, implant of a retinal prosthesis needs to be evaluated (Bloch et al., 2019). Advances in retinal prostheses increased in the last decades thanks to improved microelectronics, biomaterials and retinal surgery methodologies. Retinal prostheses can be recorded in two major groups: epiretinal prostheses and subretinal prostheses. Epiretinal prostheses are implanted on the vitreal side of the retina to stimulate retinal ganglion cells, the neurons that form the optic nerve and connect the eye to the brain. Clinical trials with hundreds of patients enrolled for the Argus II Retinal Prosthesis System (Second Sight Medical Products Inc., Sylmar, CA, USA), approved by the Food and Drug Administration in 2013, demonstrated statistically improved quality of life and functional vision tasks (Dagnelie et al., 2017; Duncan et al., 2017). Subretinal prostheses are implanted on the side of photoreceptor cells with the aim of stimulating the retinal interneurons and possibly benefit from retinal signal amplification. Clinical trials with the Alpha IMS by Retinal Implant AG (Reutlingen, Germany) reported safety and improvements in quality of life and object recognition (Kitiratschky et al., 2015).
These new biotechnological approaches are attracting a lot of attention for patients with no residual photoreceptors left and thus at a stage of complete blindness.
Patients at early stages of the disease and still presenting photoreceptor cells can aspire to a treatment to delay degeneration. Pharmacological approaches are mostly based on neuroprotection aimed at slowing the progression of the disease by interfering with inflammation or oxidative stress or apoptosis (Dias et al., 2018). The purpose of neuroprotection is the survival and maintenance of neurons and, in the case of IRD, of photoreceptors. The neuroprotective treatment is suitable for the slow degenerative progress characterizing IRD, however, at the moment, no pharmacological therapy demonstrated enough efficacy to restore vision. Some neuroprotective approaches have been attempted by injection of stem cells. Exogenous sources of stem cells are non-retinal stem cells such as mesenchymal stem cells, derived from adipose tissue, bone marrow or dental pulp. Recent studies showed that injections of these types of cells have neuroprotective effects without any replacement of photoreceptors in the murine retina (Mead et al., 2015). Genetically modified bone marrow mesenchymal stromal cells overexpressing brain derived neurotrophic factor, injected in the rd6 IRD mouse model, could effectively rescue the damaged retina by neuroprotective means (Lejkowska et al., 2019). Some clinical trials are now undergoing based on injections of autologous mesenchymal stem cells in IRD patients (Labrador-Velandia et al., 2016). We should keep in mind that the therapeutic possibilities of mesenchymal stem cells can be unpredictable because these cells are often derived from patients for autologous transplants and their ability to secrete neuroprotective molecules will vary from one individual to the other.
Neuroprotection is, otherwise, often achieved by the delivery of small molecules at specific concentrations either locally in the eye or systemically (Sieving et al., 2006; Perusek and Maeda, 2013; Scholl et al., 2015; Vighi et al., 2018). This requires prolonged administration of the drug and one treatment is usually not definitive. Therefore, appropriate delivery systems, necessary to allow the drug to reach the neural retina, need to be developed based on the chemical characteristics of the different neuroprotective molecules (Himawan et al., 2019). The promising aspects of neuroprotective approaches are based on the fact that neuroprotection is not driven by a specific mutation in one gene but aims at targeting common cell stress pathways for the treatment of a broad spectrum of patients in a mutation-independent modality. This therapeutic approach allows also combined therapies and is less dependent on the stage of the disease because it will target cells that, at the specific moment of the treatments, are facing molecular and metabolic changes associated to cell death. Production of promising neuroprotective drugs requires a deep knowledge of the physiological and metabolic changes as well as molecular pathways activated in photoreceptor cells during the degenerative process.
Cell Death Mechanisms in Rod Photoreceptor Cells
The definition of whether different genetic lesions trigger similar cell death mechanisms and the identification of the crucial players during the degenerative process are strategic matters to be addressed for the development of new treatments for this genetically heterogeneous but phenotypically similar group of diseases.
The role of apoptotic pathways engaging executioner caspases during photoreceptor degeneration has been quite controversial. Caspase 3 and caspase 7, both executioner of apoptosis, were found activated in transgenic rats and mice with a mutation in the Rhodopsin (Rho) gene (Liu et al., 1999; Gorbatyuk et al., 2010; Comitato et al., 2019b). Several data, otherwise, on different models of the disease indicated that a caspase-independent mechanism is triggered during retinal degeneration (Donovan and Cotter, 2002; Doonan et al., 2003; Comitato et al., 2019b). The limited impact of caspase 3 in IRD was confirmed by taking advantage of caspase 3-deficient mice, in which knock-out of caspase 3 provided only transient photoreceptor protection (Zeiss et al., 2004). Supporting this hypothesis were in vivo treatments with a pan-caspase inhibitor, such as Z-VAD-FMK, that offered very limited neuroprotection in murine models of IRD (Sanges et al., 2006; Comitato et al., 2019b). The focus on caspases was based on the fact that BCL2-associated X protein (BAX) had been found activated in the degenerating retinas of animal models of IRD. Our studies extensively evaluated BAX activation in at least three models of RP, the rd1 mouse bearing a recessive mutation in the Pde6b gene, the Rho knock-out mouse and a transgenic mouse expressing the P23H mutation in RHO, and demonstrated that its function is mainly related to the efflux of the apoptosis inducing factor (AIF) from mitochondria and not to caspase activation (Comitato et al., 2014). These and other studies suggested that executioner caspases may be activated but they are not critical in mediating retinal degeneration in vivo. Based on these evidences the use of the term “apoptosis” for photoreceptor cell death in IRD is, thus, considered not appropriate and scientists in the field prefer to refer to photoreceptor cell death or photoreceptor degeneration for these events (Power et al., 2019).
