

Abstract
The Tec kinase IL-2–inducible T cell kinase (ITK) regulates the expression of TCR-induced genes. Itk−/− T cell responses are impaired but not absent. ITK inhibition prevented colitis disease progression and impaired T cell migration to the colon in mice. To examine the function of ITK in T cell migration to the intestine, we examined the number of gut T cells in Itk−/− mice and then evaluated their expression of gut-homing receptors. Combined with in vitro murine T cell stimulation and in vivo migration assay using congenic B6 mice, we demonstrated an essential role for ITK in T cell migration to the intestine in mice. Reconstitution of Itk−/− mouse CD8+ T cells with IFN regulatory factor 4 restored gut-homing properties, providing mechanistic insight into the function of ITK-mediated signaling in CD8+ T cell migration to the intestinal mucosa in mice.
INTRODUCTION
IL-2–inducible T cell kinase (ITK) is a Tec-family tyrosine kinase predominantly expressed in T cells and important in modulating TCR signal strength (1–3). ITK regulates the differentiation of naive CD4+ T cells into various Th cell subsets (4–8) and is required for effector T cell responses that protect against certain infections, as well as those that contribute to allergic and autoimmune responses (4, 7, 9–11). ITK has also been implicated in CD4+ T cell trafficking to nonlymphoid tissues, such as colon and brains, in two different experimental disease models (12, 13). However, the role of ITK in CD8+ T cell trafficking, and in particular, trafficking to the intestine, has not been closely examined.
Mucosal barriers, such as skin, respiratory tract, genital tract, and intestinal tract, are common sites for pathogen invasion. Consequently, the trafficking of gut-homing T cells and the establishment of tissue-resident memory T cells (TRM) in the intestine are critical for an optimal mucosal immunity against enteric pathogens (14–16). For T cell trafficking to the gut, effector CD8+ T cells acquire the ability to enter intestinal tissue by expressing two gut-homing receptors (e.g., integrin α4β7 and CCR9) following stimulation by all-trans retinoic acid (RA)–producing dendritic cells in mesenteric lymph node (mLN) or Peyer’s patches (17–20). Effector CD8+ T cells upregulate CD69 and CD103 (integrin αE), and downregulate CCR7 and S1PR1 to prevent tissue egress, thereby adopting a TRM phenotype (21, 22). TGF-β, ubiquitously expressed throughout the intestinal tissue, promotes the expression of CD103 and thereby plays a key role in promoting T cell tissue retention (23).
The transcriptional regulator IFN regulatory factor 4 (IRF4) has a multitude of functions in CD8+ T cells (24–26). In both CD4+ and CD8+ T cells, IRF4 serves as a molecular link that translates TCR signaling strength to transcriptional changes affecting helper T cell and effector cell differentiation pathways (24, 27). In both CD4+ and CD8+ T cells, IRF4 expression levels are directly regulated by ITK signaling (8, 28). Interestingly, IRF4 is highly upregulated in CD69+ TRM from human lung tissue and also in adoptively transferred CD103-expressing CD8+ T cells in the brain after virus infection (29, 30). However, the function of IRF4 in TRM in nonlymphoid tissues or in CD8+ T cell homing to mucosal barriers has not been studied. The importance of this topic is brought to the forefront by the recent finding that a cohort of human patients with a haploinsufficiency of the IRF4 gene suffer from Whipple’s disease, a gastrointestinal disease, and more specifically, caused by impaired control of an enteric bacteria Tropheryma whipplei (31). The discovery of this human immuno-deficiency indicates a link between IRF4 expression levels and immune protection in the intestine, raising the possibility that this requirement is for high expression of IRF4 in gut T cells. Interestingly, a genetic deficiency in ITK in humans is associated with an inability to control EBV infection, ultimately leading to a fatal disease (32–35). This discovery was surprising, as studies performed in Itk−/− mice have failed to show any defects in antiviral T cell responses (36, 37). We considered the possibility that ITK signaling might be necessary for the long-term control of a persistent or latent virus infection.
To address the requirement for ITK in the regulation of intestinal CD8+ T cells, we assessed steady-state numbers of gut T cells in Itk−/− mice under homeostatic conditions. We found that gut T cell numbers were markedly reduced and, furthermore, that Itk−/− CD8+ T cells displayed defects in gut-homing receptor expression in vitro and in homing ability to the gut in vivo. Of interest, upon the challenge with mouse gammaherpesvirus 68 (MHV68), Itk−/− mice developed a lethal intestinal inflammation during latency with high viral DNA copy numbers, accompanied by a dearth of virus-specific CD8+ T cells in the intestinal tissue. Enforced expression of IRF4 in Itk−/− CD8+ T cells restored T cell trafficking to the gut, suggesting an essential role for ITK-mediated TCR signaling in T cell migration to the intestine.
MATERIALS AND METHODS
Mice
C57BL/6 wild-type (WT), Rag1−/−, Rag2−/−, congenic Ly5.1 (CD45.1), and OT-I Rag1−/− TCR transgenic mice were purchased from Taconic Biosciences and crossed with Itk−/− to generate mice used for experiments. All purchased mice were housed and maintained with Itk−/− for several generations before experiments. Irf4fl/fl × CD4-Cre were described previously (28). Mice were housed in specific pathogen-free conditions at the University of Massachusetts Medical School in accordance with Institutional Animal Care and Use Committee guidelines. All uninfected mice were analyzed at 8–10 wk of age. For MHV68 experiments, mice were infected at 8–10 wk of age and analyzed at indicated time points postinfection.
Abs and reagents for flow cytometric analyses
Cells from the spleen, mLN, lung, bone marrow, and small and large intestine were stained with anti-mouse CD3 (145-2C11), CD4 (RM4.5), CD8α (53–6.7), CD19 (6D5), CD44 (IM7), CD45.1 (A20), CD45.2 (104), CD69 (H1.2F3), CD103 (M290), CD199 (eBioCW-1.2), integrin α4β7 (DATK32), KLRG-1 (2F1), TCRβ (H57–597), TCRγδ (eBioGL3), and IRF4 (3E4) (from eBioscience, BD Biosciences, and Invitrogen). In some experiments, cells were stimulated with viral antigenic peptide ORF75c (KSLTYYKL) or the combination of PMA (50 ng/ml) and ionomycin (1.0 μg/ml) for 5 h at 37°C in the presence of GolgiStop and GolgiPlug (BD Biosciences). After stimulation, cells were fixed and permeabilized using BD Cytofix/Cytoperm Kit (BD Biosciences) to stain intracellular cytokines by using following Abs: anti-mouse IFN-γ (XMG1.2), IL-2 (JES6-5H4), TNF-α (MP6-XT22), and granzyme B (GB12) (from eBioscience, BD Biosciences, and Invitrogen). Cells were analyzed on an LSR II flow cytometer (BD Biosciences), and data were analyzed with FlowJo software (Tree Star).
Isolation of intraepithelial lymphocyte and lamina propria lymphocyte from the intestine
Intestinal lymphocytes were processed as previously described (12, 38). In brief, intestinal tissues were opened longitudinally and cut into 2–3-cm pieces. Tissues were treated with 1.0 mM DTT and 0.5 M EDTA in HBSS at 37°C for several rounds. Supernatants were collected for intraepithelial lymphocyte (IEL) isolation. For lamina propria (LP) lymphocyte (LPL) isolation, the remaining tissues were digested with a mixture of collagenase D (1.0 mg/ml; Roche), neutral protease (0.1 U/ml; Worthington Biochemical), and DNase I (1.0 U/ml; Sigma-Aldrich) for 45 min (small intestine) or 50 min (colon). Cell suspensions were layered on 40%/80% Percoll (GE Healthcare Life Sciences) density gradients for lymphocyte isolations. The viability of extracted cells was assessed using trypan blue.
Immunohistochemistry and immunofluorescence microscopy
For immunohistochemistry, dissected tissues were fixed in 10% buffered formalin (Thermo Fisher Scientific) overnight and then embedded into paraffin block for H&E staining. For immunofluorescence staining, collected tissues were embedded in Tissue-Tek O.C.T. compound (Sakura Finetek) and snap frozen on methanol mixed with dry ice. Frozen tissues were cut into 7-μm-thick sections, air-dried for 1 h at room temperature (RT), and then fixed in acetone for 10 min at 4°C. After drying for 1 h at RT, tissue sections were rehydrated with PBS with 1.0% BSA and then stained overnight with primary Abs diluted in PBS containing 1.0% BSA, 2.0% normal rat serum, and 2.0% mouse serum in a humidified chamber. Sections were washed with PBS three times and then stained for secondary for 1 h at RT. After washing, tissues were counterstained with DAPI-including ProLong Gold antifade reagent (Invitrogen) for confocal microscopy. Immunofluorescent Abs and secondaries were anti-mouse CD8α Biotin (Ly-2; BD Pharmingen); streptavidin-Cy3 (BioLegend); and anti-mouse CD11c–AlexaFluor 594 (N418; BioLegend). Images were acquired by confocal laser scanning microscope (Carl Zeiss) and analyzed by ZEN lite software (Carl Zeiss).
