

Abstract
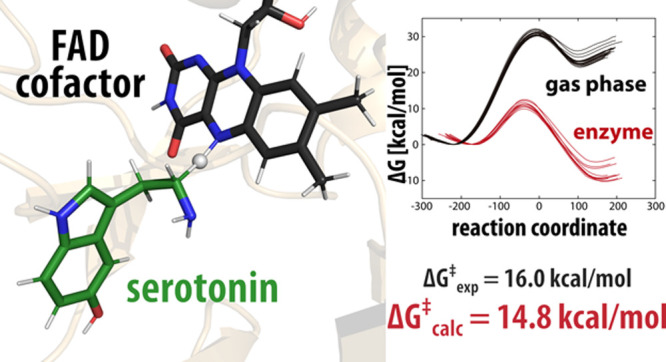
The enzyme-catalyzed degradation of the biogenic amine serotonin is an essential regulatory mechanism of its level in the human organism. In particular, monoamine oxidase A (MAO A) is an important flavoenzyme involved in the metabolism of monoamine neurotransmitters. Despite extensive research efforts, neither the catalytic nor the inhibition mechanisms of MAO enzymes are currently fully understood. In this article, we present the quantum mechanics/molecular mechanics simulation of the rate-limiting step for the serotonin decomposition, which consists of hydride transfer from the serotonin methylene group to the N5 atom of the flavin moiety. Free-energy profiles of the reaction were computed by the empirical valence bond method. Apart from the enzymatic environment, the reference reaction in the gas phase was also simulated, facilitating the estimation of the catalytic effect of the enzyme. The calculated barrier for the enzyme-catalyzed reaction of 14.82 ± 0.81 kcal mol–1 is in good agreement with the experimental value of 16.0 kcal mol–1, which provides strong evidence for the validity of the proposed hydride-transfer mechanism. Together with additional experimental and computational work, the results presented herein contribute to a deeper understanding of the catalytic mechanism of MAO A and flavoenzymes in general, and in the long run, they should pave the way toward applications in neuropsychiatry.
1. Introduction
Serotonin or 5-hydroxytryptamine is an essential monoamine neurotransmitter. It is found in the gastrointestinal tract, platelets, and the central nervous system (CNS). In the gastrointestinal tract, it is involved in the regulation of intestinal movements, while its important role in platelets is to act as a vasoconstrictor and help regulate hemostasis and blood clotting. In both cases, it is released from enterochromaffin cells.1 Only 10% of serotonin is located in the CNS, where it has several functions that are only partially understood. It is involved in the regulation of mood, sleep, appetite, and cognition.2 A depletion of serotonin in the CNS is responsible for disorders such as obsessive–compulsive disorders, depression, and anxiety,3 while its role in mediating inflammation and immunity responses4 as well as its relevance for cardiac physiology and pathology has also been proposed.5 Increasing serotonin levels is an important strategy in the pharmacological treatment of depression, and typically, the levels are increased by inhibiting the serotonin transporter or by inhibiting monoamine oxidase A (MAO A), an enzyme that breaks down serotonin.6 MAOs are flavoenzymes that catalyze the oxidative deamination of biogenic amines, producing aldehyde and hydrogen peroxide. Both products are responsible for both the formation of amyloid plaques and the degradation of neuron membranes, accompanied by massive inflammation, which manifests clinically as neurodegeneration, of which Alzheimer’s and Parkinson’s disease are the most common examples.7 As such, besides increasing monoamine levels, the inhibition of MAOs also has the potential to have a neuroprotective effect.
Despite extensive research efforts in the field of MAOs, the exact catalytic steps are still difficult to determine. MAOs convert amines into the corresponding imines by transferring two electrons and two protons from the substrate to the enzyme flavin adenine dinucleotide (FAD) cofactor, which converts the latter into its reduced FADH2 form. Although this fact is widely accepted, the rate-limiting step of the mechanistic pathway is still under debate. Several computational studies addressed this issue.8−12 We have shown in a previous study that the transfer of a hydride anion in the initial step has a much lower free activation energy than alternative mechanistic proposals.13 Until recently, all mechanistic proposals shared the common assumption that MAO A and MAO B operate by the same mechanism. However, Orru et al.14 came up with an interesting proposal that MAO A works via a polar nucleophilic mechanism involving proton transfer in the rate-limiting step, while MAO B works via a hydride mechanism. Kästner and co-workers conducted a quantum mechanics/molecular mechanics (QM/MM) study of the MAO B-catalyzed decomposition of benzylamine and proposed that the transfer of two electrons and a proton in the rate-limiting step is concerted yet asynchronous, which is in accordance with the polar nucleophilic mechanism,15 although they could not provide the evidence for the initial substrate–flavin complex originally postulated to facilitate the aforementioned proton transfer.16 One is tempted to conclude that the absence of the complex formation in Kästner’s study indirectly supports the hydride-transfer mechanism we proposed.13 Akyüz and Erdem utilized QM/MM calculations at the ONIOM level, taking into account the full dimensionality of the enzyme, and confirmed the hydride mechanism for both MAOs but proposed what they called a “slightly different hydride-transfer mechanism” among the isoforms.17 Soon after, the same group conducted a computational study on a model system to convincingly demonstrate the predominant feasibility of hydride-transfer versus proton-transfer reaction,18 followed by the MAO B-catalyzed serotonin decomposition at the ONIOM M06-2X/6-31+G(d,p):PM6 level, confirming that the rate-determining step is a hybrid of hydride and proton transfer where the hydride transfer dominates over the proton transfer.19 The mechanism of the rate-limiting step of the MAO A-catalyzed serotonin decomposition, involving hydride transfer proposed by us, is shown in Figure 1.