Accumulating evidences from our and other laboratories implied that mitochondria and the endoplasmic reticulum (ER) are major points of integration of cell death signals. These two organelles contribute to tides and ebbs in calcium ions leading to unbalance of Ca2+ fluxes triggering cell demise. Intracellular Ca2+ levels are strictly regulated because they can affect neuronal survival (Yamashima and Oikawa, 2009). Several studies on models for photoreceptor cells death reported that the molecular pathways following calcium overload differ from classical caspase mediated apoptosis and engage calpains (Paquet-Durand et al., 2006, 2019; Sanges et al., 2006; Comitato et al., 2019b; Figure 2).
Figure 2.
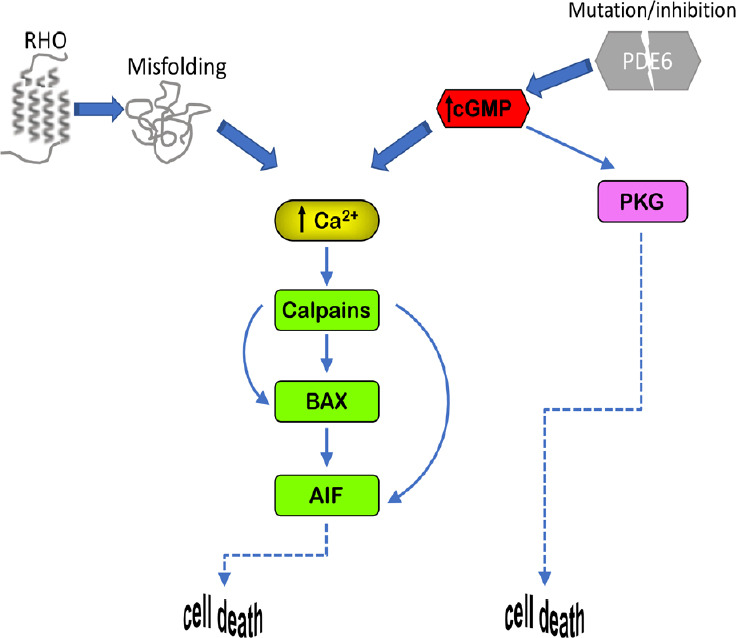
Cell death mechanisms.
The calcium-calpain pathway plays a major role in photoreceptor demise linked to IRD. Increases of intracellular calcium trigger calpain proteases which, by acting on AIF, lead to cell death through BAX activation. High intracellular Ca2+ can be caused by increases of intracellular cGMP, which can also activate PKG, as well as by protein misfolding, such as in photoreceptors bearing mutations in rhodopsin (RHO). High intracellular cGMP can be a consequence of loss of function in the phosphodiesterase 6 enzyme (PDE6), which idolizes cGMP. AIF: Apoptosis inducing factor; BAX: BCL2-associated X protein; cGMP: cyclic guanosine monophosphate; IRD: inherited retinal degeneration; PKG: cGMP-dependent protein kinase.
Calpains are cysteine proteases sensitive to intracellular calcium and are activated by increases in intracellular [Ca2+]. Inactive calpains are heterodimers, composed of an 80 kDa proteolytic subunit and a 28 kDa regulatory subunit (Ravulapalli et al., 2009). In the ER the heterodimer is associated with an endogenous calpain inhibitor called calpastatin. Calpastatin release and Ca2+ stimulated dissociation of the regulatory subunit lead to the activation of the calpain enzymes (Hood et al., 2004). Activation of calpains has been associated to cell death in IRD (Marigo, 2007; Paquet-Durand et al., 2019). Calpains do not directly cause chromatin condensation but they are proteases with a broad spectrum of substrates such as cytoskeleton components, AIF and BAX (Goll et al., 2003; Comitato et al., 2014) (Figure 2). AIF is a flavoprotein localized in the mitochondrial intermembrane space. Upon proper cell death stimuli, AIF exits the mitochondrion through BAX-formed pores and translocates to the nucleus where induces chromatin fragmentation (Arnoult et al., 2003; Comitato et al., 2014). Cleavage and release of AIF from mitochondria are regulated by calpain 1 and can occur in the absence of cytochrome c release, an event that otherwise induces apoptosis (Polster et al., 2005; Ozaki et al., 2009). In several neuronal degeneration models, including retinal degeneration, activation of AIF and its translocation to the nucleus had been observed (Cande et al., 2002; Sanges et al., 2006; Cao et al., 2007; Mizukoshi et al., 2010; Rosenbaum et al., 2010; Comitato et al., 2016, 2019b). The activated form of AIF recruits Cyclophilin A for chromatin fragmentation that culminates in cell death (Arnoult et al., 2003; Cande et al., 2004).
Dysregulation of different photoreceptor factors may cause calcium overloads leading to calpain activation. Recessive mutations in the Pde6b gene in the rd1 mouse model cause lack of PDE6 enzyme activity. PDE6 is a key enzyme in the phototransduction cascade. PDE6 hydrolyses cGMP in response to light and rhodopsin (RHO) activation. Impaired PDE6 activity causes elevated levels of cGMP (Farber and Lolley, 1974; Vighi et al., 2018). The correlation of elevated intracellular cGMP and photoreceptor cell death is quite well documented and appears to underlie photoreceptor cell demise caused by mutations in several genes linked to IRD (Power et al., 2019). In healthy photoreceptors, cGMP binds and keep open the cGMP-gated channels, channels regulating entrance of cations and, among them, Ca2+. Excessive cGMP, thus, results in elevated intracellular calcium (Sanges et al., 2006). Increased cGMP, on the other hand, can also activate cGMP-dependent protein kinase enzymes that trigger cell death mechanisms (Paquet-Durand et al., 2009; Vighi et al., 2018; Figure 2).
