

ABSTRACT
Recent studies revealed that the histone demethylase KDM2B regulates the epithelial markers E-Cadherin and ZO-1, the RhoA/B/C-small-GTPases and actin cytoskeleton organization, in DU-145 prostate- and HCT-116 colon-tumor cells. Here we addressed the role of KDM2B in the activation of Focal Adhesion Kinase (FAK)-signaling and its involvement in regulating tumor cell motility. We used RT-PCR for gene transcriptional analysis, Western blotting for the assessment of protein expression and activity and wound-healing assay for the study of cell migration. KDM2B overexpression or silencing controls the activity of FAK in DU-145 prostate- and HCT-116 colon-tumor cells without affecting gene transcription and protein expression of this kinase. Upon KDM2B overexpression in DU-145 cells, significantly enhanced migration was observed, which was abolished in cells pretreated by the specific phosphoinositide-3 kinase (PI3 K) inhibitor LY294002, implying involvement of FAK/PI3 K signaling in the migration process. In line with this, the p85-PI3 K-subunit was downregulated upon knockdown of KDM2B in DU-145 cells, while the opposite effect became evident in KDM2B-overexpressing cells. These results revealed a novel functional role of KDM2B in regulating the activation of the FAK/PI3 K signaling in prostate cancer cells that participates in the control of cell motility.
KEYWORDS: KDM2B, FAK, PI3K, migration, prostate cancer
Introduction
The epigenetic factor KDM2B is a novel oncogene that functions as a lysine-specific demethylase.1-4 The oncogenic function of KDM2B is based on its co-operation with Polycomb Repressor Complex-2 (PRC-2) that promotes the immortalization of primary mouse embryonic fibroblasts (MEFs).3,5 The basic fibroblast growth factor (bFGF) regulates KDM2B expression in various human tumors,6 governing several tumor features including migration, invasiveness, angiogenesis and actin cytoskeleton organization.7 Specifically, KDM2B was previously shown to control cell migration in HUVECs,6 ovarian cancer cells,8 cervical cancer cells,9 and DU-145 prostate cancer cells,10 while it regulated angiogenesis in HUVECs.6 In DU-145 prostate cancer cells, the regulation of cell migration by KDM2B involved actin cytoskeleton reorganization, Rho GTPase signaling and the cell-cell adhesion molecules E-cadherin and ZO-1.10 Similarly, in HCT-116 colon tumor cells, KDM2B activated actin-based signaling, including Rho-GTPases and Rac1, as well as the EMT markers E-cadherin, ZO-1, and N-cadherin.11 Since the growth and motility of various tumor cells are regulated by signaling cascades, including the FAK/PI3 K/Rac1 pathway,12-15 we addressed in the present study the possible involvement of this signaling pathway in the KDM2B actions in tumor cells and its impact on cell migration. We provide strong evidence that KDM2B activates FAK and PI3 K that control the migration potential of prostate cancer cells.
Materials and methods
RPMI 1640 medium, RPMI w/o phenol red Opti-Mem I medium, penicillin/streptomycin for cell culture, Trypsin, puromycin, SuperScript II Reverse Transcriptase, Trizol reagent for RNA extraction and dNTPs were purchased from Invitrogen/Life Technologies (Carlsbad, CA, USA). Fetal Bovine Serum (FBS) was purchased from Bioline (Bioline Reagents Limited, London, UK). Primers for RT-PCR were purchased from Eurofins MWG Operon (Ebersberg, Germany). p85PI3 K, pFAK and AKT antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). PI3 K antibody was purchased from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA). FAK antibody was purchased from BD Transduction Laboratories, Biosciences (USA). KAPA SYBR Green/ROX qPCR Master Mix was purchased from Fermentas (Maryland, USA). Anti-KDM2B antibody, Goat anti-rabbit IgG Peroxidase Conjugated, Goat anti-mouse IgG Peroxidase Conjugated, and ECL Immobilon western-Chemiluminescent HRP Substrate were purchased from Millipore Corporation (Billerica, MA, USA). Albumin Fraction V (BSA) and Dimethyl Sulfoxide DMSO (for cell culture) were purchased from PanReac Applichem, ITW Reagents (Gatersleben, Germany). LY294002 hydrochloride was purchased from Sigma-Aldrich and FAK inhibitor II (Cat#324878) was purchased from CalbioChemM. All other chemicals were obtained from usual commercial sources at the purest grade available.
Cell cultures, lentiviral and retroviral packaging, and transduction
The cell lines DU-145 and HCT-116 were cultured as previously described.10 Lentiviral and retroviral packaging and transduction in both cell lines were performed as described previously.10 The wounded area was photographed in specific time points (t0 and t24) using a LEICA inverted microscope (LEICA DMIRE2) with a 10× objective lens and was photographed with a LEICA DC 300 F digital camera. Wound width was measured using Image J Analysis Software and expressed as a percentage of the initial wound width.
Protein extraction and western blotting
Protein extraction and Western blotting of lentiviral or retroviral transduced cells were performed as previously described.10 Abs used were anti-tubulin (1:20, clone 1A2; Sigma-Aldrich), anti-FAK (1:1000, F15020, BD Transduction Laboratories, Biosciences), anti-PI3 Kp85 (1:1000, #4228, Cell Signaling Technology), anti-pAKT (1:1000, #9271, Cell Signaling Technology), anti-PI3 K (1:100, sc-377064, Santa Cruz Biotechnology), anti-pFAK (1:1000, #8556, Cell Signaling Technology), anti-JHDM1B (KDM2B) (1:1000. 09–864, Millipore). Anti-mouse and anti-rabbit horseradish peroxidase-conjugated secondary antibodies were obtained from Millipore Corporation (1:10000, Billerica, MA, USA).
RNA isolation, cDNA synthesis, and real-time PCR
cDNA synthesis and real-time PCR were performed as described previously.10 The following primers were used (5ʹ-3ʹ orientation):
Actin, forward CGGCATCGTCACCAACTG;
Actin, reverse GGCACACGCAGCTCATTG;
FAK, forward TGGGCGGAAAGAAATCCTGC;
FAK, reverse GGCTTGACACCCTCGTTGTA;
PI3 K, forward CCCGATGCGGTTAGAGCC;
PI3 K, reverse TGCCGATAGCAAAACCAATTTC;
KDM2B, forward TCTACGAGATCGAGGACAGGA;
KDM2B reverse ACCAGCACATCTCATAGTAGAAGG.
Nondirectional migration: wound-healing assay
Nondirectional migration assay was performed as described previously.10 The cells were cultured in the presence or absence of PI3 K inhibitor, LY294002.
MTT proliferation assay
DU-145 cells were grown in microplates (96 wells, flat bottom), 5000 cells per well, in a final volume of 100 μl culture medium per well, according to the media needs of the cells (RPMI 1640, RPMI w/o phenol red Opti-Mem I medium supplemented with 1% penicillin/streptomycin and 10% heat-inactivated fetal bovine serum), in a humidified atmosphere containing 5% CO2 at 37°C. The incubation period of the cell cultures was from 24 to 72 h. After the preferred incubation period, 10 μl of the MTT reagent (final concentration 0.5 mg/ml) was added to each well, in a final volume of 100 μl culture RPMI w/o phenol red Opti-Mem I medium per well. The microplate was incubated for 4 h in a humidified atmosphere. Before measurement, the MTT-medium solution was aspirated and 100 μl of DMSO was added in each well. The absorbance of the samples was estimated using a microplate (ELISA) reader. The wavelength to measure the absorbance of the formazan product was 550 nm and the reference wavelength was 655 nm. Each cell clone was done in fifth-plicate and at least 3 times.
Replicates and statistical analysis
All experiments were performed in three independent replications. The data are presented as means ± standard deviation (SD). All data were tested for significance using unpaired Student t-test. Only results with P values of 0.05 or less were considered statistically significant [n.s non statistical significance, *(p ≤ 0.05), **(p ≤ 0.01), ***(p ≤ 0.001)].
Results
We first analyzed the role of KDM2B in regulating expression and activity of FAK in DU-145 prostate- and HCT-116 colon-tumor cells. FAK gene transcription remained unaffected upon knockdown or overexpression of KDM2B in DU-145 cells (Figure 1(a)). In line with this result and in accordance with our previous observation10 the corresponding FAK-protein expression levels were also not affected (Figure 1(b,d)). Interestingly however, expression of phospho-FAK (pFAK), i.e. the active form of this kinase, was significantly reduced upon knockdown of KDM2B, while it was strongly enhanced in DU-145 cells overexpressing KDM2B (Figure 1(c,e)). These findings imply that KDM2B controls the activity of FAK without affecting expression levels of total protein. We obtained very similar results in HCT-116 colon tumor cells (Figure 2). Indeed, FAK-protein expression remained unaffected in HCT-116 cells (Figure 2(a,c)). On the other hand, pFAK expression was drastically reduced upon knockdown of KDM2B, while overexpression of the demethylase enhanced pFAK expression levels (Figure 2(b,d)). Thus, we concluded that KDM2B controls the activity of the signaling kinase FAK in various tumors.
Figure 1.
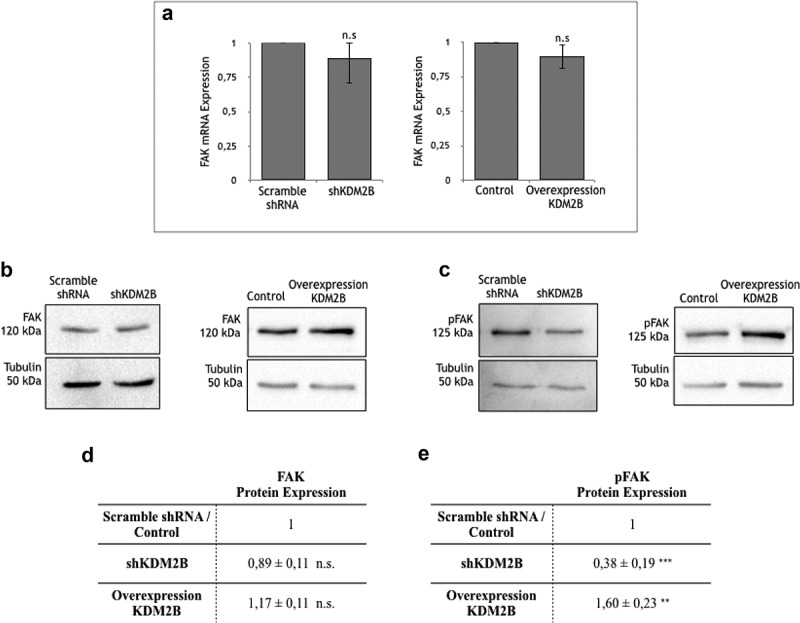
(a) RT-PCR analysis of FAK RNA extracted from DU-145 cell clone lysates. Values are normalized to β-actin and are expressed relative to those of controls (scramble shRNA and Control), which are arbitrarily settled to 1. Each value is the mean ± SD from n = 3 independent experiments, while y-axis represents the ratio between FAK and actin genes. n.s indicates nonstatistical significance.
Detection of protein expression via western blotting. Western Blot analysis of (b) FAK and (c) pFAK protein expression in DU-145 cell clone lysates. Statistical data of (d) FAK and (e) pFAK protein expression levels in DU-145 cell clone lysates. Each value is the mean ± SD from n = 4 experiments. Data are normalized using tubulin as immunoblot loading control. n.s. indicates nonstatistical significance, **(p ≤ 0.01), ***(p ≤ 0.001) indicates statistical significance.
Figure 2.
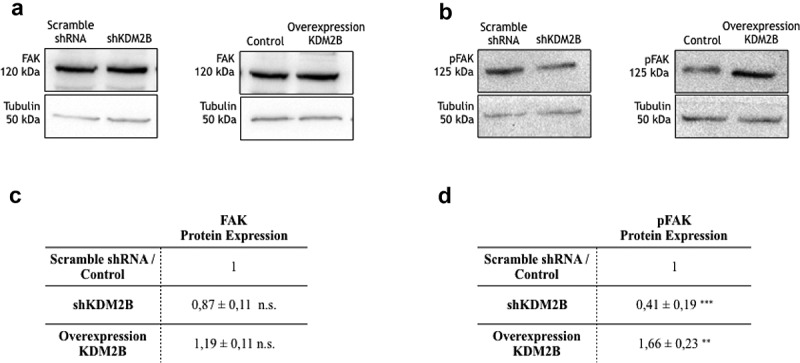
Detection of protein expression via western blotting. Western Blot analysis of (a) FAK and (b) pFAK protein expression in HCT-116 cell clone lysates. Statistical data of (c) FAK and (d) pFAK protein expression levels in HCT-116 cell clone lysates. Each value is the mean ± SD from n = 4 experiments. Data are normalized using tubulin as an immunoblot loading control. n.s. indicates nonstatistical significance, **(p ≤ 0.01), ***(p ≤ 0.001) indicates statistical significance.
Various reports established previously that FAK signaling may activate PI3 K and the downstream effectors Rac1/Cdc42 (that targets actin dynamics) and the pro-survival kinases AKT/SGK1, representing a crucial signaling pathway regulating actin cytoskeleton architecture,16-18 cell growth19 and migration.20,21 Accordingly, we further addressed the possible involvement of this kinase-signaling cascade in the reported KDM2B effects on the migration potential of tumor cells.6,8-10 We focused on DU-145 prostate cancer cells, since KDM2B does not influence the migration potential in HCT-116 cells due to its low expression levels as reported previously.11 Initial experiments using the FAK inhibitor II failed to provide clear results (data not shown), most probably due to the well-established toxicity of those molecules.22 Thus, we used the specific inhibitor LY294002 targeting PI3 K. As shown in Figure 3(a,c), the enhancement of cell migration observed in KDM2B overexpressing cells was virtually abrogated when cells were pretreated with the PI3 K inhibitor LY294002, while in KDM2B-knockdown cells a significant reduction in cell migration was observed; similarly, to what observed in the presence of the PI3 K inhibitor LY294002, suggesting that KDM2B silencing recapitulates the effect of PI3 K inhibition (Figure 3(b,d)). Interestingly, the addition of LY294002 to KDM2B-knockdown cells determined a further decrease of cell migration, suggesting that LY294002 inhibited the residual PI3 K activity in cells upon KDM2B knockdown.
Figure 3.
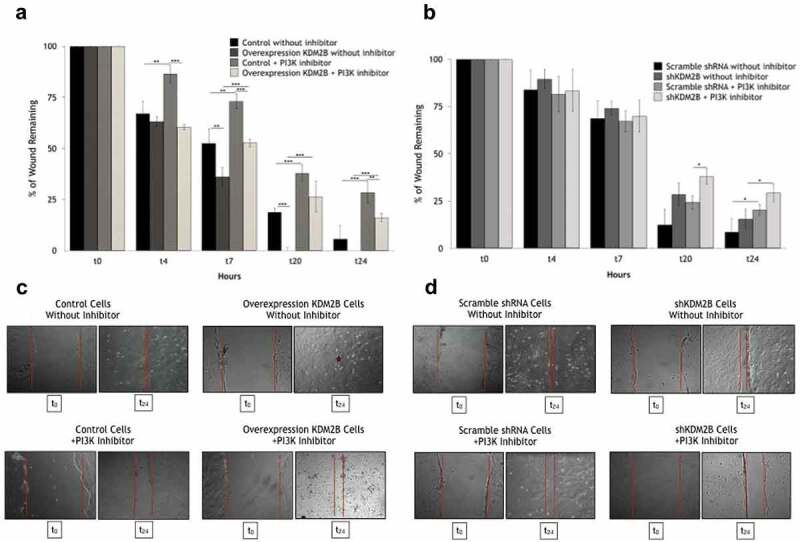
In vitro scratch assay. At time = 0 h confluent untreated and pretreated with PI3 K inhibitor LY294002 DU-145 cell clones were scratched (100% area remaining). Wound area, at the indicated time points, was expressed as a percentage of the initial wound area. The cell migration capability in DU-145 untreated cell clones is obviously higher than pre-treated with the PI3 K inhibitor LY294002 cell clones. (c) (d) Microscopic monitoring of wound closure at the indicated times (t0, t24 hours). Arithmetic means ± SD, from n = 3 independent experiments, of the percentage migrated DU-145 cells in the absence and presence of LY294002; *(p ≤ 0.05), **(p ≤ 0.01), ***(p ≤ 0.001) indicates statistical significance.
These results suggest that the FAK/PI3 K signaling is implicated in cell motility of DU-145 prostate cancer cells. To further confirm this hypothesis, we addressed the role of KDM2B on PI3 K activity profiles in those cells. In line with the observations for FAK, knockdown, or overexpression of KDM2B did not significantly affect gene transcription (Figure 4(a)) and protein expression of PI3 K (Figure 4(b,d)). However, activity of this FAK-downstream acting kinase was significantly reduced upon knockdown of KDM2B, as indicated by the significant decrease of the expression level of p85 subunit of PI3 K. On the other hand, overexpression of KDM2B enhanced the p85 subunit, clearly implying activation of PI3 K (Figure 4(c,e)). As already indicated for FAK activation, these findings are in line with previous observations in DU-145 cells.10
Figure 4.
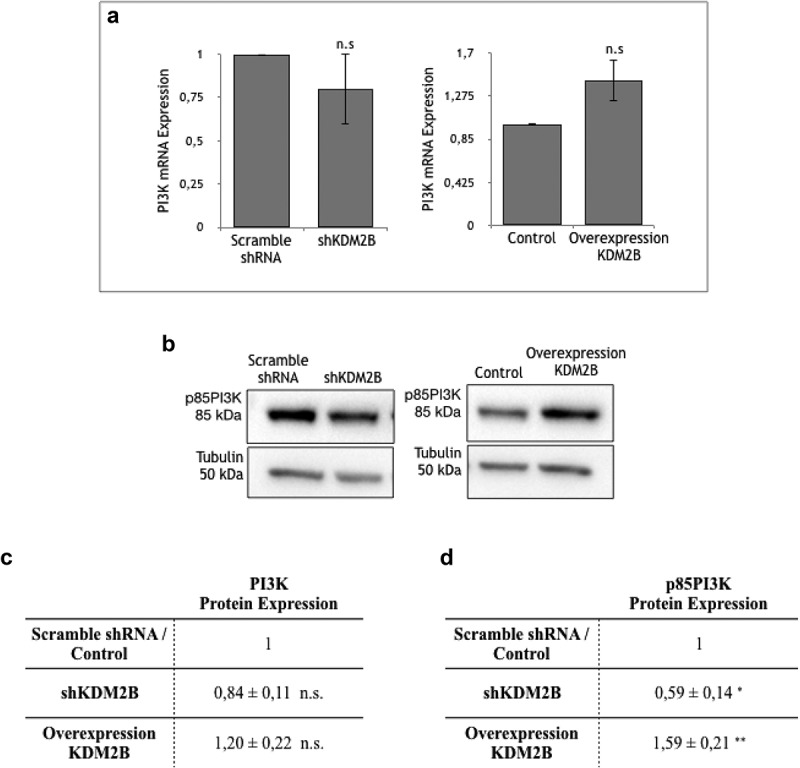
(a) RT-PCR analysis of PI3 K RNA extracted from DU-145 cell clone lysates. Values are normalized to β-actin and are expressed relative to those of control (Scramble shRNA and Control), which are arbitrarily settled to 1. Each value is the mean ± SD from n = 3 independent experiments, while y-axis represents the ratio between PI3 K and actin genes. n.s indicates nonstatistical significance.
Detection of protein expression via western blotting. Western Blot analysis of (b) PI-3 K and (c) p85PI3 K protein expression in DU-145 cell clone lysates. Statistical data of (d) PI3 K and (e) p85PI3 K protein expression levels in DU-145 cell clone lysates. Each value is the mean ± SD from n = 4 experiments. Data are normalized using tubulin as immunoblot loading control. n.s. indicates nonstatistical significance, *(p ≤ 0.05), **(p ≤ 0.01) indicates statistical significance.
We finally addressed the role of KDM2B in regulating additional downstream effectors of FAK/PI3 K signaling, focusing on the pro-survival kinase AKT. Interestingly, phosphorylated Akt expression profiles in KDM2B-overexpressing or KDM2B-knockdown DU-145 cells showed only moderate, statistically non-significant changes (Figure 5(a,b)). In addition, MTT assay, a colorimetric assay designed to assess cell metabolic activity, revealed only a tendency to moderate changes in short-term proliferation profiles (up to 72 h) not reaching statistical significance in KDM2B-overexpressing or KDM2B-knockdown DU-145 cells (Figure 5(c)).
Figure 5.
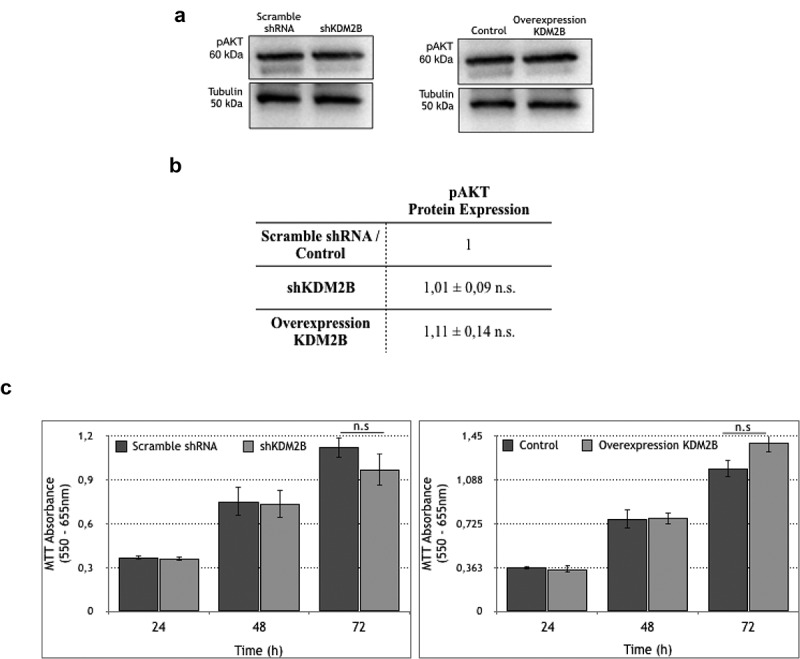
Western Blot analysis of pAKT protein expression in DU-145 cell clone lysates. Statistical data of (b) pAKT protein expression levels in DU-145 cell clone lysates. Each value is the mean ± SD from n = 3 experiments. Data are normalized using tubulin as an immunoblot loading control. n.s. indicates nonstatistical significance. (c) Proliferation rate of DU-145 cells upon knockdown and overexpression of KDM2B. The graphs represent the MTT absorbance in specific time points in scramble shRNA, shKDM2B (left panel) and control overexpression, overexpression of KDM2B (right panel) DU-145 cells; n.s indicates nonstatistical significance.
Discussion
The present study reveals for the first time a significant role of the epigenetic factor KDM2B in regulating FAK/PI3 K signaling. We provide clear evidence that KDM2B activates via phosphorylation both FAK and PI3 K kinases, without affecting protein expression or transcription of the corresponding genes. This is an interesting finding suggesting that this histone demethylase may trigger transcriptional activation of factors that promote the activity of signaling effectors, as this was reported for chromatin-remodeling SWI/SNF complexes,23 or natural histone acetyltransferase inhibitors.24 Moreover, activation of this signaling pathway contributes to the KDM2B stimulation of cell migration, since inhibition of FAK/PI3 K signaling blocks the KDM2B effects. These findings support previous reports suggesting that epigenetic mechanisms regulate cellular signaling pathways in cancer cells10,11,25 that may involve actin cytoskeleton restructuring genes10 or EMT11 and, thus, are crucial mediators conferring cancer cells with migratory and invasive characteristics.17,26-31 Further experiments are now required to establish whether the KDM2B-initiated activation step targets initially FAK phosphorylation, followed by activation of the downstream acting PI3 K,16,21,32 or whether the opposite activation cascade may as well be implicated.33
Regulation of cell migration in tumors is highly intricate and involves the cross talk of various signaling pathways and effectors.34-40 FAK signaling represents one major pathway. Indeed, activated FAK interacts with Src kinases and this complex triggers through phosphorylation multiple downstream signaling cascades that govern different cellular responses including migration.36 The FAK/PI3 K signaling and the downstream acting effectors Rac1/Cdc42, or the Src-Rho-ROCK signaling, are crucial actors in governing actin redistribution and cell motility in various tumors.16,21,41-44 Our findings showing the interaction of KDM2B with this pathway manifested by the activation of FAK and downstream acting effectors such as PI3 K [this study] that contributes to cell motility control, or Rac1,11 suggest that FAK-signaling is involved in the observed regulation of cell migration by KDM2B in tumors. On the other hand, our previous studies established that additional signaling pathways, such as the Rho/ROCK signaling and actin redistribution that are known to control cell movement35,37,45,46 participate in KDM2B-induced regulation of cell motility.10 Taken together, our results indicate that KDM2B may trigger various pathways, and use in parallel differential signaling effectors, to control cell migration in tumors. Additional studies are now needed to investigate the coordination of those parallel mechanisms. Moreover, further analysis should elucidate whether KDM2B elicits additional signaling machineries that may as well control motility, such as the Ca2+-related Orai1/STIM1 signaling cascade.40,47,48
Our present results showed that the KDM2B-governed activation of the FAK/PI3 K signaling did not significantly alter the phosphorylation profile of AKT in DU-145 cells. Moreover, in these cells, KDM2B knockdown or overexpression failed to significantly affect cell proliferation. Even though this observation is consistent with the observed Akt-unaffected activation profile, it is rather an unexpected finding that needs further analysis. Since previous studies reported KDM2B-governed regulation of cell growth in various cell models (including Lung carcinomas, T-cell lymphomas, gastric tumor cells, as well as in mouse embryonic fibroblasts),6,49,50 our present observations indicate that KDM2B either acts differentially in prostate cancer cells, controlling migration rather than cell proliferation, or influences cell growth only in a long-term manner. The molecular basis of these observations deserves further intensive examination, including analysis in various tumors that is now in progress in our laboratory.
To conclude, our findings uncover novel molecular targets, namely FAK and PI3 K, which are activated by the epigenetic factor KDM2B in prostate (and colon) cancer cells. They further establish a crucial participation of the FAK/PI3 K signaling pathway in regulating cell migration in prostate cancer cells. Moreover, since KDM2B targets both Rho-GTPases signaling and actin cytoskeleton organization10 that similarly contribute to the control of cell motility, we assume that this histone demethylase may govern tumor cell migration through concomitant coordination of distinct signaling flows.
Funding Statement
Authors extend their appreciation to the International Scientific Partnership Program ISPP at the King Saud University for funding this work through ISPP#009.
Data availability statement
Our data are available.
References
- 1.Kampranis SC, Tsichlis PN.. Histone demethylases and cancer. Adv Cancer Res. 2009;102:103–169. doi: 10.1016/S0065-230X(09)02004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tzatsos A, Paskaleva P, Lymperi S, Contino G, Stoykova S, Chen Z, Wong KK, Bardeesy N. Lysine-specific demethylase 2b (kdm2b)-let-7-enhancer of zester homolog 2 (ezh2) pathway regulates cell cycle progression and senescence in primary cells. J Biol Chem. 2011;286:33061–33069. doi: 10.1074/jbc.M111.257667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tzatsos A, Pfau R, Kampranis SC, Tsichlis PN. Ndy1/kdm2b immortalizes mouse embryonic fibroblasts by repressing the ink4a/arf locus. Proc Natl Acad Sci U S A. 2009;106:2641–2646. doi: 10.1073/pnas.0813139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He J, Nguyen AT, Zhang Y. Kdm2b/jhdm1b, an h3k36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood. 2011;117:3869–3880. doi: 10.1182/blood-2010-10-312736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kottakis F, Foltopoulou P, Sanidas I, Keller P, Wronski A, Dake BT, Ezell SA, Shen Z, Naber SP, Hinds PW, et al. Ndy1/kdm2b functions as a master regulator of polycomb complexes and controls self-renewal of breast cancer stem cells. Cancer Res. 2014;74:3935–3946. doi: 10.1158/0008-5472.CAN-13-2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN. Fgf-2 regulates cell proliferation, migration, and angiogenesis through an ndy1/kdm2b-mir-101-ezh2 pathway. Mol Cell. 2011;43:285–298. doi: 10.1016/j.molcel.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fajol A, Honisch S, Zhang B, Schmidt S, Alkahtani S, Alarifi S, Lang F, Stournaras C, Foller M. Fibroblast growth factor (fgf) 23 gene transcription depends on actin cytoskeleton reorganization. FEBS Lett. 2016;590:705–715. doi: 10.1002/1873-3468.12096. [DOI] [PubMed] [Google Scholar]
- 8.Kuang Y, Lu F, Guo J, Xu H, Wang Q, Xu C, Zeng L, Yi S. Histone demethylase kdm2b upregulates histone methyltransferase ezh2 expression and contributes to the progression of ovarian cancer in vitro and in vivo. Onco Targets Ther. 2017;10:3131–3144. doi: 10.2147/OTT.S134784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peta E, Sinigaglia A, Masi G, Di Camillo B, Grassi A, Trevisan M, Messa L, Loregian A, Manfrin E, Brunelli M, et al. Hpv16 e6 and e7 upregulate the histone lysine demethylase kdm2b through the c-myc/mir-146a-5p axys. Oncogene. 2018;37:1654–1668. doi: 10.1038/s41388-017-0083-1. [DOI] [PubMed] [Google Scholar]
- 10.Zacharopoulou N, Tsapara A, Kallergi G, Schmid E, Tsichlis PN, Kampranis SC, Stournaras C. The epigenetic factor kdm2b regulates cell adhesion, small rho gtpases, actin cytoskeleton and migration in prostate cancer cells. Biochim Biophys Acta Mol Cell Res. 2018;1865:587–597. doi: 10.1016/j.bbamcr.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Zacharopoulou N, Tsapara A, Kallergi G, Schmid E, Alkahtani S, Alarifi S, Tsichlis PN, Kampranis SC, Stournaras C. The epigenetic factor kdm2b regulates emt and small GTPases in colon tumor cells. Cell Physiol Biochem. 2018;47:368–377. doi: 10.1159/000489917. [DOI] [PubMed] [Google Scholar]
- 12.Schwock J, Dhani N, Hedley DW. Targeting focal adhesion kinase signaling in tumor growth and metastasis. Expert Opin Ther Targets. 2010;14:77–94. doi: 10.1517/14728220903460340. [DOI] [PubMed] [Google Scholar]
- 13.Lee BY, Timpson P, Horvath LG, Daly RJ. Fak signaling in human cancer as a target for therapeutics. Pharmacol Ther. 2015;146:132–149. doi: 10.1016/j.pharmthera.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Liu G, Honisch S, Liu G, Schmidt S, Pantelakos S, Alkahtani S, Toulany M, Lang F, Stournaras C. Inhibition of sgk1 enhances mar-induced apoptosis in mcf-7 breast cancer cells. Cancer Biol Ther. 2015;16:52–59. doi: 10.4161/15384047.2014.986982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao XK, Cheng Y, Liang Cheng M, Yu L, Mu M, Li H, Liu Y, Zhang B, Yao Y, Guo H, et al. Focal adhesion kinase regulates fibroblast migration via integrin beta-1 and plays a central role in fibrosis. Sci Rep. 2016;6:19276. doi: 10.1038/srep19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papakonstanti EA, Kampa M, Castanas E, Stournaras C. A rapid, nongenomic, signaling pathway regulates the actin reorganization induced by activation of membrane testosterone receptors. Mol Endocrinol. 2003;17:870–881. doi: 10.1210/me.2002-0253. [DOI] [PubMed] [Google Scholar]
- 17.Papakonstanti EA, Stournaras C. Cell responses regulated by early reorganization of actin cytoskeleton. FEBS Lett. 2008;582:2120–2127. doi: 10.1016/j.febslet.2008.02.064. [DOI] [PubMed] [Google Scholar]
- 18.Gu S, Kounenidakis M, Schmidt EM, Deshpande D, Alkahtani S, Alarifi S, Foller M, Alevizopoulos K, Lang F, Stournaras C. Rapid activation of fak/mtor/p70s6k/pak1-signaling controls the early testosterone-induced actin reorganization in colon cancer cells. Cell Signal. 2013;25:66–73. doi: 10.1016/j.cellsig.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Panera N, Crudele A, Romito I, Gnani D, Alisi A. Focal adhesion kinase: insight into molecular roles and functions in hepatocellular carcinoma. Int J Mol Sci. 2017;18:99. doi: 10.3390/ijms18010099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA. Focal adhesion kinase: A regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19:5606–5613. doi: 10.1038/sj.onc.1203877. [DOI] [PubMed] [Google Scholar]
- 21.Kallergi G, Agelaki S, Markomanolaki H, Georgoulias V, Stournaras C. Activation of fak/pi3k/rac1 signaling controls actin reorganization and inhibits cell motility in human cancer cells. Cell Physiol Biochem. 2007;20:977–986. doi: 10.1159/000110458. [DOI] [PubMed] [Google Scholar]
- 22.Schultze A, Fiedler W. Therapeutic potential and limitations of new fak inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2010;19:777–788. doi: 10.1517/13543784.2010.489548. [DOI] [PubMed] [Google Scholar]
- 23.Sethuraman A, Brown M, Seagroves TN, Wu ZH, Pfeffer LM, Fan M. Smarce1 regulates metastatic potential of breast cancer cells through the hif1a/ptk2 pathway. Breast Cancer Res. 2016;18:81. doi: 10.1186/s13058-016-0738-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Y, He L, Zhang L, Chen J, Yi Z, Zhang J, Liu M, Pang X. Anacardic acid (6-pentadecylsalicylic acid) inhibits tumor angiogenesis by targeting src/fak/rho GTPases signaling pathway. J Pharmacol Exp Ther. 2011;339:403–411. doi: 10.1124/jpet.111.181891. [DOI] [PubMed] [Google Scholar]
- 25.Gari HH, DeGala GD, Ray R, Lucia MS, Lambert JR. Prl-3 engages the focal adhesion pathway in triple-negative breast cancer cells to alter actin structure and substrate adhesion properties critical for cell migration and invasion. Cancer Lett. 2016;380:505–512. doi: 10.1016/j.canlet.2016.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katsantonis J, Tosca A, Koukouritaki SB, Theodoropoulos PA, Gravanis A, Stournaras C. Differences in the g/total actin ratio and microfilament stability between normal and malignant human keratinocytes. Cell Biochem Funct. 1994;12:267–274. doi: 10.1002/cbf.290120407. [DOI] [PubMed] [Google Scholar]
- 27.Stournaras C, Gravanis A, Margioris AN, Lang F. The actin cytoskeleton in rapid steroid hormone actions. Cytoskeleton (Hoboken). 2014;71:285–293. doi: 10.1002/cm.21172. [DOI] [PubMed] [Google Scholar]
- 28.Vignjevic D, Montagnac G. Reorganisation of the dendritic actin network during cancer cell migration and invasion. Semin Cancer Biol. 2008;18:12–22. doi: 10.1016/j.semcancer.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Insall RH, Machesky LM. Actin dynamics at the leading edge: from simple machinery to complex networks. Dev Cell. 2009;17:310–322. doi: 10.1016/j.devcel.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 30.Gu S, Papadopoulou N, Nasir O, Foller M, Alevizopoulos K, Lang F, Stournaras C. Activation of membrane androgen receptors in colon cancer inhibits the prosurvival signals akt/bad in vitro and in vivo and blocks migration via vinculin/actin signaling. Mol Med. 2011;17:48–58. doi: 10.2119/molmed.2010.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vega FM, Thomas M, Reymond N, Ridley AJ. The RHo GTPase RHoB regulates cadherin expression and epithelial cell-cell interaction. Cell Commun Signal. 2015;13:6. doi: 10.1186/s12964-015-0085-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen HC, Appeddu PA, Isoda H, Guan JL. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:26329–26334. doi: 10.1074/jbc.271.42.26329. [DOI] [PubMed] [Google Scholar]
- 33.Saito Y, Mori S, Yokote K, Kanzaki T, Saito Y, Morisaki N. Phosphatidylinositol 3-kinase activity is required for the activation process of focal adhesion kinase by platelet-derived growth factor. Biochem Biophys Res Commun. 1996;224:23–26. doi: 10.1006/bbrc.1996.0978. [DOI] [PubMed] [Google Scholar]
- 34.Koukouritaki SB, Margioris AN, Gravanis A, Hartig R, Stournaras C. Dexamethasone induces rapid actin assembly in human endometrial cells without affecting its synthesis. J Cell Biochem. 1997;65:492–500. doi: 10.1002/()1097-4644. [DOI] [PubMed] [Google Scholar]
- 35.Vardouli L, Vasilaki E, Papadimitriou E, Kardassis D, Stournaras C. A novel mechanism of TGFbeta-induced actin reorganization mediated by smad proteins and RHo GTPases. Febs J. 2008;275:4074–4087. doi: 10.1111/j.1742-4658.2008.06549.x. [DOI] [PubMed] [Google Scholar]
- 36.Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63:610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haga RB, Ridley AJ. Rho gtpases: regulation and roles in cancer cell biology. Small GTPases. 2016;7:207–221. doi: 10.1080/21541248.2016.1232583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Devreotes PN, Bhattacharya S, Edwards M, Iglesias PA, Lampert T, Miao Y. Excitable signal transduction networks in directed cell migration. Annu Rev Cell Dev Biol. 2017;33:103–125. doi: 10.1146/annurev-cellbio-100616-060739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kourtidis A, Lu R, Pence LJ, Anastasiadis PZ. A central role for cadherin signaling in cancer. Exp Cell Res. 2017;358:78–85. doi: 10.1016/j.yexcr.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anguita E, Villalobo A. Ca(2+) signaling and src-kinases-controlled cellular functions. Arch Biochem Biophys. 2018;650:59–74. doi: 10.1016/j.abb.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 41.Lang F, Alevizopoulos K, Stournaras C. Targeting membrane androgen receptors in tumors. Expert Opin Ther Targets. 2013;17:951–963. doi: 10.1517/14728222.2013.806491. [DOI] [PubMed] [Google Scholar]
- 42.Gu S, Honisch S, Kounenidakis M, Alkahtani S, Alarifi S, Alevizopoulos K, Stournaras C, Lang F. Membrane androgen receptor down-regulates c-src-activity and beta-catenin transcription and triggers GSK-3beta-phosphorylation in colon tumor cells. Cell Physiol Biochem. 2014;34:1402–1412. doi: 10.1159/000366346. [DOI] [PubMed] [Google Scholar]
- 43.Mondal B, Patil V, Shwetha SD, Sravani K, Hegde AS, Arivazhagan A, Santosh V, Kanduri M, Somasundaram K. Integrative functional genomic analysis identifies epigenetically regulated fibromodulin as an essential gene for glioma cell migration. Oncogene. 2017;36:71–83. doi: 10.1038/onc.2016.176. [DOI] [PubMed] [Google Scholar]
- 44.Nomikou E, Livitsanou M, Stournaras C, Kardassis D. Transcriptional and post-transcriptional regulation of the genes encoding the small GTPases RhoA, RhoB, and rhoc: implications for the pathogenesis of human diseases. Cell Mol Life Sci. 2018;75:2111–2124. doi: 10.1007/s00018-018-2787-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tseliou M, Al-Qahtani A, Alarifi S, Alkahtani SH, Stournaras C, Sourvinos G. The role of RhoA, RhoB and RhoC GTPases in cell morphology, proliferation and migration in human cytomegalovirus (hcmv) infected glioblastoma cells. Cell Physiol Biochem. 2016;38:94–109. doi: 10.1159/000438612. [DOI] [PubMed] [Google Scholar]
- 46.Lawson CD, Ridley AJ. Rho GTPase signaling complexes in cell migration and invasion. J Cell Biol. 2018;217:447–457. doi: 10.1083/jcb.201612069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt S, Schneider S, Yang W, Liu G, Schmidt EM, Schmid E, Mia S, Brucker S, Stournaras C, Wallwiener D, et al. Tgfbeta1 and sgk1-sensitive store-operated ca2+ entry and orai1 expression in endometrial ishikawa cells. Mol Hum Reprod. 2014;20:139–147. doi: 10.1093/molehr/gat066. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt S, Liu G, Liu G, Yang W, Honisch S, Pantelakos S, Stournaras C, Honig A, Lang F. Enhanced orai1 and stim1 expression as well as store operated ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget. 2014;5:4799–4810. doi: 10.18632/oncotarget.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao E, Tang C, Jiang X, Weng X, Zhong X, Zhang D, Hou J, Wang F, Huang M, Cui H. Inhibition of cell proliferation and induction of autophagy by KDM2B/FBXL10 knockdown in gastric cancer cells. Cell Signal. 2017;36:222–229. doi: 10.1016/j.cellsig.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 50.He J, Kallin EM, Tsukada Y, Zhang Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b). Nat Struct Mol Diol. 2008;15:1169–1175. doi: 10.1038/nsmb.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Our data are available.