Figure 1.

Mechanism of the rate-limiting step of MAO A-catalyzed serotonin decomposition, involving hydride transfer from the serotonin methylene group to the N5 atom of the FAD cofactor. Please note that the intermediate formally consists of two ionic species and the reaction in this respect is strongly dependent on the polar environment. It is worth to emphasize that in the following reactions, which do not represent the rate-limiting steps, the formed imine is deprotonated by FADH– and hydrolyzed, while the reduced flavin is reoxidized back to FAD.
Enzyme catalysis is the speed up of the enzymatic reaction compared to the corresponding reaction in aqueous solution.20 Warshel and co-workers clearly showed that the majority of the catalytic power of enzymes originates from the electrostatic preorganization of the active site.21,22 Recently, our group demonstrated the significant role of electrostatics in the catalytic function of the MAO A enzyme.23 Therefore, because of the long-range nature of the electrostatic interactions, it is essential to include a large part of the enzyme in the modeling of an enzymatic reaction, correctly assign the protonation states of the ionizable residues, and obtain the converged free activation energy through intensive thermal averaging. To evaluate the catalytic effect of the enzymatic environment, it is also essential to consider the reference reaction. From a practical point of view, a hierarchical treatment of the reactive system at the QM/MM level should be used, and computationally inexpensive quantum chemistry should allow for converged free-energy calculations. The Empirical Valence Bond (EVB) approach developed by Warshel is the method of choice for computational enzymology because it is computationally inexpensive and elegantly incorporates the parameters for the reference reaction.
In this article, we calculated the activation free energy for the serotonin decomposition by MAO A. The calculations were performed using the multiscale QM/MM approach, where the quantum subsystem was described by the EVB methodology in conjunction with an all-atom classical representation of the hydrated enzyme. In order to properly calibrate the EVB treatment, the thermodynamic and kinetic parameters of the reference reaction (in the aqueous solution or in the gas phase) need to be evaluated. For the amine oxidation reaction studied herein, experimental assessment of these parameters is not practical because in the solution, the reaction proceeds at vanishingly slow rates. Therefore, the parameters of the reference reaction were obtained by quantum calculations, which is standard practice. Presently, we computed the reaction energy and barrier at the M06-2X/6-31+G(d,p) level of theory in line with our previous reports.24−27
2. Materials and Methods
2.1. Gas-Phase Calculations
At the initial stage, the reference reaction in the gas phase was characterized by density functional theory calculations employing the M06-2X functional (developed by Zhao and Truhlar) that proved to be accurate for the calculation of the barriers of organic reactions.28 In conjunction with the M06-2X functional, the 6-31+G(d,p) basis set was used for geometry optimization of the reacting moiety in the state of reactants, in the transition state, and in the state of products. Because in the gas phase, the FADH– anion and the serotonin cation form an adduct immediately upon the hydride transfer, we approximated the products by selecting a structure of the FADH– group that matches the geometry of the gas-phase-optimized isolated FADH–. This approach was applied and rationalized in our previous work.24,27 All quantum computations were carried out by the Gaussian16 software package.29 The gas-phase model consisted of a serotonin molecule accompanied by lumiflavin, the latter representing the FAD prosthetic group located in the active site of MAO A. Vibrational analysis of the optimized species resulted in all real harmonic frequency values for reactants and products and one imaginary frequency value for the transition state (νimag = 1240i cm–1). Visualization of the corresponding eigenvector revealed that the imaginary mode mainly corresponds to the reactive C–H stretching motion. Using the energies of the optimized species corrected for the vibrational zero-point energy, the reaction energy and barrier were computed and subsequently used in the calibration of the EVB protocol (see below), which is standard practice.30 The calculated gas-phase barrier was ΔGgas⧧ = 30.90 kcal mol–1, and the reaction energy ΔGgas = 22.93 kcal mol–1.