MHV68 strains, infection, and LPS-induced viral reactivation
Mice were infected with MHV68 via i.p. injection (106 PFU) or, where indicated, by intranasal (i.n.) infection (103 PFU). For reactivation from latency, a single subclinical dose of LPS (20 μg) was injected i.p. MHV68-M3-OVA was engineered to express chicken OVA (39). All viral stocks were propagated in Vero or NIH 3T12 cells, and viral titers were determined by plaque assay as previously described (40).
Viral DNA copy number measurement via quantitative PCR
To examine viral replication, organs were washed with PBS and cut into 2–3-cm pieces. Tissues were digested with lysis buffer (100 mM Tris-HCl [pH 8], 0.5 mM EDTA, 0.2% SDS, and 200 mM NaCl) mixed with proteinase K (20 μg/ml) (Sigma-Aldrich) at 55°C overnight with rotation, and DNA was isolated after phenol: chloroform extraction. For viral copy number determination, DNA samples were subjected to quantitative PCR specific for viral gene ORF75c, and values were compared with a standard curve generated using a plasmid containing ORF75c. PCR primers were 5′-AAA TGG TGA AAG CCA TTT TGA-3′ (forward) and 5′-CCA CCA TCG CAT AAC AGT TG-3′ (reverse).
OT-I adoptive transfer to MHV68-M3-OVA–infected hosts
Splenic CD8+ T cells of OT-I WT (CD45.2) and Itk−/− (CD45.1) uninfected donor mice were isolated using EasySep Mouse CD8+ T Cell Isolation Kit (STEMCELL Technologies) according to the manufacturer’s instructions. A total of 5.0 × 105 cells were i.v. injected into congenic hosts that were then infected with MHV68-M3-OVA–infected (1.0 × 106 PFU, i.p.). Mice were sacrificed at day 7 postinfection for analysis.
RNA isolation, reverse transcription, and quantitative PCR
Total RNA was isolated with TRIzol (Life Technologies) following the manufacturer’s instructions. cDNA was synthesized with iScript cDNA Synthesis Kit (Bio-Rad), and quantitative PCR was performed with iQSYBR Green PCR Supermix (Bio-Rad). Primer sequences used for quantitative PCR were as follows: Tgfbr2, forward primer (FP), 5′-AGC ATC ACG GCC ATC TGT G-3′; Tgfbr2 reverse primer (RP), 5′-TGG CAA ACC GTC TCC AGA GT-3′; Rara FP, 5′-TCA TGA AGT GTG ACG TTG ACA TCC GT-3′; Rara RP, 5′-TTG GCA AGG CAA AGA C-3′; ActB FP, 5′-CGC CAC CAG TTC GCC ATG G-3′; ActB RP, 5′-TAC AGC CCG GGG AGC ATC GT-3′.
Chromatin immunoprecipitation sequencing data analysis
Original chromatin immunoprecipitation sequencing data were obtained from National Center for Biotechnology Information Gene Expression Omnibus Database (accession GSM1309509 and GSM1309511; GSM2259177 and GSM2259178) (27). Data were analyzed in Integrative Genomics Viewer (Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA) (41).
Retroviral transduction
pMigR-IRES-EGFP retrovirus (RV) vectors (empty vector and IRF4) and pCL-10A1 packaging vector were cotransfected into Platinum-E RV packaging cells (Cell Biolabs) using TransIT-293 transfection reagent according to the manufacturer’s protocol (Mirus). Isolated spleen OT-I WT and Itk−/− CD8+ T cells were stimulated with anti-CD3 (1.0 μg/ml; eBioscience) and anti-CD28 (4.0 μg/ml; eBioscience) in the presence of IL-2 (20 ng/ml; eBioscience) for 24 h before RV transduction. Viral supernatants from RV-transfected cells were harvested after 48 h and then transduced to prestimulated OT-I T cells in the presence of polybrene (8.0 μg/ml) (AmericanBio) using spin infection for 90 min at RT. OT-I CD8+ T cells were replated and then stimulated with anti-CD3/CD28 plus IL-2 for 24 h. Transduced CD8+ T cells were primed in the presence of RA and TGF-β for 48 h and then adoptively transferred to congenic WT naive hosts for further analyses.
Statistical analyses
Data are represented as mean ± SEM. All statistical analyses were performed using Prism 8 GraphPad Software. Differences between individual groups were analyzed for statistical significance using unpaired or paired two-tailed Student t tests (*p < 0.05, **p < 0.01, ***p < 0.001).
RESULTS
Itk−/− mice have a steady-state reduction in numbers of gut-resident T cells
Our previous study demonstrated that a small molecule inhibitor of ITK could interfere with T cell–mediated colitis and that disease prevention was associated with reduced numbers of T cells in the colon (12). These findings suggested a role for ITK signaling in T cell gut homing, prompting us to examine steady-state gut-resident T cells in Itk−/− mice. Confocal imaging of small intestine (duodenum) showed the expected localization of CD8+ T cells in the intestinal epithelium and LP compartments of uninfected WT mice (Fig. 1A, 1B). Strikingly, uninfected Itk−/− mice had noticeably fewer CD8+ T cells in both the LP and intestinal epithelium compartments (Fig. 1A, 1B). These findings were confirmed by flow cytometry of dissociated intestinal tissues from WT and Itk−/− mice; in contrast, no differences in T cell numbers were observed in the spleen (Fig. 1C, 1D). To determine whether there was any alteration in the T cell subsets found in the intestinal compartments of WT versus Itk−/− mice, we examined IELs in the small intestine for CD8αα, CD8αβ, CD4+ CD8αβ−, and CD4− CD8αβ− subsets of TCRβ+ or TCRγδ+ cells. This analysis revealed no profound differences in specific IEL subpopulations in Itk−/− mice and, instead, confirmed a global reduction in intestinal T cell numbers in the absence of ITK (Supplemental Fig. 1A, 1B). Overall, these data are consistent with a role for ITK in gut T cells under steady-state homeostatic conditions.
FIGURE 1. Itk−/− mice have a steady-state reduction in gut-resident T cells.
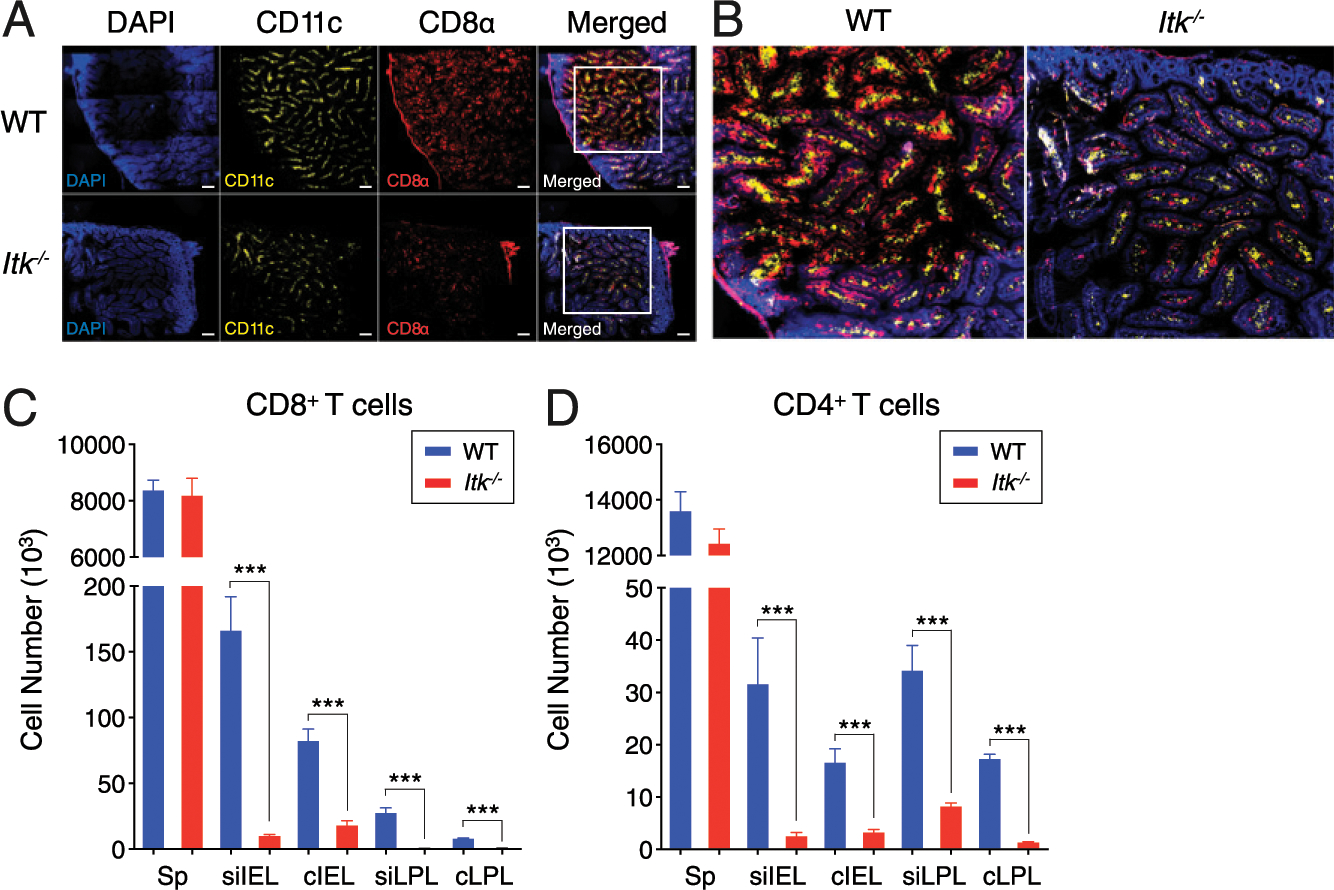
(A and B) Confocal microscopy of intestinal tissue (duodenum) from uninfected WT and Itk−/− mice. Staining with DAPI (blue), anti-CD11c (yellow), anti-CD8 (red), and merged images are shown. Images in (A) were taken at original magnification ×20 and eight tiled (3 × 3) sections were created using Carl Zeiss ZEN lite software. (B) Merged insets [white boxes from (A), far right] are shown at a magnification of ×4.0. Scale bar, 100 μm. (C and D) Compilation data of CD4+ (C) and CD8+ (D) T cell numbers in the spleen (Sp) and intestinal tissues (siIEL, cIEL, siLPL, and cLPL) are shown. Statistical significance between individual groups was analyzed using unpaired or paired two-tailed Student t tests (***p < 0.001).