Mutations that cause misfolding of the RHO protein have been also associated with increased intracellular Ca2+ in rod photoreceptors (Shinde et al., 2016; Comitato et al., 2019b). Dominant mutations in RHO account for 20–25% of the dominant forms of RP and most of these mutations lead to misfolding of the protein and retention in the ER (Behnen et al., 2018). Dominant mutations in RHO have been studied for several years and numerous murine models are available as transgenic or knock-in mice. Activation of ER resident sensors, such as IRE (inositol-requiring enzyme 1), ATF6 (activating transcription factor-6) and PERK (protein kinase R-like ER protein kinase) have been reported but the functions of these sensors in the unfolded protein response to activate protective mechanisms or in ER-stress leading to cell death are still not completely defined in degenerating photoreceptors (Lin et al., 2007; Kunte et al., 2012; Chiang et al., 2015; Athanasiou et al., 2017; Comitato et al., 2018). In transgenic mice, in which expression of mutant RHO is combined with RHO protein overexpression, ER-stress sensors linked to apoptosis are activated (Gorbatyuk et al., 2010; Kunte et al., 2012; Comitato et al., 2016). Differently, in RHO recessive mutations, as in the knock-out mouse of the Rho gene, no ER-stress could be revealed but high [Ca2+] was reported (Comitato et al., 2016). Interestingly, in murine models of IRD with an equal gene dosage of wild type and mutant proline 23 to histidine (P23H) Rho, such as in the P23H knock-in mouse (RhoP23H/+), activation of the ER resident sensors, i.e. phosphorylated IRE1 and PERK, could be detected but this activation appeared to be related to ER-associated protein degradation and unfolded protein response and not to cell death (Chiang et al., 2015; Comitato et al., 2019b). Specifically, we defined that activation of PERK leads to phosphorylation of the nuclear factor erythroid 2-related factor 2 transcription factor, which is a mediator of the antioxidant response and possibly a protective mechanism during photoreceptor degeneration (Comitato et al., 2019b). On this line of evidences, inhibition of the PERK pathway revealed to be detrimental, suggesting that PERK sustains unfolded protein response and is a compensatory response in the degenerating retina (Athanasiou et al., 2017; Comitato et al., 2019b). These data are relevant for the studies on IRD because the RhoP23H/+ knock-in mouse models the degenerative progression of photoreceptors similarly to what found in RP patients bearing the P23H mutation in RHO, i.e., with a slow degeneration starting from the ventral side of the retina (Sakami et al., 2011).
Altogether the studies suggest common mechanisms of cell demise in dominant and recessive forms of RP caused by mutations in the PDE6B and RHO genes. In murine models of these types of RP intracellular increase of calcium ions and activation of calpains have been pinpointed as key players triggering photoreceptor degeneration.
Targeting Cell Death Mechanisms for Neuroprotection of Degenerating Photoreceptors
Several studies demonstrated that targeting calpains can be a promising therapeutic neuroprotective option for the degenerating retina. We showed that, in the rd1 mouse model with a recessive mutation in the Pde6b gene, calpain 1 appears to play a major role in the activation of the cell death pathway leading to AIF nuclear translocation (Comitato et al., 2014). Several calpain inhibitors have been tested in this mouse model of IRD and many of them showed neuroprotective effect in short-term delivery (Paquet-Durand et al., 2006, 2010; Sanges et al., 2006). However, some calpain inhibitors, such as CX295 and SJA6017, demonstrated to be toxic when the retina was exposed for a prolong time period to the inhibiting compounds (Paquet-Durand et al., 2010). In healthy tissue, calpain 1 and calpain 2 are maintained in an inactive state by binding to calpastatin, a highly specific endogenous inhibitor (Hood et al., 2004). A peptide derived from the natural inhibitor calpastatin was tested in the degenerating eyes and provided neuroprotection in recessive and dominant models of IRD after short and prolonged exposure times in vitro, on retinal explants, and in vivo after intravitreal injection (Paquet-Durand et al., 2010; Comitato et al., 2014, 2016). Neuroprotection by in vivo injection in the eye of the calpastatin peptide gave variable results in different models of IRD, with effects ranging from 30% to 80% reduction of dying cells (Paquet-Durand et al., 2010; Comitato et al., 2014, 2019a). A possible explanation is the high specificity of calpastatin for two types of calpains, i.e., calpain 1 and calpain 2. In case other calpains are activated, calpastatin cannot block them. In fact, while in the knock-in mouse RhoP23H/+ we found 80% correlation of calpain activation with the cell death marker terminal deoxynucleotidyl transferase dUTP nick end labeling at the peak of degeneration, calpastatin peptide could reduce cell death only by 30% (Comitato et al., 2019a). We reasoned that other calpains, aside calpain 1 and calpain 2, might be activated during retinal degeneration caused by the P23H mutation in RHO and found that a different calpain inhibitor (PD150606), which can target the majority of calpain types, could protect the retina from cell death by 65%. The strong neuroprotective activity of PD150606 suggests that different calpains are activated in retinas bearing different mutations leading to IRD. Nevertheless, the common mechanism activated by changes in [Ca2+] appears to be shared by several models of the disease and, thus, lowering calcium ions should also be evaluated as new therapeutic avenue (Comitato et al., 2019b).
In a recent study we confirmed the hypothesis that decreasing intracellular calcium can be neuroprotective in models of IRD. We and others showed that the pigment epithelium derived factor (PEDF) can preserve the degenerating retina of recessive and dominant models of IRD (Holekamp et al., 2002; Wang et al., 2013; Kenealey et al., 2015; Polato and Becerra, 2016; Comitato et al., 2018). Short-term treatments by intravitreal injection of human recombinant PEDF restrained cell death in the rd1 mutant retina by binding the PEDF receptor encoded by the PNPLA2 gene (Kenealey et al., 2015) and a small peptide of 17 amino acids (17mer) was identified as the neurotrophic domain of the protein. PEDF or the 17mer were demonstrated to act by targeting the PMCA pumps at the plasma membrane of photoreceptors and, thus, by favoring calcium efflux with a consequent reduction of intracellular [Ca2+] below toxic levels (Comitato et al., 2018). Interestingly, the PEDF receptor is an integral membrane protein with a phospholipase A2 activity stimulating the release of the omega-3 fatty acid docosahexaenoic acid from phospholipids (Subramanian et al., 2013; Pham et al., 2017). Docosahexaenoic acid was proven in myocytes and cardiomyocytes to support PMCA pumps and to interfere L-type Ca2+ channels counteracting calcium overload (Pepe et al., 1994; Mączewski et al., 2016). We, thus, propose that PEDF neuroprotective activity for degenerating photoreceptors acts by releasing intracellular docosahexaenoic acid, which increases PMCA pump activity to extrude calcium ions. The decrease of intracellular [Ca2+] induced by PEDF attenuate the cell death mechanism with lowered calpain activation and reduced mitochondrial BAX and nuclear translocation of AIF (Comitato et al., 2018). Altogether, the central cell death mechanism triggered by high intracellular [Ca2+] is diminished by PEDF. The open question that needs to be addressed is whether long-term exposure to PEDF can support photoreceptor survival or may have undesired side effects which will preclude the use of PEDF in therapy.
Perspectives and Conclusions
A limiting aspect in designing a cure for IRD is the high genetic heterogeneity found by molecular diagnosis in patients and the high percentage of isolated instances. General and common factors activated by mutations in different genes can be keystones for translational research. We identified activation of calpains, engaged by high intracellular Ca2+, as features shared by several murine models of IRD. In order to plan effective treatments to stop cell death during retinal degeneration we need specific studies aimed at defining whether high Ca2+ and calpains activate all the downstream catastrophic events leading to cell death or whether they cooperate with other proteases. The identification and characterization of molecules acting on these events needs also to be complemented by the development of appropriate delivery systems for the retina (Himawan et al., 2019). In fact, the different chemico-physical properties of neuroprotective agents tested in mice by short-term delivery will require specific and differentiated delivery systems. A second challenge will be a specific delivery either to rod or to cone photoreceptors to avoid side effects, such as bioconjugated compounds, in case of synthetic molecules (Wadhawan et al., 2019) or viral pseudotypes and rod-specific promoters in case of gene therapy approaches (Auricchio et al., 2001; Mussolino et al., 2011). Treatments with neuroprotectants targeting cell death mechanisms can delay photoreceptor degeneration but may also be of interest for combined treatments. In fact, gene therapy for recessive forms of IRD is in the clinic but appears to be more effective in young individuals and thus on cells at an early stage of degeneration (Trapani and Auricchio, 2019). A healthier photoreceptor appears to be a better target for gene therapy. Neuroprotection could, thus, be envisaged as a treatment to prolong sight but also for combined therapies to enhance the effectiveness of gene therapy approaches. Similarly, we may expect that also cell transplantation may have more chance of integration in a retinal tissue with a limited stressed status and low inflammation.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: This work was supported by grants from the Telethon Foundation (GGP14180, GGP19113) and the European Union (LSHG-CT-2005-512036 and transMed, MSCA-ITN-2017-765441) (all to VM).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This work was supported by grants from the Telethon Foundation (GGP14180, GGP19113) and the European Union (LSHG-CT-2005-512036 and transMed, MSCA-ITN-2017-765441) (all to VM).
C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. Embo J. 2003;22:4385–4399. doi: 10.1093/emboj/cdg423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Athanasiou D, Aguila M, Bellingham J, Kanuga N, Adamson P, Cheetham ME. The role of the ER stress-response protein PERK in rhodopsin retinitis pigmentosa. Hum Mol Genet. 2017;26:4896–4905. doi: 10.1093/hmg/ddx370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auricchio A, Kobinger G, Anand V, Hildinger M, O’Connor E, Maguire AM, Wilson JM, Bennett J. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: the retina as a model. Hum Mol Genet. 2001;10:3075–3081. doi: 10.1093/hmg/10.26.3075. [DOI] [PubMed] [Google Scholar]
- 4.Behnen P, Felline A, Comitato A, Salvo MT Di, Raimondi F, Gulati S, Kahremany S, Palczewski K, Marigo V, Fanelli F. A small chaperone improves folding and routing of rhodopsin mutants linked to inherited blindness. Science. 2018;4:1–19. doi: 10.1016/j.isci.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berry MH, Holt A, Salari A, Veit J, Visel M, Levitz J, Aghi K, Gaub BM, Sivyer B, Flannery JG, Isacoff EY. Restoration of high-sensitivity and adapting vision with a cone opsin. Nat Commun. 2019;10:1221. doi: 10.1038/s41467-019-09124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertolotti E, Neri A, Camparini M, Macaluso C, Marigo V. Stem cells as source for retinal pigment epithelium transplantation. Prog Retin Eye Res. 2014;42:130–144. doi: 10.1016/j.preteyeres.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Bloch E, Luo Y, da Cruz L. Advances in retinal prosthesis systems. Ther Adv Ophthalmol. 2019;11:2515841418817501. doi: 10.1177/2515841418817501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broadgate S, Yu J, Downes SM, Halford S. Unravelling the genetics of inherited retinal dystrophies: present and future. Prog Retin Eye Res. 2017;59:53–96. doi: 10.1016/j.preteyeres.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018;62:24–37. doi: 10.1016/j.preteyeres.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Cande C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death. J Cell Sci. 2002;115:4727–4734. doi: 10.1242/jcs.00210. [DOI] [PubMed] [Google Scholar]
- 11.Cande C, Vahsen N, Kouranti I, Schmitt E, Daugas E, Spahr C, Luban J, Kroemer RT, Giordanetto F, Garrido C, Penninger JM, Kroemer G. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene. 2004;23:1514–1521. doi: 10.1038/sj.onc.1207279. [DOI] [PubMed] [Google Scholar]
- 12.Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiang WC, Kroeger H, Sakami S, Messah C, Yasumura D, Matthes MT, Coppinger JA, Palczewski K, LaVail MM, Lin JH. Robust endoplasmic reticulum-associated degradation of rhodopsin precedes retinal degeneration. Mol Neurobiol. 2015;52:679–695. doi: 10.1007/s12035-014-8881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coles BLK, Angenieux B, Inoue T, Rio-Tsonis K Del, Spence JR, McInnes RR, Arsenijevic Y, Kooy D van der. Facile isolation and the characterization of human retinal stem cells. Proc Natl Acad Sci U S A. 2004;101:15772–15777. doi: 10.1073/pnas.0401596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comitato A, Salvo MT Di, Turchiano G, Montanari M, Sakami S, Palczewski K, Marigo V. Dominant and recessive mutations in rhodopsin activate different cell death pathways. Hum Mol Genet. 2016;25:2801–2812. doi: 10.1093/hmg/ddw137. [DOI] [PubMed] [Google Scholar]
- 16.Comitato A, Sanges D, Rossi A, Humphries MM, Marigo V. Activation of bax in three models of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2014;55:3555–3562. doi: 10.1167/iovs.14-13917. [DOI] [PubMed] [Google Scholar]
- 17.Comitato A, Schiroli D, La Marca C, Marigo V. Differential contribution of calcium-activated proteases and ER-stress in three mouse models of retinitis pigmentosa expressing P23H mutant RHO. Adv Exp Med Biol. 2019a;1185:311–316. doi: 10.1007/978-3-030-27378-1_51. [DOI] [PubMed] [Google Scholar]
- 18.Comitato A, Schiroli D, Montanari M, Marigo V. Calpain activation is the major cause of cell death in photoreceptors expressing a rhodopsin misfolding mutation. Mol Neurobiol. 2019b doi: 10.1007/s12035-019-01723-5. doi: 101007/s12035-019-01723-5. [DOI] [PubMed] [Google Scholar]
- 19.Comitato A, Subramanian P, Turchiano G, Montanari M, Becerra SP, Marigo V. Pigment epithelium-derived factor hinders photoreceptor cell death by reducing intracellular calcium in the degenerating retina. Cell Death Dis. 2018;9:560. doi: 10.1038/s41419-018-0613-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cremers FPM, Boon CJF, Bujakowska K, Zeitz C. Special issue introduction: inherited retinal disease: novel candidate genes, genotype-phenotype, correlations, and inheritance models. Genes (Basel) 2018;9:215. doi: 10.3390/genes9040215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dagnelie G, Christopher P, Arditi A, da Cruz L, Duncan JL, Ho AC, Olmos de Koo LC, Sahel JA, Stanga PE, Thumann G, Wang Y, Arsiero M, Dorn JD, Greenberg RJ Argus® II Study Group. Performance of real-world functional vision tasks by blind subjects improves after implantation with the Argus® II retinal prosthesis system. Clin Exp Ophthalmol. 2017;45:152–159. doi: 10.1111/ceo.12812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84:132–141. doi: 10.1111/cge.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demontis GC, Aruta C, Comitato A, Marzo A De, Marigo V. Functional and molecular characterization of rod-like cells from retinal stem cells derived from the adult ciliary epithelium. PLoS One. 2012;7:e33338. doi: 10.1371/journal.pone.0033338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dias MF, Joo K, Kemp JA, Fialho SL, Silva Cunha A da, Woo SJ, Kwon YJ. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog Retin Eye Res. 2018;63:107–131. doi: 10.1016/j.preteyeres.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Donovan M, Cotter TG. Caspase-independent photoreceptor apoptosis in vivo and differential expression of apoptotic protease activating factor-1 and caspase-3 during retinal development. Cell Death Differ. 2002;9:1220–1231. doi: 10.1038/sj.cdd.4401105. [DOI] [PubMed] [Google Scholar]
- 26.Doonan F, Donovan M, Cotter TG. Caspase-independent photoreceptor apoptosis in mouse models of retinal degeneration. J Neurosci. 2003;23:5723–5731. doi: 10.1523/JNEUROSCI.23-13-05723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duncan JL, Richards TP, Arditi A, da Cruz L, Dagnelie G, Dorn JD, Ho AC, Olmos de Koo LC, Barale PO, Stanga PE, Thumann G, Wang Y, Greenberg RJ. Improvements in vision-related quality of life in blind patients implanted with the Argus II Epiretinal Prosthesis. Clin Exp Ophthalmol. 2017;100:144–150. doi: 10.1111/cxo.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farber DB, Lolley RN. Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science. 1974;186:449–451. doi: 10.1126/science.186.4162.449. [DOI] [PubMed] [Google Scholar]
- 29.Fortuny C, Flannery JG. Mutation-independent gene therapies for rod-cone dystrophies. Adv Exp Med Biol. 2018;1074:75–81. doi: 10.1007/978-3-319-75402-4_10. [DOI] [PubMed] [Google Scholar]
- 30.Frasson M, Sahel JA, Fabre M, Simonutti M, Dreyfus H, Picaud S. Retinitis pigmentosa: rod photoreceptor rescue by a calcium-channel blocker in the rd mouse. Nat Med. 1999;5:1183–1187. doi: 10.1038/13508. [DOI] [PubMed] [Google Scholar]
- 31.Gasparini SJ, Llonch S, Borsch O, Ader M. Transplantation of photoreceptors into the degenerative retina: Current state and future perspectives. Prog Retin Eye Res. 2019;69:1–37. doi: 10.1016/j.preteyeres.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Giannelli SG, Demontis GC, Pertile G, Rama P, Broccoli V. Adult human muller glia cells are a highly efficient source of rod photoreceptors. Stem Cells. 2011;29:344–356. doi: 10.1002/stem.579. [DOI] [PubMed] [Google Scholar]
- 33.Giordano F, De Marzo A, Vetrini F, Marigo V. Fibroblast growth factor and epidermal growth factor differently affect differentiation of murine retinal stem cells in vitro. Mol Vis. 2007;13:1842–1850. [PubMed] [Google Scholar]
- 34.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;1990:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 35.Gorbatyuk MS, Knox T, LaVail MM, Gorbatyuk OS, Noorwez SM, Hauswirth WW, Lin JH, Muzyczka N, Lewin AS. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107:5961–5966. doi: 10.1073/pnas.0911991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gust J, Reh TA. Adult donor rod photoreceptors integrate into the mature mouse retina. Invest Ophthalmol Vis Sci. 2011;52:5266–5272. doi: 10.1167/iovs.10-6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Himawan E, Ekström P, Buzgo M, Gaillard P, Stefánsson E, Marigo V, Loftsson T, Paquet-Durand F. Drug delivery to retinal photoreceptors. Drug Discov Today. 2019;24:1637–1643. doi: 10.1016/j.drudis.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holekamp NM, Bouck N, Volpert O. Pigment epithelium-derived factor is deficient in the vitreous of patients with choroidal neovascularization due to age-related macular degeneration. Am J Ophthalmol. 2002;134:220–227. doi: 10.1016/s0002-9394(02)01549-0. [DOI] [PubMed] [Google Scholar]
- 39.Hood JL, Brooks WH, Roszman TL. Differential compartmentalization of the calpain/calpastatin network with the endoplasmic reticulum and Golgi apparatus. J Biol Chem. 2004;279:43126–43135. doi: 10.1074/jbc.M408100200. [DOI] [PubMed] [Google Scholar]
- 40.Jayakody SA, Gonzalez-Cordero A, Ali RR, Pearson RA. Cellular strategies for retinal repair by photoreceptor replacement. Prog Retin Eye Res. 2015;46:31–66. doi: 10.1016/j.preteyeres.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Kenealey J, Subramanian P, Comitato A, Bullock J, Keehan L, Polato F, Hoover D, Marigo V, Becerra SP. Small retinoprotective peptides reveal a receptor-binding region on pigment epithelium-derived factor. J Biol Chem. 2015;290:25241–25253. doi: 10.1074/jbc.M115.645846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kitiratschky VBD, Stingl K, Wilhelm B, Peters T, Besch D, Sachs H, Gekeler F, Bartz-Schmidt KU, Zrenner E. Safety evaluation of “retina implant alpha IMS”—a prospective clinical trial. Graefes Arch Clin Exp Ophthalmol. 2015;253:381–387. doi: 10.1007/s00417-014-2797-x. [DOI] [PubMed] [Google Scholar]
- 43.Kunte MM, Choudhury S, Manheim JF, Shinde VM, Miura M, Chiodo VA, Hauswirth WW, Gorbatyuk OS, Gorbatyuk MS. ER stress is involved in T17M rhodopsin-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2012;53:3792–3800. doi: 10.1167/iovs.11-9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Labrador-Velandia S, Alonso-Alonso ML, Alvarez-Sanchez S, González- Zamora J, Carretero-Barrio I, Pastor JC, Fernandez-Bueno I, Srivastava GK. Mesenchymal stem cell therapy in retinal and optic nerve diseases: An update of clinical trials. World J Stem Cells. 2016;8:376–383. doi: 10.4252/wjsc.v8.i11.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lakowski J, Baron M, Bainbridge J, Barber AC, Pearson RA, Ali RR, Sowden JC. Cone and rod photoreceptor transplantation in models of the childhood retinopathy Leber congenital amaurosis using flow-sorted Crx-positive donor cells. Hum Mol Genet. 2010;19:4545–4559. doi: 10.1093/hmg/ddq378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lamba DA, Gust J, Reh TA. Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell. 2009;4:73–79. doi: 10.1016/j.stem.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamba DA, McUsic A, Hirata RK, Wang PR, Russell D, Reh TA. Generation purification and transplantation of photoreceptors derived from human induced pluripotent stem cells. PLoS One. 2010;5:e8763. doi: 10.1371/journal.pone.0008763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langhe R, Pearson RA. Rebuilding the retina: prospects for Müller glial-mediated self-repair. Curr Eye Res. 2019;2:1–12. doi: 10.1080/02713683.2019.1669665. [DOI] [PubMed] [Google Scholar]
- 49.Lejkowska R, Kawa MP, Pius-Sadowska E, Rogińska D, Łuczkowska K, Machaliński B, Machalińska A. Preclinical evaluation of long-term neuroprotective effects of BDNF-engineered mesenchymal stromal cells as intravitreal therapy for chronic retinal degeneration in Rd6 mutant mice. Int J Mol Sci. 2019;20:1–25. doi: 10.3390/ijms20030777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, LaVail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu C, Li Y, Peng M, Laties AM, Wen R. Activation of caspase-3 in the retina of transgenic rats with the rhodopsin mutation s334ter during photoreceptor degeneration. J Neurosci. 1999;19:4778–4785. doi: 10.1523/JNEUROSCI.19-12-04778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacLaren RE, Pearson RA, MacNeil A, Douglas RH, Salt TE, Akimoto M, Swaroop A, Sowden JC, Ali RR. Retinal repair by transplantation of photoreceptor precursors. Nature. 2006;444:203–207. doi: 10.1038/nature05161. [DOI] [PubMed] [Google Scholar]
- 53.Mączewski M, Duda M, Marciszek M, Kołodziejczyk J, Dobrzyń P, Dobrzyń A, Mackiewicz U. Omega-3 fatty acids do not protect against arrhythmias in acute nonreperfused myocardial infarction despite some antiarrhythmic effects. J Biol Chem. 2016;117:2570–2582. doi: 10.1002/jcb.25550. [DOI] [PubMed] [Google Scholar]
- 54.Mandai M, Watanabe A, Kurimoto Y, Hirami Y, Morinaga C, Daimon T, Fujihara M, Akimaru H, Sakai N, Shibata Y, Terada M, Nomiya Y, Tanishima S, Nakamura M, Kamao H, Sugita S, Onishi A, Ito T, Fujita K, Kawamata S, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376:1038–1046. doi: 10.1056/NEJMoa1608368. [DOI] [PubMed] [Google Scholar]
- 55.Marigo V. Programmed cell death in retinal degeneration. Cell Cycle. 2007;6:652–655. doi: 10.4161/cc.6.6.4029. [DOI] [PubMed] [Google Scholar]
- 56.Marigo V, Casarosa S. Photoreceptor transplantation and regeneration. Vertebrate Photoreceptors: Functional Molecular Bases. 2014 doi: 101007/978-4-431-54880-5_12. [Google Scholar]
- 57.Mead B, Berry M, Logan A, Scott RAH, Leadbeater W, Scheven BA. Stem cell treatment of degenerative eye disease. Stem Cell Res. 2015;14:243–257. doi: 10.1016/j.scr.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mizukoshi S, Nakazawa M, Sato K, Ozaki T, Metoki T, Ishiguro S. Activation of mitochondrial calpain and release of apoptosis-inducing factor from mitochondria in RCS rat retinal degeneration. Exp Eye Res. 2010;91:353–361. doi: 10.1016/j.exer.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 59.Mussolino C, della Corte M, Rossi S, Viola F, Di Vicino U, Marrocco E, Neglia S, Doria M, Testa F, Giovannoni R, Crasta M, Giunti M, Villani E, Lavitrano M, Bacci ML, Ratiglia R, Simonelli F, Auricchio A, Surace EM. AAV-mediated photoreceptor transduction of the pig cone-enriched retina. Gene Ther. 2011;18:637–645. doi: 10.1038/gt.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Osakada F, Ikeda H, Mandai M, Wataya T, Watanabe K, Yoshimura N, Akaike A, Sasai Y, Takahashi M. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat Biotechnol. 2008;26:215–224. doi: 10.1038/nbt1384. [DOI] [PubMed] [Google Scholar]
- 61.Osakada F, Jin ZB, Hirami Y, Ikeda H, Danjyo T, Watanabe K, Sasai Y, Takahashi M. In vitro differentiation of retinal cells from human pluripotent stem cells by small-molecule induction. J Cell Sci. 2009;122:3169–3179. doi: 10.1242/jcs.050393. [DOI] [PubMed] [Google Scholar]
- 62.Ostrovsky MA, Kirpichnikov MP. Prospects of optogenetic prosthesis of the degenerative retina of the eye. Biochemstry (Mosc) 2019;84:479–490. doi: 10.1134/S0006297919050031. [DOI] [PubMed] [Google Scholar]
- 63.Ozaki T, Yamashita T, Ishiguro S. ERp57-associated mitochondrial μ-calpain truncates apoptosis-inducing factor. Biochim Biophys Acta. 2009;1793:1848–1859. doi: 10.1016/j.bbamcr.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 64.Paquet-Durand F, Azadi S, Hauck SM, Ueffing M, van Veen T, Ekström P. Calpain is activated in degenerating photoreceptors in the rd1 mouse. J Neurochem. 2006;96:802–814. doi: 10.1111/j.1471-4159.2005.03628.x. [DOI] [PubMed] [Google Scholar]
- 65.Paquet-Durand F, Ekström P, Marigo V. Modulation of calcium overload and calpain activity. In RSC Drug Discovery Series. 2019:48–60. [Google Scholar]
- 66.Paquet-Durand F, Hauck SM, van Veen T, Ueffing M, Ekström P. PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J Neurochem. 2009;108:796–810. doi: 10.1111/j.1471-4159.2008.05822.x. [DOI] [PubMed] [Google Scholar]
- 67.Paquet-Durand F, Sanges D, McCall J, Silva J, van Veen T, Marigo V, Ekström P. Photoreceptor rescue and toxicity induced by different calpain inhibitors. J Neurochem. 2010;115:930–940. doi: 10.1111/j.1471-4159.2010.06983.x. [DOI] [PubMed] [Google Scholar]
- 68.Pardue MT, Allen RS. Neuroprotective strategies for retinal disease. Prog Retin Eye Res. 2018;65:50–76. doi: 10.1016/j.preteyeres.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pepe S, Bogdanov K, Hallaq H, Spurgeon H, Leaf A, Lakatta E. Omega 3 polyunsaturated fatty acid modulates dihydropyridine effects on L-type Ca2+ channels, cytosolic, Ca2+, and contraction in adult rat cardiac myocytes. Proc Natl Acad Sci U S A. 1994;91:8832–8836. doi: 10.1073/pnas.91.19.8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Perusek L, Maeda T. Vitamin A derivatives as treatment options for retinal degenerative diseases. Nutrients. 2013;5:2646–2666. doi: 10.3390/nu5072646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pham TL, He J, Kakazu A, Jun B, Bazan NG, Bazan HEP. Defining a mechanistic link between pigment epithelium-derived factor, docosahexaenoic acid and corneal nerve regeneration. J Biol Chem. 2017;292:18486–18499. doi: 10.1074/jbc.M117.801472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Polato F, Becerra SP. Pigment epithelium-derived factor a protective factor for photoreceptors in vivo. Adv Exp Med Biol. 2016;854:699–706. doi: 10.1007/978-3-319-17121-0_93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- 74.Power M, Das S, Schütze K, Marigo V, Ekström P, Paquet-Durand F. Cellular mechanisms of hereditary photoreceptor degeneration – Focus on cGMP. Prog Retin Eye Res. 2019 doi: 10.1016/j.preteyeres.2019.07.005. doi: 101016/jpreteyeres201907005. [DOI] [PubMed] [Google Scholar]
- 75.Ravulapalli R, Campbell RL, Gauthier SY, Dhe-Paganon S, Davies PL. Distinguishing between calpain heterodimerization and homodimerization. FEBS J. 2009;276:973–982. doi: 10.1111/j.1742-4658.2008.06833.x. [DOI] [PubMed] [Google Scholar]
- 76.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sahel JA, Léveillard T. Maintaining cone function in rod-cone dystrophies. Adv Exp Med Biol. 2018;1074:499–509. doi: 10.1007/978-3-319-75402-4_62. [DOI] [PubMed] [Google Scholar]
- 78.Sakami S, Maeda T, Bereta G, Okano K, Golczak M, Sumaroka A, Roman AJ, Cideciyan A V, Jacobson SG, Palczewski K. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem. 2011;286:10551–10567. doi: 10.1074/jbc.M110.209759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanges D, Comitato A, Tammaro R, Marigo V. Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc Natl Acad Sci U S A. 2006;103:17366–17371. doi: 10.1073/pnas.0606276103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scholl HP, Moore AT, Koenekoop RK, Wen Y, Fishman GA, van den Born LI, Bittner A, Bowles K, Fletcher EC, Collison FT, Dagnelie G, Degli Eposti S, Michaelides M, Saperstein DA, Schuchard RA, Barnes C, Zein W, Zobor D, Birch DG, Mendola JD. Safety and proof-of-concept study of oral QLT091001 in retinitis pigmentosa due to inherited deficiencies of retinal pigment epithelial 65 protein (RPE65) or kecithin:retinol acyltransferase (LRAT) PLoS One. 2015;10:e0143846. doi: 10.1371/journal.pone.0143846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schwartz SD, Regillo CD, Lam BL, Eliott D, Rosenfeld PJ, Gregori NZ, Hubschman JP, Davis JL, Heilwell G, Spirn M, Maguire J, Gay R, Bateman J, Ostrick RM, Morris D, Vincent M, Anglade E, Del Priore LV, Lanza R. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet. 2015;385:509–516. doi: 10.1016/S0140-6736(14)61376-3. [DOI] [PubMed] [Google Scholar]
- 82.Shinde V, Kotla P, Strang C, Gorbatyuk M. Unfolded protein response-induced dysregulation of calcium homeostasis promotes retinal degeneration in rat models of autosomal dominant retinitis pigmentosa. Cell Death Dis. 2016;7:e2085. doi: 10.1038/cddis.2015.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sieving PA, Caruso RC, Tao W, Coleman HR, Thompson DJS, Fullmer KR, Bush RA. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: Phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci U S A. 2006;103:3896–3901. doi: 10.1073/pnas.0600236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stefanov A, Novelli E, Strettoi E. Inner retinal preservation in the photoinducible I307N rhodopsin mutant mouse, a model of autosomal dominant retinitis pigmentosa. J Comp Neurol. 2019 doi: 10.1002/cne.24838. doi: 101002/cne24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Subramanian P, Locatelli-Hoops S, Kenealey J, Desjardin J, Notari L, Becerra SP. Pigment Epithelium-derived Factor (PEDF) Prevents Retinal Cell Death via PEDF Receptor (PEDF-R): identification of a functional ligand binding site. J Biol Chem. 2013;288:23928–23942. doi: 10.1074/jbc.M113.487884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Trapani I, Auricchio A. Has retinal gene therapy come of age? From bench to bedside and back to bench. Hum Mol Genet. 2019;28:R108–118. doi: 10.1093/hmg/ddz130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tropepe V, Coles BL, Chiasson BJ, Horsford DJ, Elia AJ, McInnes RR, van der Kooy D. Retinal stem cells in the adult mammalian eye. Science. 2000;287:2032–2036. doi: 10.1126/science.287.5460.2032. [DOI] [PubMed] [Google Scholar]
- 88.Tucker BA, Mullins RF, Stone EM. Stem cells for investigation and treatment of inherited retinal disease. Hum Mol Genet. 2014;23:R9–R16. doi: 10.1093/hmg/ddu124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, Hoyng CB, Roepman R, Klevering BJ. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–186. doi: 10.1016/j.preteyeres.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 90.Vighi E, Trifunović D, Veiga-Crespo P, Rentsch A, Hoffmann D, Sahaboglu A, Strasser T, Kulkarni M, Bertolotti E, van den Heuvel A, Peters T, Reijerkerk A, Euler T, Ueffing M, Schwede F, Genieser HG, Gaillard P, Marigo V, Ekström P, Paquet-Durand F. Combination of cGMP analogue and drug delivery system provides functional protection in hereditary retinal degeneration. Proc Natl Acad Sci U S A. 2018;115:E2997–3006. doi: 10.1073/pnas.1718792115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wadhawan A, Chatterjee M, Singh G. Present scenario of bioconjugates in cancer therapy: A review. Int J Mol Sci. 2019;20:5243. doi: 10.3390/ijms20215243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Y, Subramanian P, Shen D, Tuo J, Becerra SP, Chan CC. Pigment epithelium-derived factor reduces apoptosis and pro-inflammatory cytokine gene expression in a murine model of focal retinal degeneration. ASN Neuro. 2013;5:309–319. doi: 10.1042/AN20130028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wubben TJ, Zacks DN, Besirli CG. Retinal neuroprotection: current strategies and future directions. Curr Opin Ophthalmol. 2019;30:199–205. doi: 10.1097/ICU.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 94.Yamashima T, Oikawa S. The role of lysosomal rupture in neuronal death. Prog Neurobiol. 2009;89:343–358. doi: 10.1016/j.pneurobio.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 95.Zeiss CJ, Neal J, Johnson EA. Caspase-3 in postnatal retinal development and degeneration. Invest Ophthalmol Vis Sci. 2004;45:964–970. doi: 10.1167/iovs.03-0439. [DOI] [PubMed] [Google Scholar]