2.2. Free Energy of Serotonin Deprotonation
Serotonin is a weak base with an experimental pKa value of the amino group of 10.02 in aqueous solution,31 which indicates that the vast majority of serotonin is protonated at a physiological pH of 7.4, yet our mechanistic proposal (Figure 1) requires the metabolizing amine to be unionized in order to undergo MAO degradation. Our previous research shows that protonated monoamines enter the reaction with MAO enzymes extremely slowly, and therefore, the reaction channel with protonated species does not contribute to the kinetics.13 In this respect, it is a good strategy to calculate the free energy for serotonin deprotonation and add this value to the reaction barrier calculated for neutral serotonin.24−27 In addition, based on our previous studies, we concluded that the pKa value of dopamine does not change during transfer from water to the MAO B active site.32 Because the MAO A and MAO B active sites are very similar in terms of structure and electric field,33 we can safely assume that the serotonin pKa value would also not change significantly during transfer from water to the MAO A active site.
With this in mind, the free energy for the deprotonation of a Brønsted acid with a particular pKa value to a bulk solution with a certain pH value is given as
| 1 |
where kBT is 0.59 kcal mol–1 at room temperature and the value of ln(10) is 2.303.
At a pKa value of 10.02 for serotonin and a physiological pH of 7.4, we obtain the free energy for serotonin deprotonation of 3.56 kcal mol–1. According to our assumptions, this value is the same for the enzyme-catalyzed reaction as well as for the reaction in water, while it is zero for the gas phase.
2.3. EVB Calculations
A high-resolution crystal structure of MAO A was obtained from the Protein Data Bank (accession code 2Z5X). The protein chain and FAD cofactor were retained, while the inhibitor and water molecules were removed from the crystal structure. The serotonin molecule was built and manually docked into the active site by utilizing the UCSF Chimera program package.34 Special attention was paid to the protonation states—the serotonin molecule was neutral and the protonation states of the ionizable MAO A residues were selected based on their pKa values,33 calculated with the help of the PROPKA server.35 The obtained protonation state of MAO A residues was the same as that described in our previous studies.36
Simulation of the enzyme-catalyzed reaction was based on the EVB description of the reactive subsystem consisting of lumiflavin and the neutral serotonin molecule embedded in the classically treated solvated enzyme. Two EVB states were used: the first corresponding to the reacting Michaelis complex and the second to the intermediate state in which the hydride is already transferred to the N5 atom of the lumiflavin moiety. The atomic charges for both EVB states were calculated by fitting to the electrostatic potential calculated at the HF/6-31G(d) level of theory according to the RESP scheme. The enzyme–substrate complex was solvated in a spherical cell with a radius of 30 Å, centered on the N5 atom of the flavin moiety, encompassing 1649 water molecules in total, all described with the OPLS-AA force field.37 The structure of MAO A with the serotonin molecule in the active site is shown in Figure 2.
Figure 2.
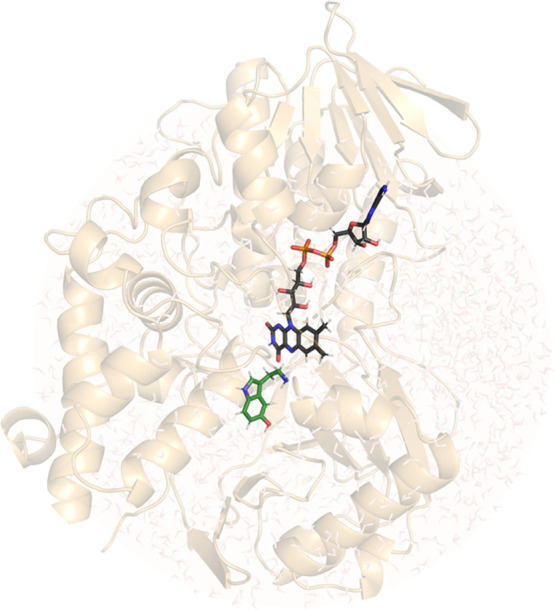
Structure of MAO A with serotonin in the active site. The flavin and serotonin moieties are represented using colored sticks, serotonin carbon atoms are depicted in green, and flavin carbon atoms are depicted in black.
All simulations and free-energy calculations were performed with the Q5(38) software package. The system was first equilibrated by slowly raising the temperature from 1 to 300 K while at the same time increasing the time step for integration from 0.1 to 1 fs and gradually releasing the applied positional restraints. The classical molecular dynamics (MD) trajectories for the reaction step were obtained using a mapping potential21,39,40 of the type
| 2 |
where the force field of the reactants (ε1) was gradually transformed into the force field of the products (ε2) via the coupling parameter λ. We simulated a total of 10 replicas using 51 λ-frames, each 10 ps long, resulting in 5.1 ns of MD. The same protocol was employed in our previous work.26 The production runs were performed at a temperature of 300 K, with a time step of 1 fs. A spherical cutoff of 10 Å was used for protein–protein, protein–water, and water–water interactions, and the local reaction field was applied for long-range interactions beyond 10 Å. All interactions between the EVB region (serotonin and FAD, truncated to the lumiflavin molecule LFN) and the solvated protein were included. The structure of the MAO A active site with the serotonin molecule (corresponding to the reactants, transition state, and products) is shown in Figure 3, with a clear indication of the hydride ion being transferred from the methylene group on serotonin to the N5 atom of the FAD cofactor (Figure 1).
Figure 3.
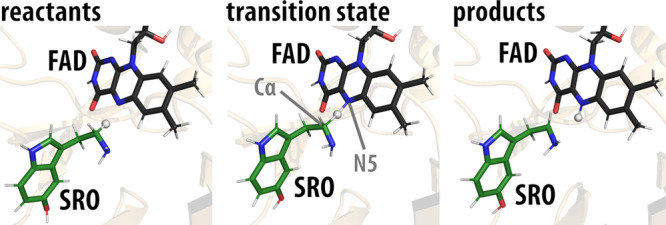
Reactant (left), transition-state (middle), and product (right) structures of the MAO A active site with the reacting serotonin molecule. The serotonin moiety is denoted by SRO and the flavin moiety by FAD. Serotonin carbon atoms are depicted in green, and flavin carbon atoms are depicted in black. Please note that in the transition state, the transferring hydride ion is located about halfway between the reactive carbon Cα atom of serotonin and the flavin N5 atom. The averaged distances between the reactive carbon Cα atom and the flavin N5 atom are 3.01, 2.67, and 3.21 Å for the reactants, transition state, and products, respectively.
The corresponding free-energy profiles were then computed from these simulations by using the well-established Free Energy Perturbation/Umbrella Sampling approach,21,39,40 as described in our previous work.36,41
In addition to the enzymatic environment, the same reaction was also simulated in the gas phase and in the aqueous solution. The free-energy profile in the gas phase was fitted to the quantum chemically calculated barrier height of ΔGgas⧧ = 30.90 kcal mol–1 and the reaction energy of ΔGgas = 22.93 kcal mol–1. The mapping yielded calibrated EVB parameters, namely, the off-diagonal matrix element Hij of 44.28 kcal mol–1 and the gas-phase shift α of 103.94 kcal mol–1. By using identical values of the parameters Hij and α, mapping was performed for reactions in the enzyme and in water. Visualization of the trajectories was performed with the VMD program.42
3. Results
The reaction profiles for the reaction in the gas phase and in the MAO A enzyme are shown in Figure 4.
Figure 4.
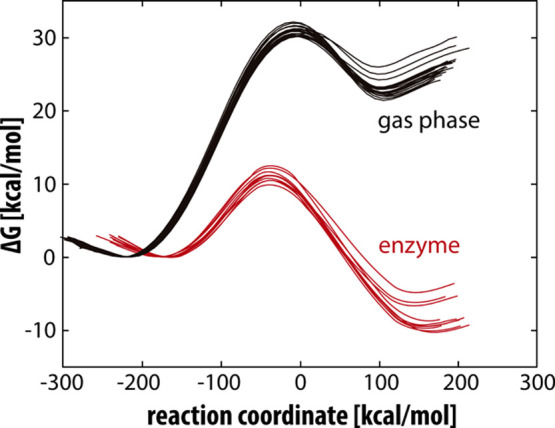
Reaction profiles for the decomposition of neutral serotonin. The gas-phase profiles are depicted in black, while the MAO A-catalyzed profiles are in red. The reaction coordinate is defined as the energy difference between EVB states 2 and 1 and is commonly used in displaying EVB free-energy profiles.
The calculated barrier for the neutral serotonin decomposition of 11.26 ± 0.81 kcal mol–1 was subsequently corrected for the free-energy cost of serotonin deprotonation of 3.56 kcal mol–1 at a pH value of 7.4, resulting in an overall barrier of 14.82 ± 0.81 kcal mol–1, which is in very good agreement with the experimental value of 16.0 kcal mol–1.43 The error of the calculated activation energy was estimated as the standard deviation for the barrier obtained from 10 replicas. The reaction free energy for the enzyme-catalyzed reaction is exergonic at −8.46 ± 1.88 kcal mol–1. The experimental and calculated kinetic parameters for the enzyme-catalyzed reaction are listed in Table 1.
Table 1. Experimental and Calculated Kinetic Parameters for the MAO A-Catalyzed Decomposition of Serotonina.
| kexpcat (s–1) | ΔGexp⧧ (kcal mol–1) | ΔGcalc⧧ (kcal mol–1) |
|---|---|---|
| 3.0 | 16.0 | 14.82 ± 0.81 |
Please note that the experimental barrier value ΔGexp⧧ was calculated from the experimental rate constant kexp(43) by using the Eyring–Polanyi equation.