Impaired expression of gut-homing receptors on CD8+ T cells in Itk−/− mice
We next assessed expression of homing receptors known to be important in migration and/or tissue retention in the intestine. We compared WT to Itk−/− CD8+ T cells isolated from the intestinal epithelium and LP compartments of intestinal tissue under steady-state conditions for the expression of markers associated with tissue-resident T cells, CD69 and CD103. As shown in Fig. 2A and 2B, of the few CD8+ T cells that were recovered from all compartments of the intestinal tissue in Itk−/− mice, a smaller proportion of Itk−/− small intestine IEL (siIEL) and small intestine LPL (siLPL) cells coexpressed CD69 and CD103 compared with WT counterparts. In the colon IEL (cIEL), we observed a modest decrease in the proportions of CD69+ CD103+ cells in Itk−/− mice, whereas no differences were found in the colon LPL (cLPL) compartment when comparing WT and Itk−/− mice. Examination of gut-homing receptors CCR9 and integrin α4β7 expression also revealed a significant defect in the expression of these gut-homing molecules on small intestinal IEL and LPL from Itk−/− mice compared with WT mice, a defect that was seen for both CD69+ and CD69− cell subsets (Fig. 2C–F). These data were consistent with a substantial reduction in the proportion of Itk−/− CD8+ T cells expressing CD103, CCR9, and integrin α4β7 in the mLN (Fig. 2G, 2H). These data strongly suggest that the reduced numbers of gut CD8+ T cells in Itk−/− mice are associated with impaired expression of gut tissue homing and retention molecules and that this defect may arise during T cell priming in the mLN.
FIGURE 2. Gut-resident and mLN CD8+ T cells in Itk−/− mice have reduced gut-homing receptor expression.
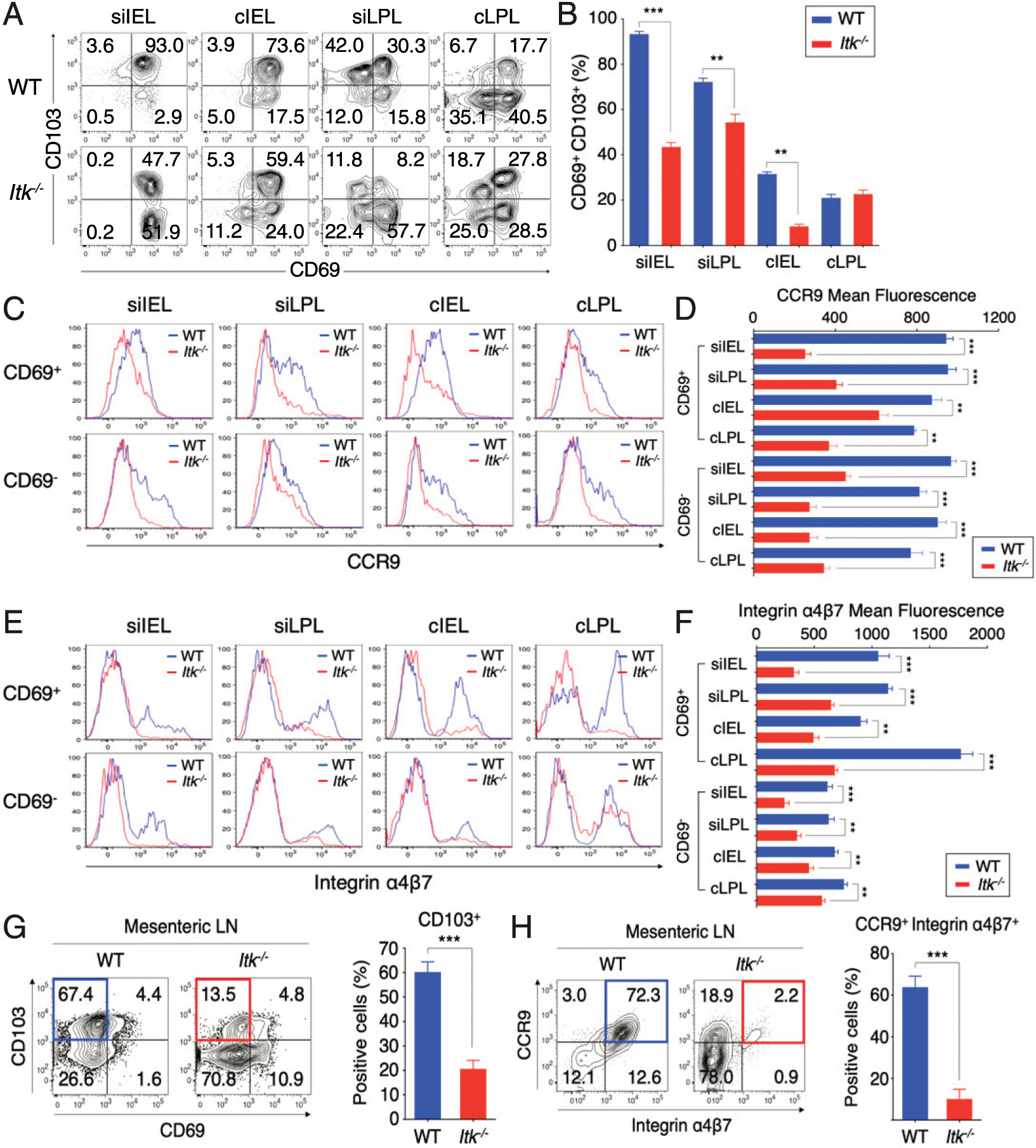
(A and B) Expression of CD69 versus CD103 on CD8+ T cells from intestinal tissues (siIEL, cIEL, siLPL, and cLPL) of uninfected WT and Itk−/− mice. Graphs show compilation of percentages of CD8+ T cells that are CD69+ CD103+. (C–F) Expression of CCR9 (C and D) and integrin α4β7 (E and F) gated on CD69+ or CD69− CD8+ T cells from intestinal tissues of uninfected WT and Itk−/− mice. Graphs show compilation of mean fluorescence intensities of CCR9 and integrin α4β7 from CD69+ or CD69− CD8+ T cells from WT and Itk−/− mice. (G and H) Expression of CD69 versus CD103 (G) and CCR9 versus integrin α4β7 (H) on CD8+ T cells from the mLN of naive WT and Itk−/− mice with compiled proportion of CD103+ and CCR9+ integrin α4β7+ cells. Data shown are representative of five mice of each genotype (mean ± SEM). Statistical significance between individual groups was analyzed using unpaired or paired two-tailed Student t tests (**p < 0.01, ***p < 0.001).
ITK is required for optimal expression of gut-homing receptors and migration to the intestinal mucosa
The data presented above suggest that ITK is required for optimal upregulation of gut-homing migration and retention molecules following TCR activation and that a defect in this process contributes to impaired migration or retention of Itk−/− CD8+ T cells in this tissue. To test this, we stimulated WT and Itk−/− OT-I T cells with anti-CD3 plus RA or TGF-β for 48 h in vitro as previously described (42) and then examined the cells for expression of CD103, CCR9, and integrin α4β7. In the absence of TCR stimulation, neither WT nor Itk−/− OT-I T cells expressed these molecules. Following TCR stimulation alone, both WT and Itk−/− OT-I T cells exhibited a modest upregulation of these receptors, but no difference was seen between cells of the two genotypes. When these cells were stimulated with anti-CD3 plus RA or TGF-β, Itk−/− cells showed reduced expression of CD103, or CCR9 and integrin α4β7, respectively, compared with WT cells (Fig. 3A, 3B). The impaired upregulation of gut-homing receptors observed on Itk−/− cells was not a consequence of choosing a single dose of RA or TGF-β, as extensive titrations of these ligands failed to alter the responses of Itk−/− cells (Supplemental Fig. 2A–F). We also assessed expression of TGF-βRII and RARα to determine whether impaired receptor expression might account for the reduced responses of Itk−/− cells to these compounds. Although we observed an interesting dose-response effect of RA on the expression of Tgfbr2, in the presence of the standard doses of RA (10 nM) and TGF-β (5.0 ng/ml) used in our functional studies, no significant differences in Tgfbr2 or Rara mRNA levels were seen when comparing WT and Itk−/− OT-I cells (Supplemental Fig. 2G, 2H). Overall, these data confirm that Itk−/− OT-I cells have impaired responses to TCR stimulation plus RA or TGF-β, leading to suboptimal expression of gut-homing receptors.
FIGURE 3. ITK regulates CD8+ T cell migration to the intestinal mucosa.
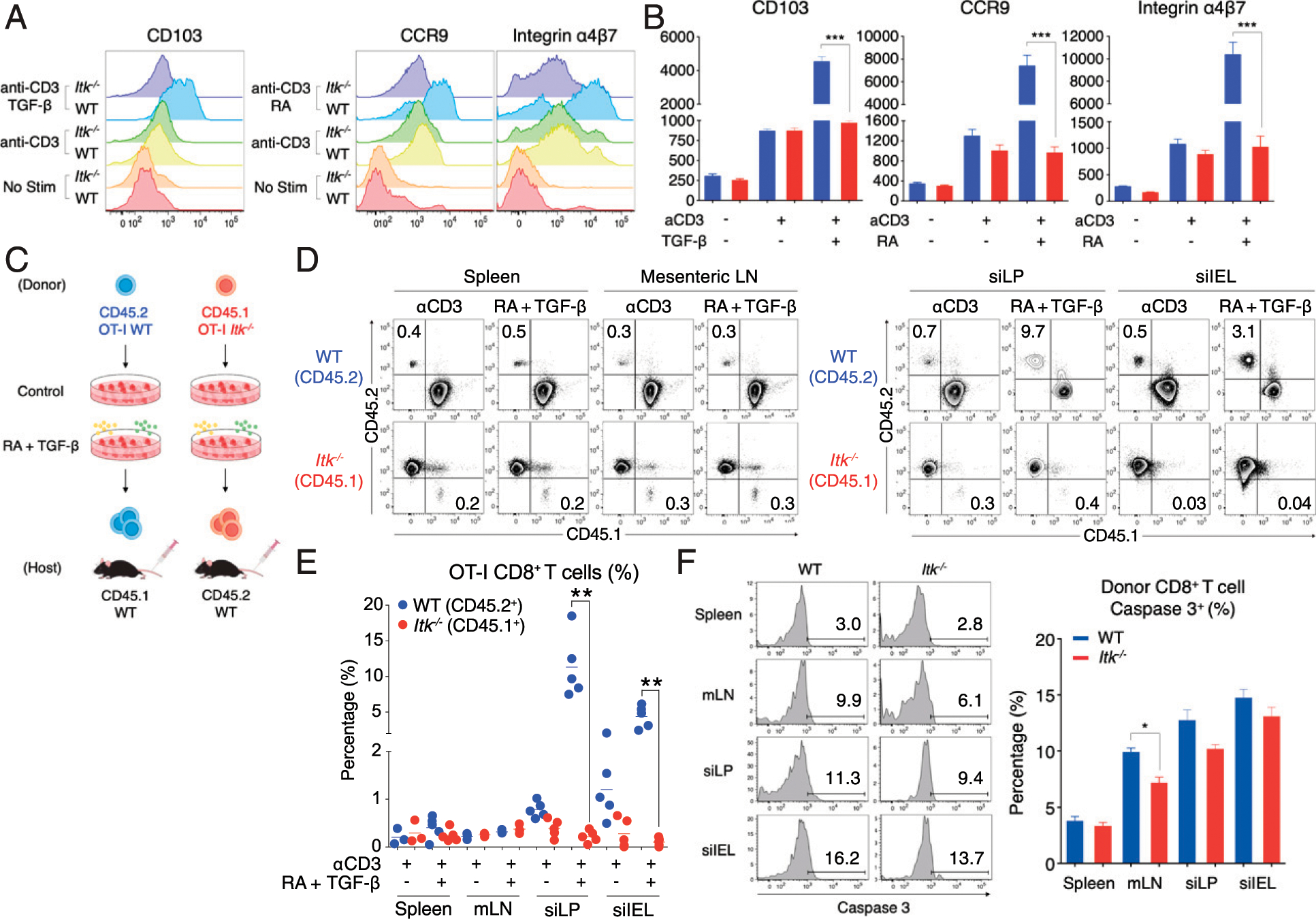
(A and B) Congenically labeled OT-I WT (CD45.2) and Itk−/− (CD45.1) CD8+ T cells were stimulated with anti-CD3 (1.0 μg/ml) in the presence of TGF-β (5.0 ng/ml) or RA (10 nM) for 48 h, and CD103, CCR9, and integrin α4β7 expression were examined. Compilation of data from three independent experiments is shown. (C–E) Congenic WT or Itk−/− OT-I cells (6.0 × 106 cells) were then adoptively transferred to uninfected congenic WT hosts and donor T cells in the spleen, mLN, and small intestine (LP or epithelium) were analyzed at day 3 posttransfer. Compilation of data from two independent experiments is shown; each data point represents an individual recipient. (F) Donor OT-I cells were harvested from the spleen, mLN, siLPL, and siIEL of congenic WT hosts at day 3 posttransfer. Activated caspase 3 expression on WT and Itk−/− cells were examined. Caspase 3+ proportions were compiled from two independent experiments and are shown. Statistical significance between individual groups was analyzed using unpaired or paired two-tailed Student t tests (*p < 0.05, **p < 0.01, ***p < 0.001).
Next, we tested whether this defect in gut-homing receptors would lead to a homing defect in vivo. We stimulated WT or Itk−/− OT-I cells with anti-CD3 in the presence of RA (10 nM) and TGF-β (5.0 ng/ml) for 48 h as above and then adoptively transferred the cells to congenic naive WT recipients (Fig. 3C). Three days after cell transfer, recipient mice were sacrificed, and OT-I T cell proportions in the spleen, mLN, and small intestine LPL and IEL compartments were determined. No differences were seen between WT and Itk−/− OT-I T cells in the spleen or mLN, regardless of stimulation conditions (Fig. 3D, 3E). When recipient mice received WT OT-I cells that had been stimulated with anti-CD3 plus RA and TGF-β, substantial populations of transferred cells were detected in the intestinal tissue, particularly in the siLPL compartment, compared with mice that received WT OT-I cells that had been primed in vitro with anti-CD3 stimulation alone. Recipients injected with Itk−/− T cells had few transferred cells in the intestinal tissue, regardless of whether RA and TGF-β were present during their initial stimulation. To determine whether the low numbers of Itk−/− T cells recovered from the recipient mice were a result of enhanced cell death in the absence of ITK, we examined activated caspase 3 expression in the donor cells recovered following transfer of WT or Itk−/− cells. Although a modest reduction in caspase 3+ Itk−/− cells was seen in mLN, no other differences were detected when comparing WT to Itk−/− cells in the spleen or the small intestinal mucosa (Fig. 3F). These data are consistent with a defect in Itk−/− T cell migration into the small intestine LP and IEL, rather than a substantial increase in Itk−/− T cell death after adoptive transfer. Overall, these findings indicate that ITK is required for CD8+ T cells to acquire gut-homing properties in response to TCR plus RA and TGF-β stimulation and that this function correlates with ITK signaling functioning in the upregulation of gut-homing receptors during T cell priming.
Latent MHV68 infection in Itk−/− mice results in failure to control viral replication in the intestine
Several clinical studies reported a lethal lymphoproliferation in EBV patients with ITK mutations due to the failure to control viral replication (32, 33). To investigate possible functional consequences of the T cell gut-homing defect observed in Itk−/− mice, we infected WT and Itk−/− mice with 106 PFU of MHV68 via i.p. injection. We first examined the CD8+ T cell response in the spleen, a major site of virus replication. This analysis revealed a reduced frequency of MHV68-specific CD8+ T cells in Itk−/− mice compared with controls at both day 7 and day 14 postinfection (Fig. 4A). Upon ex vivo stimulation with the immunodominant epitope ORF75c (43), a smaller percentage of Itk−/− CD8+ T cells produced IFN-γ, and in addition, the frequencies of ORF75c-specific CD8+ T cells were reduced in Itk−/− compared with WT infected mice, a difference that persisted to day 60 postinfection (Fig. 4B–D). MHV68 viral DNA copy numbers in the spleens were higher in infected Itk−/− mice than in WT mice at day 7 postinfection, but by day 14 and later, no further differences in splenic viral DNA copy number were observed (Fig. 4E). This modestly impaired antiviral immune response by Itk−/− mice is consistent with previous studies of other acute virus infections (36, 37). Despite the reduced magnitude of the antiviral CD8+ T cell response, Itk−/− mice ultimately cleared each of these acute viral infections and showed no defect in the control of acute MHV68 infection in the spleen.
FIGURE 4. MHV68-infected Itk−/− mice exhibit impaired viral control and enhanced pathology in the gastrointestinal tract.

(A) Scheme of MHV68 infection (i.p.) of WT and Itk−/− mice. (B) Isolated splenocytes at day 7 and day 14 postinfection were stimulated with the MHV68-ORF75c peptide for 5 h and stained for intracellular IFN-γ. Data are representative of two to four experiments per timepoint. Numbers on dot-plots indicate mean ± SEM of IFN-γ producing cells. (C and D) The frequencies (C) and absolute numbers (D) of MHV68-specific CD8+ splenocytes were determined by tetramer staining. (E) Viral copy number of MHV68 DNA in the spleen from three to four experiments per timepoint. (F) Survival of WT (n = 24) and Itk−/− (n = 24) mice after MHV68 infection. (G and H) Compilation of viral DNA copy numbers in sections of small and large intestine (Col, Colon; Duo, duodenum; Ile, Ileum; Jej, Jejunum) at D180 (G) and day 7 and day 14 (H) postinfection. (I) Lymphocytes were harvested from the spleen, siLPL, and siIEL of MHV68-infected WT and Itk−/− mice at day 7 postinfection. Cells were stimulated with MHV68-ORF75c peptide for 5 h and stained for intracellular granzyme B in CD8+ T cells. Granzyme B–producing cell proportions were compiled from three experiments and are shown. (J) WT and Itk−/− mice were infected with MHV68 (i.p. or i.n.) for 60 d and then analyzed 3 d after LPS injection. Histology of duodenum is shown; images are at original magnification ×10. Scale bar, 75 μm. Statistical significance between individual groups was analyzed using unpaired or paired two-tailed Student t tests (*p < 0.05, **p < 0.01, ***p < 0.001).
Additional cohorts of WT and Itk−/− mice were infected with MHV68 as above and were assessed for their ability to control latent MHV68. In contrast to WT mice, which remained healthy throughout the time course of these studies, Itk−/− mice showed signs of gastrointestinal distress and began to succumb to the infection after long-term latency (>day 100 postinfection) (Fig. 4F). The impaired survival of latently infected Itk−/− mice was accompanied by high viral loads in the small intestine (Fig. 4G). These findings prompted us to examine viral DNA copy numbers in the intestine at early timepoints postinfection (days 7 and 14), revealing that viral DNA copy numbers were significantly higher in the small intestines of Itk−/− mice compared with WT controls at these times (Fig. 4H). Consistent with a failure of virus control, Itk−/− CD8+ T cells in both the spleen and the intestine showed an impaired granzyme B production compared with WT CD8+ T cells at day 7 of postinfection in response to a viral peptide stimulation (Fig. 4I). These data suggest that Itk−/− mice fail to control viral replication in the intestine from the early to late phase of infection, which leads to viral persistence.
We also tested whether latently infected Itk−/− mice displayed exacerbated pathology following viral reactivation induced by TLR agonists, such as LPS or CpG DNA (44). WT and Itk−/− mice were infected with MHV68 and then treated with a single i.p. injection of a subclinical dose of LPS (20 μg/mouse) at varying time points postinfection (days 30, 60, 90, and 100) (Supplemental Fig. 3A). Interestingly, Itk−/− mice harboring latent MHV68 and treated with LPS exhibited severe gut inflammation; furthermore, the severity of the inflammation increased as the latency period increased, such that the majority (11 out of 18, 61.1%) of Itk−/− mice given LPS at day 90–100 postinfection succumbed within 1–2 d of treatment (Supplemental Fig. 3B, 3C). In contrast, latently infected WT mice given LPS, naive WT and Itk−/− mice, and both WT and Itk−/− mice given LPS at the early time point (day 7) were all devoid of detectable gut pathology and severe inflammation (Supplemental Fig. 3D–F). A similar set of findings was also observed in Itk−/− mice given their primary MHV68 infection by the i.n. route (1.0 × 103 PFU per mouse). When these latently infected mice were treated with a single i.p. injection of LPS at day 60 postinfection, severe gut pathology was observed (Fig. 4J). Examination of the lungs of these mice showed only a modest level of inflammation and no obvious differences between WT and Itk−/− responses (Supplemental Fig. 3G). Collectively, these results suggest that ITK is required for the control of latent MHV68 infection specifically in the intestine.
Antiviral CD8+ T cells require ITK for migration to the intestinal tissue
In light of the intestinal pathology observed in MHV68-infected Itk−/− hosts, we considered whether these mice had a defect in generating antiviral CD8+ T cell responses in the intestine. We assessed MHV68-specific CD8+ T cells in the spleen, mLN, IEL, and LPL compartments from both small intestine and colon tissue at day 7 postinfection in WT and Itk−/− mice. Itk−/− mice had significantly reduced numbers of total CD8+ T cells in the mLN, IEL, and LPL, and consequently, had very few virus-specific cells in these compartments (Fig. 5A). Although the numbers of total splenic CD8+ T cells in infected WT versus Itk−/− mice were comparable, fewer CD8+ T cells were found in the intestine of Itk−/− mice (Fig. 5B); furthermore, both tetramer-positive CD8+ T cell proportions and absolute numbers were significantly reduced in all tissues of Itk−/− mice, with the largest difference seen in siLPL (Fig. 5A, 5C).
FIGURE 5. Effector CD8+ T cell numbers in the intestinal tissue of MHV68-infected mice are reduced in the absence of ITK.
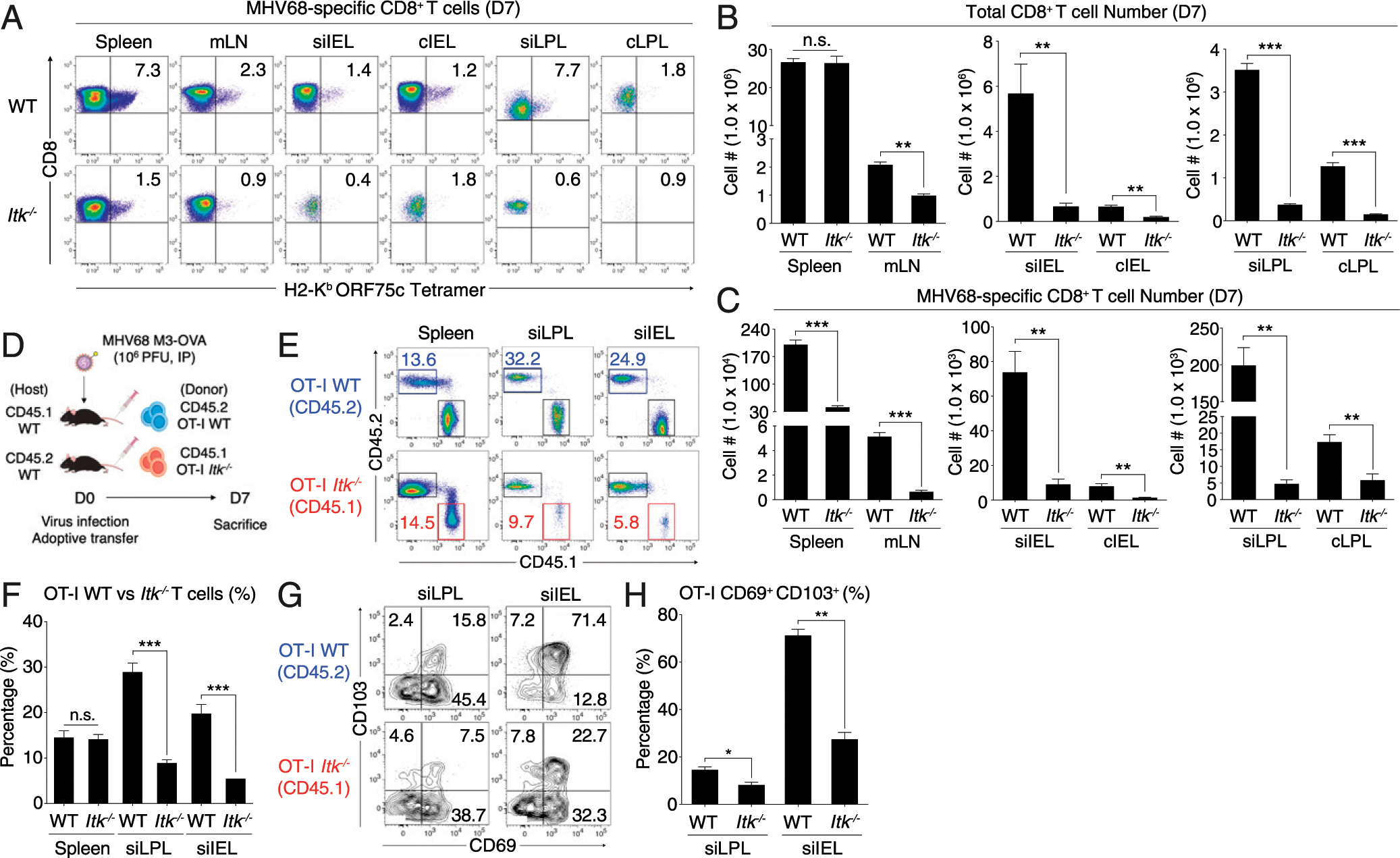
(A) MHV68-specific CD8+ T cells in various organs at day 7 postinfection were identified by tetramer staining. Data are representative of three experiments. (B and C) Compilations of absolute numbers of total (B) and MHV68-specific (C) CD8+ T cells in various organs at day 7 postinfection. (D–F) Congenically labeled OT-I WT (CD45.2, blue box) or Itk−/− (CD45.1, red box) CD8+ T cells (5.0 × 105 cells) were transferred into congenic WT recipients. Recipient mice were infected with MHV68-M3-OVA and OT-I cells were analyzed at day 7 postinfection (E). Compilation of data from four to six infected recipients per group is shown (F). (G and H) OT-I cells shown in (A) from the small intestine were analyzed for CD69 and CD103 expression (G). Compilation of percentages of CD69+ CD103+ OT-I T cells from flow cytometric data are shown (H). Statistical significance between individual groups was analyzed using unpaired or paired two-tailed Student t tests (*p < 0.05, **p < 0.01, ***p < 0.001).
To determine if the defect in Itk−/− CD8+ T cell numbers in the intestine is a feature intrinsic to a lack of ITK in these cells or to defects caused by altered environmental factors in Itk−/− mice, we performed adoptive transfer studies. Recipient mice received congenically marked WT (CD45.2) or Itk−/− (CD45.1) OT-I TCR transgenic (Rag−/−) CD8+ T cells (5.0 × 105), followed by infection with an MHV68 strain expressing OVA (MHV68-M3-OVA) (Fig. 5D). Despite the reduced percentages of MHV68-specific CD8+ T cells in the spleens of MHV68-infected intact Itk−/− compared with WT mice, no such difference was observed for WT versus Itk−/− OT-I T cells in the spleens of adoptive transfer recipients at day 7 postinfection. In contrast, the frequencies of Itk−/− OT-I T cells in the small IEL and LP were significantly reduced compared with those of WT OT-I cells (Fig. 5E, 5F). The Itk-deficient OT-I T cells in these tissues also had smaller proportions expressing CD69 and CD103 compared with WT OT-I cells (Fig. 5G, 5H), consistent with a potential defect in Itk−/− CD8+ T cell migration and/or tissue retention in the intestine. Taken together, these data indicate that ITK is required in CD8+ T cells for efficient homing and/or retention of these cells in the intestinal tissue in response to virus.
Impaired IRF4 expression leads to the defect in gut-homing receptor expression
Based on the data presented above, we questioned how ITK signaling might regulate gut-homing properties of effector CD8+ T cells. One likely candidate to mediate this function is a transcription factor that is highly responsive to TCR signaling in an ITK-dependent manner, such as IRF4 (28). IRF4 is also known to bind to regulatory regions of gut-homing receptor genes, as examination of published datasets revealed IRF4/basic leucine zipper ATF-like transcription factor (BATF) complex binding sites on the Ccr9, Itgae, Itga4, and Itgb7 loci in activated T cells (27, 44). As an example (see Supplemental Fig. 4A), both Ccr9 and Itga4 appear to have prominent IRF4/BATF binding sites. To test whether IRF4 expression levels might be important in programming activated CD8+ T cells for gut-homing, we assessed steady-state T cell numbers in the small intestine of Irf4 heterozygous mice (Irf4+/fl × CD4-Cre; hereafter referred to as Irf4+/fl), in which TCR-dependent IRF4 expression levels are reduced but not abolished (24, 45). Because of a dysregulated immune system in homozygous Irf4fl/fl mice (28, 46), mice bearing T cells lacking all IRF4 expression were not examined. In Irf4+/fl mice, steady-state numbers of CD4+ T cells in the small IEL and LP were comparable to wild-type littermate controls. In contrast, we observed reduced proportions and numbers of Irf4+/fl CD8+ T cells in the IEL and LPL compared with controls (Fig. 6A, 6B). When assessed for CD103, CCR9, and integrin α4β7 expression, we also found that CD8+ T cells from the small intestine of Irf4+/fl had reduced expression of CD103 compared with wild-type cells but no change in expression of CCR9 or integrin α4β7 (Fig. 6C, 6D).
FIGURE 6. Impaired IRF4 expression in CD8+ T cell leads to a defect in gut-homing receptor expression.
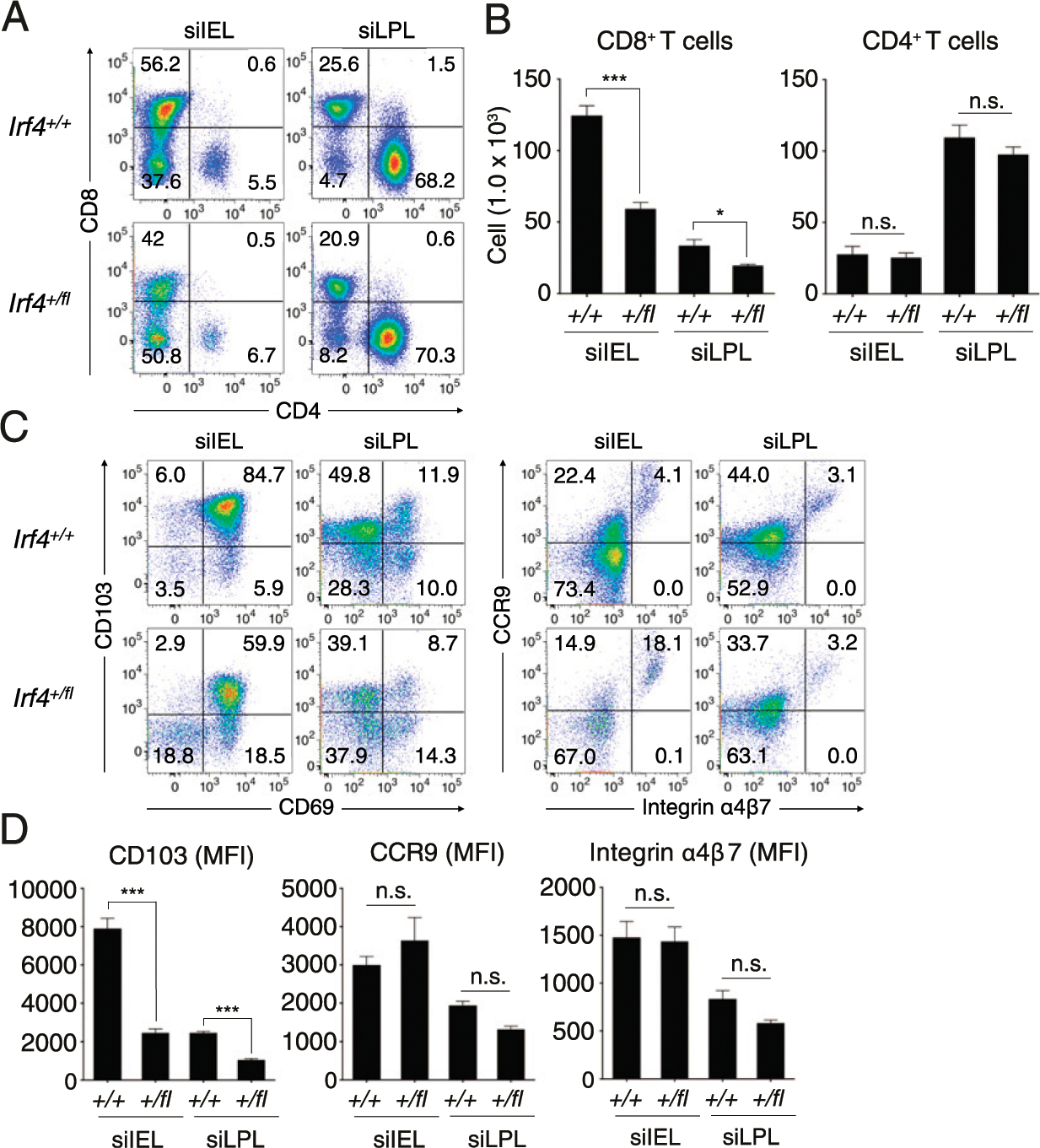
(A and B) The proportions (A) and numbers (B) of CD8+ and CD4+ T lymphocytes (gated on TCRβ+) from the siIEL and siLPL of Irf4+/+ and Irf4+/fl mice. (C and D) Expression of CD69 versus CD103 or integrin α4β7 versus CCR9 from CD8+ T cells from siIEL and siLPL of Irf4+/+ and Irf4+/fl mice (C). Mean fluorescence of each molecule was enumerated at the bottom (D). Data shown are compilations of five to seven mice of each genotype from two to three independent experiments. Statistical significance between individual groups was analyzed using unpaired or paired two-tailed Student t tests (*p < 0.05, **p < 0.01, ***p < 0.001).
To assess more directly whether optimal expression of IRF4 is important in regulating gut-homing receptor expression during T cell activation, we tested Irf4+/fl CD8+ T cells for their in vitro responses to stimulation in the presence of RA or TGF-β. Consistent with the data shown above for Irf4+/fl CD8+ T cells analyzed directly ex vivo from the small intestine, we observed reduced expression of CD103 on Irf4+/fl CD8+ T cells compared with control T cells in response to varying doses (0.5–5.0 ng/ml) of TGF-β (Supplemental Fig. 4B). Parallel studies examining the responsiveness of CCR9 and integrin α4β7 to RA revealed reduced CCR9 expression by Irf4+/fl CD8+ T cells compared with WT controls in the presence of low levels of RA (Supplemental Fig. 4C) but no consistent differences in integrin α4β7 expression (Supplemental Fig. 4D). These correlative data suggested a mechanistic link between ITK signaling and IRF4 upregulation in the priming of efficient gut-homing CD8+ effector T cells.
Enforced expression of IRF4 in Itk−/− CD8+ T cells restores migration to the intestine
The findings with Irf4+/fl CD8+ T cells prompted us to test whether enforced expression of IRF4 could restore efficient gut-homing properties to Itk−/− CD8+ T cells. For these studies, WT and Itk−/− OT-I CD8+ T cells were activated in vitro and then transduced with a RV carrying GFP alone (empty vector) or GFP plus IRF4 (IRF4). Following this, transduced T cells were cultured with RA and TGF-β for 48 h and then adoptively transferred into congenic naive WT hosts. Three days after transfer, recipient mice were analyzed for OT-I T cells in the small intestine (Fig. 7A). As shown, WT and Itk−/− OT-I cells were expressing comparable levels of GFP from the empty vector or the IRF4 virus, respectively (Fig. 7B). We then assessed the expression levels of IRF4 in both WT and Itk−/− OT-I T cells (Fig. 7C). As expected, Itk−/− T cells transduced with empty virus expressed lower levels of IRF4 compared with WT T cells, and in both cases, IRF4 expression levels were increased in cells transduced with the IRF4-expressing virus (Fig. 7C).
FIGURE 7. Enforced expression of IRF4 restores Itk−/− CD8+ T cell migration to the intestine.
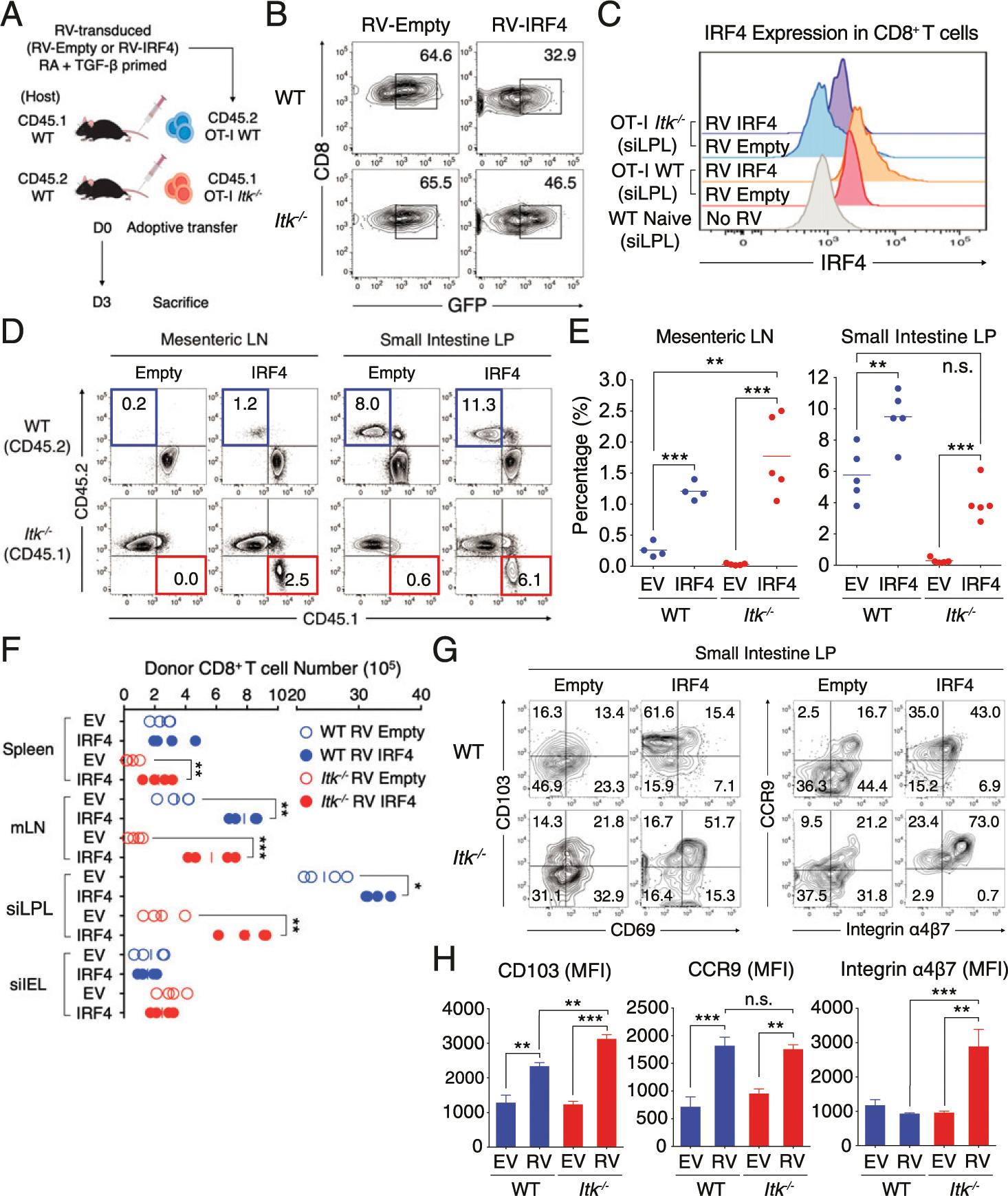
(A) RV-transduced (Empty vector or IRF4) congenically labeled WT (CD45.2) and Itk−/− (CD45.1) CD8+ T cells were primed with RA and TGF-β for 48 h and then adoptively transferred (4.0 × 107) to congenic WT naive host. (B and C) RV transduction efficiency (GFP+) (B) and IRF4 expression (C) of WT and Itk−/− CD8+ T cells from small intestine LP at day 3 of posttransfer are shown. No RV control shows the level of IRF4 expression in naive WT CD8+ T cells from siLPL. (D–F) Transferred T cells (WT, blue; Itk−/−, red) were isolated from mLN (left panel) and small intestinal LP (right panel) at day 3 posttransfer (D). Compilation data from four to five mice per each group from two to three independent experiments are shown at the right (E). Cell numbers of RV-transduced (Empty vector or IRF4) and RA- and TGF-β–primed donor OT-I cells in the spleen, mLN, siLPL and siIEL were enumerated in the congenic naive WT hosts at day 3 posttransfer (F). (G and H) The expression of CD69 versus CD103 (left) or integrin α4β7 versus CCR9 (right) in transferred T cells in small intestinal LP is shown (G). Mean fluorescence of each molecule was enumerated in the graph at the right (H). Statistical significance between individual groups was analyzed using unpaired or paired two-tailed Student t tests (*p < 0.05, **p < 0.01, ***p < 0.001).
Next, we assessed the localization of the transferred cells in the recipients. We first observed increased proportions and absolute numbers of both IRF4 transduced WT and Itk−/− CD8+ T cells in the mLN and small intestine LP of hosts compared with cells transduced with empty vector (Fig. 7D–F). Examination of transferred cells in these sites indicated substantial improvement in the gut-homing capacity of Itk−/− CD8+ T cells when transduced with the IRF4 virus compared with the empty vector; in these studies, enforced IRF4 expression restored the gut-homing of Itk−/− CD8+ T cells to that seen for empty virus-transduced WT T cells. Further supporting a key role for IRF4 in regulating gut-homing properties of CD8+ T cells, even WT T cells transduced with the IRF4 virus showed improved gut-homing compared with cells transduced with empty vector. Interestingly, this improved gut-homing capability via IRF4 overexpression in Itk−/− T cells was correlated with enhanced gut-homing receptor expression upon RV transduction (Fig. 7G, 7H). Together, these data confirm that optimal IRF4 expression is critical for gut-homing of effector CD8+ T cells and support the conclusion that IRF4 expression levels provide the link between TCR signaling mediated by ITK to the migration of CD8+ T cells to the intestine by inducing optimal expression of gut-homing receptors.
DISCUSSION
In this study, we show that mice lacking ITK have a dramatic reduction in tissue-resident T cells in the intestinal mucosa and that as a consequence, Itk−/− mice are unable to mount a protective immune response to a viral infection in the gastrointestinal tract. Previous studies have shown that long-term maintenance of CD8+ TRM in mucosal tissues provides for an immediate protective response against reinfection by the same virus or due to reactivation of a latent virus (47–50). TRM are also known to express constitutively high levels of cytolytic molecules, such as granzyme B and perforin, despite the absence of Ag after viral clearance (51). These data support the notion that the lack of gut CD8+ T cells in Itk−/− mice is strongly associated with the ongoing uncontrolled replication of MHV68 in this tissue. Consistent with this, the pathology showing tissue disruption was only observed in the intestine but not in other organs (e.g., spleen or lung) of infected Itk−/− mice.
Although Itk−/− mice have a steady-state deficiency in gut-resident T cells, we do not observe spontaneous gastrointestinal disease in uninfected Itk−/− mice. Furthermore, administration of a single dose of LPS (i.p.) into uninfected Itk−/− mice also caused no tissue pathology, despite its known effect on intestinal epithelial permeability (52, 53). However, LPS treatment of latently MHV68-infected Itk−/− mice caused a massive disruption of IEL regardless of the route of infection. We reasoned that long-term persistence of virus in the intestine caused a low level of chronic tissue damage, as has previously been reported in mice infected with CMV (54). Thus, when these mice are treated with LPS, the increase in intestinal epithelial permeability induced by the LPS triggers an irreparable level of acute tissue damage that is fatal in long-term latently infected Itk−/− mice.
To test the whether the defect in Itk−/− CD8+ T cells in the intestine was due to an intrinsic role for ITK in these T cells, we transferred congenically marked WT or Itk−/− OT-I CD8+ T cells into recipients. Although a cotransfer of WT and Itk−/− cells would have been preferable, we felt that the added time and expense of breeding CD45.1/CD45.2 heterozygous mice needed to perform the cotransfer experiment were not warranted in this case, as our separate adoptive transfer studies revealed a profound defect in gut homing/persistence of transferred Itk−/− OT-I T cells.
Our data also indicated that production of effector molecules (e.g., IFN-γ and granzyme B) is broadly impaired in Itk−/− CD8+ T cells from both the spleen and the intestine after viral infection (Fig. 4B, 4I). Furthermore, Itk−/− mice also show an overall reduction in the numbers of virus-specific CD8+ T cells postinfection (Fig. 5A, 5C). Therefore, one component of the impaired control of MHV68 in the intestines of Itk−/− mice is likely to be the overall reduced antiviral T cell response that is visible in all tissues of the mice. In this regard, we also have not directly investigated whether ITK is important in antiviral CD4+ T cells. Our previous studies examining CD4+ T cell–mediated colitis in the adoptive transfer model have indicated that CD4+ T cells may also require ITK signaling for efficient gut-homing (12) and thus may also be required for efficient viral control in the intestine.
Although further studies are warranted to fully elucidate a mechanism for the gut-specific failure of virus control in Itk−/− mice, we reason that a population of TRM is critical for the control of virus replication in the intestine, the largest mucosal immune tissue in the host. To support this notion, the impaired viral control in the small intestine of Itk−/− mice correlates with the more substantial T cell deficit in this tissue site (Figs. 1, 5). Although we did not examine antiviral immunity of Itk−/− mice to other enteric viruses, our data suggest that effective protection against gut-specific viral persistence is likely to be associated with the successful generation of TRM in the intestinal barrier, along with the functional competence of these effector cells in promoting antiviral immunity.
The current study reinforces previous data demonstrating an important role for ITK signaling in the upregulation of IRF4 in T cells stimulated through the TCR. We previously reported that a small molecule inhibitor of ITK could potently inhibit IRF4 expression in stimulated OT-I CD8+ T cells (28). In this report, we confirm this finding in genetically ITK-deficient CD8+ T cells and show that impaired IRF4 expression in Itk−/− OT-I T cells correlates with reduced T cell numbers in the small intestine after adoptive transfer (Fig. 7C). ITK has also been shown to be important for IRF4 expression in CD4+ T cells (8). To date, the precise pathway from ITK to IRF4 upregulation is not fully known. Studies of IRF4 transcriptional regulation have implicated NFAT (25), a transcription factor that is known to be ITK dependent for its activation in T cells (4). Whether a defect in NFAT activation alone accounts for the impaired IRF4 upregulation in Itk−/− T cells or whether other factors contribute to IRF4 expression will be the subject of future studies of this signaling pathway.
We observed enhanced gut homing and/or retention of both WT and Itk−/− CD8+ T cells after retroviral transduction of IRF4 in these cells. It is also possible that IRF4 is mediating enhanced survival of activated CD8+ T cells after adoptive transfer. Although we did not see significant increases in both WT and Itk−/− transferred T cells in the spleens after adoptive transfer of IRF4-transduced cells, we did find increases in cell numbers in the mLNs as well as the intestinal tissue (Fig. 7F). Additionally, the increased numbers of IRF4-transduced cells in the small intestinal LP correlated with increased expression of gut-homing receptors. Thus, these data support a role for IRF4 in regulating gut homing and/or retention but do not definitively rule out an additional role for IRF4 in T cell survival, as previously reported (24, 25).
Our work establishes a defect in gut-homing and/or gut tissue retention of Itk−/− CD8+ T cells. In an autoimmune disease setting, the migratory function of CD4+ T cells to multiple organs, such as liver and lung, is severely impaired in the absence of ITK (9). We previously examined the status of Itk−/− T cells in the lung, as this is an important route of MHV68 infection (38). Unlike the cellular defect in the gut, Itk−/− mice do not show any significant reduction in T cell numbers in the lung at steady-state. Consistently, previous studies showed that Itk−/− mice elicit a comparable antiviral response in the lung relative to WT mice, following influenza A virus infection (55). More comprehensive studies examining the migratory capacity of Itk−/− T cells to other organs and various tissue sites are clearly warranted in the future.
Our data indicate that T cell priming in the mLN fails to upregulate high levels of CD103, integrin α4β7, and CCR9 on Itk−/− CD8+ T cells. This defect can be recapitulated by in vitro stimulation of naive Itk−/− OT-I CD8+ T cells in the presence of TGF-β and RA. In addition, transfer of these in vitro–activated T cells into WT recipients confirms their gut-homing defect. Impaired expression of CD103 on primed Itk−/− CD8+ T cells can be explained by the demonstrated cooperativity of TGF-β–induced Smad2/3 and TCR-induced NFAT1 activation for optimal CD103 induction (42), as ITK signaling is known to be required for robust TCR-induced NFAT1 activation (4). However, examination of integrin α4β7 and CCR9 upregulation on cells stimulated with varying doses of TGF-β and/or RA indicates that Itk−/− CD8+ T cells have a more global defect in responding to TGF-β and RA. We tested the possibility that this might result from impaired expression of TGF-βRII and/or RARα in Itk−/− CD8+ T cells. Although we did observe reduced Tgfbr2 mRNA in Itk−/− CD8+ T cells stimulated with low concentrations of RA, no impairment in Tgfbr2 expression was seen in Itk−/− CD8+ T cells when stimulations were performed in the presence of TGF-β plus RA. For Rara mRNA expression, we did observe modest reductions (2–3-fold) in Itk−/− compared with WT CD8+ T cells after stimulation through the TCR plus TGF-β and RA, but we have no direct evidence that this difference is functionally relevant. As an alternative, we suggest that Itk−/− CD8+ T cells may lack other factors (e.g., Smad3, other RAR isoforms, or retinoid X receptor [RXR]) required for optimal responses to TGF-β and RA.
Transcriptome analyses of tissue-resident T cells in the brain or lung tissues of humans and mice have revealed that IRF4 is one of the most highly upregulated transcription factors in these cells (29, 30, 56). However, detailed functional studies of IRF4 in T cell homing to mucosal barriers have not been reported, although one study examined IRF4 in homing of Foxp3+ regulatory T cells to visceral adipose tissue (57). Thus, to our knowledge, our data provide new insights into how TCR signaling via the ITK–IRF4 axis controls the migration of CD8+ T cells to the intestine. Interestingly, Kurachi et al. (58) and others have found that IRF4/BATF bind directly to the 5′ upstream regions of gut-homing molecule genes (e.g., Ccr9 and Itga4) (27, 59). Furthermore, BATF, the most important binding partner of IRF4, is known to be required for the expression of CCR9 and integrin α4β7 in response to RA in CD4+ T cells, and BATF-deficient T cells are greatly impaired in trafficking to the small intestine (44). Although we have not tested whether enforced expression of BATF would also restore gut-homing potential to Itk−/− T cells, we reasoned that IRF4 was a more likely candidate in our system because of the greater magnitude of Irf4 induction after TCR stimulation compared with Batf (ImmGen Consortium Database).
Our previous data connect ITK signaling to the magnitude of IRF4 upregulation (28). Importantly, a recent study reported that IRF4 haploinsufficiency in humans leads to severe gastrointestinal inflammation due to an impaired control of T. whipplei (31). Although this clinical report did not directly assess whether patients exhibited impaired T cell homing the intestine, our data provide a potential mechanism to account for this surprising susceptibility in patients carrying a single allele deficiency in IRF4.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the Berg Laboratory, especially Regina White-head and Sharlene Hubbard, for technical assistance. We also thank the University of Massachusetts Medical School’s Department of Animal Medicine for the maintenance of mouse colonies. We thank Samuel H. Speck (Emory Vaccine Center) and Philip G. Stevenson (The University of Queensland, Australia) for permission to use MHV68 and its engineered strains. This work benefitted from data assembled by the ImmGen Consortium (www.immgen.org).
This work was supported by National Institutes of Allergy and Infectious Disease Grant AI32417.
Abbreviations used in this article:
- BATF
basic leucine zipper ATF-like transcription factor
- cIEL
colon IEL
- cLPL
colon LPL
- FP
forward primer
- IEL
intraepithelial lymphocyte
- i.n.
intranasal
- IRF4
IFN regulatory factor 4
- ITK
IL-2–inducible T cell kinase
- LP
lamina propria
- LPL
lamina propria lymphocyte
- MHV68
mouse gammaherpesvirus 68
- mLN
mesenteric lymph node
- RA
all-trans retinoic acid
- RP
reverse primer
- RV
retrovirus
- siIEL
small intestine IEL
- siLPL
small intestine LPL
- TRM
tissue-resident memory T cell
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
DISCLOSURES
The authors have no financial conflicts of interest.
REFERENCES
- 1.Berg LJ, Finkelstein LD, Lucas JA, and Schwartzberg PL. 2005. Tec family kinases in T lymphocyte development and function. