
FIG. 1.
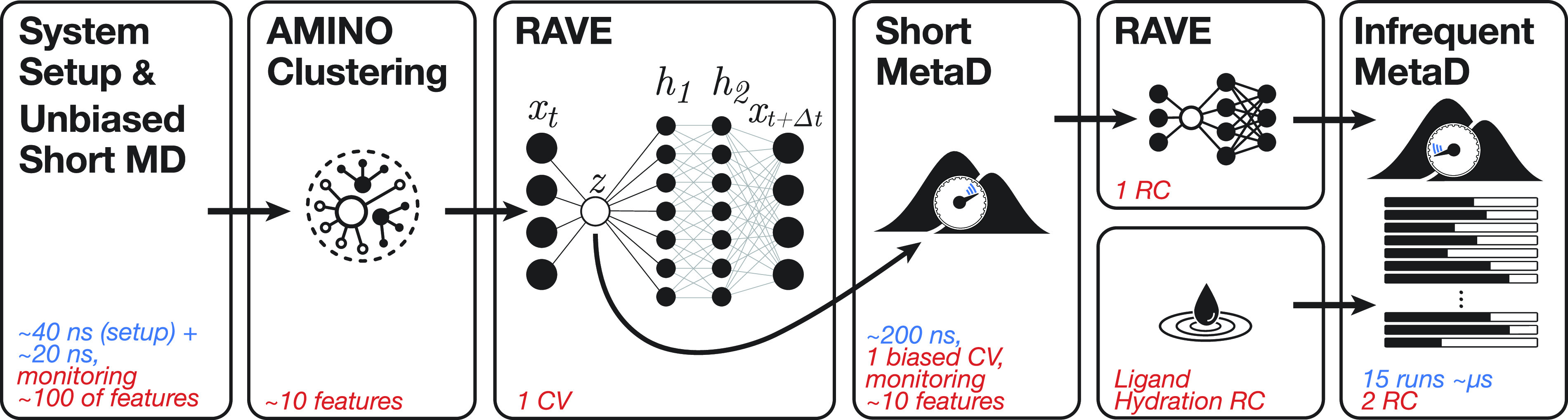
Overall schematic of the computation protocol. The first panel indicates that the protocol starts with the system setup followed by a short unbiased MD simulation in which we track the time evolution of 145 molecular features for about 20 ns. The second panel represents the use of AMINO to cluster these molecular features into 10–20 representatives that best describe ligand unbinding from the receptor. The third panel indicates that RAVE is used to learn a first 1D collective variable (CV) or trial reaction coordinate (RC) from the AMINO-derived features. The fourth panel indicates that a metadynamics simulation is run with bias deposited along the first RAVE-based CV while tracking the time evolution of the AMINO-derived features. In the top half of the fifth panel, the final RAVE-based RC is generated using the metadynamics data collected during training, while in the bottom half the ligand’s hydration state is defined as another RC. The final panel indicates the production-grade infrequent metadynamics simulations that were run by biasing the potential along these two RCs.