In the aqueous environment, the corresponding reaction barrier is computed to be 18.53 ± 0.56 kcal mol–1, resulting in a barrier value of 22.09 ± 0.56 kcal mol–1, after applying the correction for the free energy of serotonin deprotonation (3.56 kcal mol–1). The reaction free energy for the reaction in water is −5.27 ± 0.38 kcal mol–1. Interestingly, this value is by 3.2 kcal mol–1 less exergonic than that for the MAO A active site, suggesting that the charged intermediates, following the hydride ion transfer, are better stabilized within the enzyme than in the aqueous solution. Comparison of the activation free energies between the aqueous and enzymatic environments reveals that MAO A lowers the barrier by 7.27 kcal mol–1 relative to water. By employing the transition-state theory, the corresponding increase in the reaction rate constant can therefore be estimated to be about 5 orders of magnitude at room temperature, implying that the reaction in the enzymatic environment proceeds much faster as compared to the reaction in the aqueous solution. In contrast to the aqueous and enzymatic environments, the same reaction in the gas phase is much less favorable both kinetically and thermodynamically, the latter being even highly endergonic at ΔGgasr = 22.93 kcal mol–1, as demonstrated by quantum calculations. With the present results at hand, it is interesting to observe that our earlier report for the MAO B-catalyzed dopamine degradation24 showed that the catalytic effect of this enzyme is 12.3 kcal mol–1, which analogously corresponds to 9 orders of magnitude increase in the rate constant relative to the reference reaction in the aqueous solution. This indicates a higher catalytic efficiency of MAO B as compared to that of MAO A, likely hinting at its predominance in the CNS, where a fast and efficient regulation and clearance of the brain monoamines is of great importance.
4. Conclusions
In this article, we conducted a multiscale study of the catalytic step of the serotonin decomposition catalyzed by MAO A by using the state-of-the-art EVB treatment. By properly including a fully featured enzymatic environment, well-converged free-energy profiles were obtained. The calculated free activation energy of 14.82 ± 0.81 kcal mol–1 reasonably reproduces the experimental free activation energy of 16.0 kcal mol–1, which gives very strong evidence for the validity of Vianello’s hydride-transfer mechanism13 for the MAO A-catalyzed serotonin decomposition as well.
By comparing the activation free energy between the enzymatic and aqueous environments, the catalytic effect (barrier lowering) provided by MAO A is estimated to be 7.27 kcal mol–1, which ranks MAO A among the enzymes with mid-range proficiency. According to Warshel, the catalytic effect of enzymes is mainly attributed to preorganized electrostatics,30 whereas other factors, including steric strain or dynamical effects, are less relevant. The assumed decisive role of electrostatics can also be devised for the presently studied reaction, that is, the reaction involves the formation of two charged species as a result of the hydride transfer from serotonin to lumiflavin—because of this, the reaction profiles are strongly dependent on the environment. The water environment significantly reduces the gas-phase barrier and enables the reaction thermodynamically, while the preorganized enzyme electrostatics provides an additional (and for physiological purposes probably decisive) contribution to the barrier lowering.
The present study represents one of several attempts to elucidate the catalytic effect of MAO enzymes. A number of open questions to be addressed in future research remain, including consecutive steps in the catalytic process, MAO A versus MAO B selectivity, and effects of point mutations. Point mutations lead to altered enzyme activity by changing the electrostatic stabilization of the reactants and the transition state, which results in a changed free activation energy and thus in a changed turnover rate. A particular challenge is the application of the MAO A function to genomic medicine. MAO A activity can be modulated by the level of its expression and by altered activities of its mutants. The effects of point mutations can be studied experimentally and theoretically (see ref (36)) and can be obtained relatively easily from the genomic information, whereas the level of MAO A expression is not easily linked to the genomic information. High MAO A activity, along with highly active transporters for its reuptake (serotonin transporter—SERT), leads to decreased serotonin and noradrenalin levels, typically followed by pathologies such as depression, anxiety, and other mood disorders.44−46 Low MAO A activity leads to increased serotonin and noradrenaline levels, which have pathological effects on brain plasticity in the process of prenatal neurogenesis. In psychiatry, the term “warrior gene” is established for the gene that encodes a less active MAO A. The Brunner syndrome is a well-established pathology that can be caused by the complete absence of MAO A, truncation of the enzyme, or point mutations, as shown in clinical studies and animal models.47−50 Summarizing this, it remains a major challenge for the future to develop a macroscopic model that combines genomic information, experimental kinetic data from MAOs, and experimental data on monoamine transporters (such as the serotonin or dopamine transporters) with molecular simulation data to predict neuropsychiatric pathologies such as depression. The method of choice in this case is a description of the synapse using a system of ordinary differential equations, as we did in our recent study concerning the effects of cocaine and amphetamines on dopamine autoxidation and therewith associated induction of Parkinsonism.51
Acknowledgments
We would like to thank Dr. Matej Repič, Krka Ltd., Novo mesto, for many stimulating discussions.
Author Contributions
A.P. and M.P. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
A.P., J.S., and J.M. would like to thank the Slovenian Research Agency for financial support in the framework of the program group P1-0012. R.V. acknowledges the financial support from the Croatian Science Foundation.
The authors declare no competing financial interest.
References
- Rudnick G.; Sandtner W. Serotonin transport in the 21st century. J. Gen. Physiol. 2019, 151, 1248–1264. 10.1085/jgp.201812066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman L. S.; Brunton L. L.; Chabner B.; Knollmann B. C.. Goodman & Gilman’s Pharmacological Basis of Therapeutics; McGraw-Hill: New York, USA, 2011. [Google Scholar]
- Sadock B. J.; Kaplan H. I.; Sadock V. A.. Kaplan & Sadock’s Synopsis of Psychiatry: Behavioral Sciences/Clinical Psychiatry; Lippincott Williams & Wilkins: Philadelphia, USA, 2007. [Google Scholar]
- Wu H.; Denna T. H.; Storkersen J. N.; Gerriets V. A. Beyond a neurotransmitter: The role of serotonin in inflammation and immunity. Pharmacol. Res. 2019, 140, 100–114. 10.1016/j.phrs.2018.06.015. [DOI] [PubMed] [Google Scholar]
- Mialet-Perez J.; Santin Y.; Parini A. Monoamine oxidase-A, serotonin and norepinephrine: synergistic players in cardiac physiology and pathology. J. Neural. Transm. 2018, 125, 1627–1634. 10.1007/s00702-018-1908-y. [DOI] [PubMed] [Google Scholar]
- Naoi M.; Maruyama W.; Shamoto-Nagai M. Type A monoamine oxidase and serotonin are coordinately involved in depressive disorders: from neurotransmitter imbalance to impaired neurogenesis. J. Neural. Transm. 2018, 125, 53–66. 10.1007/s00702-017-1709-8. [DOI] [PubMed] [Google Scholar]
- Pavlin M.; Repič M.; Vianello R.; Mavri J. The Chemistry of Neurodegeneration: Kinetic Data and Their Implications. Mol. Neurobiol. 2016, 53, 3400–3415. 10.1007/s12035-015-9284-1. [DOI] [PubMed] [Google Scholar]
- Erdem S. S.; Karahan Ö.; Yıldız İ.; Yelekçi K. A computational study on the amine-oxidation mechanism of monoamine oxidase: Insight into the polar nucleophilic mechanism. Org. Biomol. Chem. 2006, 4, 646–658. 10.1039/b511350d. [DOI] [PubMed] [Google Scholar]
- Akyüz M. A.; Erdem S. S.; Edmondson D. E. The aromatic cage in the active site of monoamine oxidase B: effect on the structural and electronic properties of bound benzylamine and p-nitrobenzylamine. J. Neural. Transm. 2007, 114, 693–698. 10.1007/s00702-007-0670-3. [DOI] [PubMed] [Google Scholar]
- Abad E.; Zenn R. K.; Kästner J. Reaction Mechanism of Monoamine Oxidase from QM/MM Calculations. J. Phys. Chem. B 2013, 117, 14238–14246. 10.1021/jp4061522. [DOI] [PubMed] [Google Scholar]
- Zapata-Torres G.; Fierro A.; Barriga-González G.; Salgado J. C.; Celis-Barros C. Revealing monoamine oxidase B catalytic mechanisms by means of the quantum chemical cluster approach. J. Chem. Inf. Model. 2015, 55, 1349–1360. 10.1021/acs.jcim.5b00140. [DOI] [PubMed] [Google Scholar]
- Fierro A.; Edmondson D. E.; Celis-Barros C.; Rebolledo-Fuentes M.; Zapata-Torres G. Why p-OMe- and p-Cl-beta-Methylphenethylamines Display Distinct Activities upon MAO-B Binding. PLoS One 2016, 11, e0154989 10.1371/journal.pone.0154989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vianello R.; Repič M.; Mavri J. How are biogenic amines metabolized by monoamine oxidases?. Eur. J. Org. Chem. 2012, 2012, 7057–7065. 10.1002/ejoc.201201122. [DOI] [Google Scholar]
- Orru R.; Aldeco M.; Edmondson D. E. Do MAO A and MAO B utilize the same mechanism for the C-H bond cleavage step in catalysis? Evidence suggesting differing mechanisms. J. Neural. Transm. 2013, 120, 847–851. 10.1007/s00702-013-0991-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenn R. K.; Abad E.; Kästner J. Influence of the environment on the oxidative deamination of p-substituted benzylamines in monoamine oxidase. J. Phys. Chem. B 2015, 119, 3678–3686. 10.1021/jp512470a. [DOI] [PubMed] [Google Scholar]
- Binda C.; Li M.; Hubalek F.; Restelli N.; Edmondson D. E.; Mattevi A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 9750–9755. 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akyüz M. A.; Erdem S. S. Computational modeling of the direct hydride transfer mechanism for the MAO catalyzed oxidation of phenethylamine and benzylamine: ONIOM (QM/QM) calculations. J. Neural. Transm. 2013, 120, 937–945. 10.1007/s00702-013-1027-8. [DOI] [PubMed] [Google Scholar]
- Atalay V. E.; Erdem S. S. A comparative computational investigation on the proton and hydride transfer mechanisms of monoamine oxidase using model molecules. Comput. Biol. Chem. 2013, 47, 181–191. 10.1016/j.compbiolchem.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Cakir K.; Erdem S. S.; Atalay V. E. ONIOM calculations on serotonin degradation by monoamine oxidase B: insight into the oxidation mechanism and covalent reversible inhibition. Org. Biomol. Chem. 2016, 14, 9239–9252. 10.1039/c6ob01175f. [DOI] [PubMed] [Google Scholar]
- Warshel A.Computer modeling of chemical reactions in enzymes and solutions; Wiley: New York, USA, 1997. [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Xiang Y.; Liu H.; Olsson M. H. M. Electrostatic basis for enzyme catalysis. Chem. Rev. 2006, 106, 3210–3235. 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- Kamerlin S. C. L.; Haranczyk M.; Warshel A. Progress in ab initio QM/MM free-energy simulations of electrostatic energies in proteins: accelerated QM/MM studies of pKa, redox reactions and solvation free energies. J. Phys. Chem. B 2009, 113, 1253–1272. 10.1021/jp8071712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prah A.; Frančišković E.; Mavri J.; Stare J. Electrostatics as the Driving Force Behind the Catalytic Function of the Monoamine Oxidase A Enzyme Confirmed by Quantum Computations. ACS Catal. 2019, 9, 1231–1240. 10.1021/acscatal.8b04045. [DOI] [Google Scholar]
- Repič M.; Vianello R.; Purg M.; Duarte F.; Bauer P.; Kamerlin S. C. L.; Mavri J. Empirical valence bond simulations of the hydride transfer step in the monoamine oxidase B catalyzed metabolism of dopamine. Proteins: Struct., Funct., Bioinf. 2014, 82, 3347–3355. 10.1002/prot.24690. [DOI] [PubMed] [Google Scholar]
- Oanca G.; Stare J.; Vianello R.; Mavri J. Multiscale simulation of monoamine oxidase catalyzed decomposition of phenylethylamine analogs. Eur. J. Pharmacol. 2017, 817, 46–50. 10.1016/j.ejphar.2017.05.061. [DOI] [PubMed] [Google Scholar]
- Oanca G.; Stare J.; Mavri J. How fast monoamine oxidases decompose adrenaline? Kinetics of isoenzymes A and B evaluated by empirical valence bond simulation. Proteins: Struct., Funct., Bioinf. 2017, 85, 2170–2178. 10.1002/prot.25374. [DOI] [PubMed] [Google Scholar]
- Poberžnik M.; Purg M.; Repič M.; Mavri J.; Vianello R. Empirical valence bond simulations of the hydride-transfer step in the monoamine oxidase A catalyzed metabolism of noradrenaline. J. Phys. Chem. B 2016, 120, 11419–11427. 10.1021/acs.jpcb.6b09011.s001. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Warshel A. Multiscale modeling of biological functions: from enzymes to molecular machines (Nobel Lecture). Angew. Chem. 2014, 53, 10020–10031. 10.1002/anie.201403689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezençon J.; Wittwer M. B.; Cutting B.; Smieško M.; Wagner B.; Kansy M.; Ernst B. pKa determination by (1)H NMR spectroscopy - an old methodology revisited. J. Pharm. Biomed. Anal. 2014, 93, 147–155. 10.1016/j.jpba.2013.12.014. [DOI] [PubMed] [Google Scholar]
- Borštnar R.; Repič M.; Kamerlin S. C. L.; Vianello R.; Mavri J. Computational study of the pK(a) values of potential catalytic residues in the active site of monoamine oxidase B. J. Chem. Theory Comput. 2012, 8, 3864–3870. 10.1021/ct300119u. [DOI] [PubMed] [Google Scholar]
- Repič M.; Purg M.; Vianello R.; Mavri J. Examining electrostatic preorganization in monoamine oxidases A and B by structural comparison and pKa calculations. J. Phys. Chem. B 2014, 118, 4326–4332. 10.1021/jp500795p. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Olsson M. H. M.; Søndergaard C. R.; Rostkowski M.; Jensen J. H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pK(a) Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Oanca G.; Purg M.; Mavri J.; Shih J. C.; Stare J. Insights into enzyme point mutation effect by molecular simulation: phenylethylamine oxidation catalyzed by monoamine oxidase A. Phys. Chem. Chem. Phys. 2016, 18, 13346–13356. 10.1039/c6cp00098c. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. 10.1021/ja9621760. [DOI] [Google Scholar]
- Marelius J.; Kolmodin K.; Feierberg I.; Åqvist J. Q: a molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graphics Modell. 1998, 16, 213–225. 10.1016/s1093-3263(98)80006-5. [DOI] [PubMed] [Google Scholar]
- Hwang J. K.; King G.; Creighton S.; Warshel A. Simulation of Free-Energy Relationships and Dynamics of Sn2 Reactions in Aqueous-Solution. J. Am. Chem. Soc. 1988, 110, 5297–5311. 10.1021/ja00224a011. [DOI] [Google Scholar]
- Villà J.; Warshel A. Energetics and Dynamics of Enzymatic Reactions. J. Phys. Chem. B 2001, 105, 7887–7907. 10.1021/jp011048h. [DOI] [Google Scholar]
- Pregeljc D.; Jug U.; Mavri J.; Stare J. Why does the Y326I mutant of monoamine oxidase B decompose an endogenous amphetamine at a slower rate than the wild type enzyme? Reaction step elucidated by multiscale molecular simulations. Phys. Chem. Chem. Phys. 2018, 20, 4181–4188. 10.1039/c7cp07069a. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics Modell. 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Nandigama R. K.; Miller J. R.; Edmondson D. E. Loss of serotonin oxidation as a component of the altered substrate specificity in the Y444F mutant of recombinant human liver MAO A. Biochemistry 2001, 40, 14839–14846. 10.1021/bi011113d. [DOI] [PubMed] [Google Scholar]
- Du L.; Faludi G.; Palkovits M.; Sotonyi P.; Bakish D.; Hrdina P. D. High activity-related allele of MAO-A gene associated with depressed suicide in males. Neuroreport 2002, 13, 1195–1198. 10.1097/00001756-200207020-00025. [DOI] [PubMed] [Google Scholar]
- Gutiérrez B.; Arias B.; Gastó C.; Catalán R.; Papiol S.; Pintor L.; Fañanás L. Association analysis between a functional polymorphism in the monoamine oxidase A gene promoter and severe mood disorders. Psychiatr. Genet. 2004, 14, 203–208. 10.1097/00041444-200412000-00007. [DOI] [PubMed] [Google Scholar]
- Lim L. C. C.; Powell J. F.; Murray R.; Gill M. Monoamine oxidase A gene and bipolar affective disorder. Am. J. Hum. Genet. 1994, 54, 259–263. 10.1002/ajmg.1320540314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner H.; Nelen M.; Breakefield X.; Ropers H.; van Oost B. Abnormal-behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science 1993, 262, 578–580. 10.1126/science.8211186. [DOI] [PubMed] [Google Scholar]
- Palmer E. E.; Leffler M.; Rogers C.; Shaw M.; Carroll R.; Earl J.; Cheung N. W.; Champion B.; Hu H.; Haas S. A.; Kalscheuer V. M.; Gecz J.; Field M. New insights into Brunner syndrome and potential for targeted therapy. Clin. Genet. 2016, 89, 120–127. 10.1111/cge.12589. [DOI] [PubMed] [Google Scholar]
- Piton A.; Poquet H.; Redin C.; Masurel A.; Lauer J.; Muller J.; Thevenon J.; Herenger Y.; Chancenotte S.; Bonnet M.; Pinoit J.-M.; Huet F.; Thauvin-Robinet C.; Jaeger A.-S.; Le Gras S.; Jost B.; Gérard B.; Peoc’h K.; Launay J.-M.; Faivre L.; Mandel J.-L. 20 ans apres: a second mutation in MAOA identified by targeted high-throughput sequencing in a family with altered behavior and cognition. Eur. J. Hum. Genet. 2014, 22, 776–783. 10.1038/ejhg.2013.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases O.; Seif I.; Grimsby J.; Gaspar P.; Chen K.; Pournin S.; Muller U.; Aguet M.; Babinet C.; Shih J.; et al. Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science 1995, 268, 1763–1766. 10.1126/science.7792602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pregeljc D.; Teodorescu-Perijoc D.; Vianello R.; Umek N.; Mavri J. How Important Is the Use of Cocaine and Amphetamines in the Development of Parkinson Disease? A Computational Study. Neurotox. Res. 2020, 37, 724–731. 10.1007/s12640-019-00149-0. [DOI] [PubMed] [Google Scholar]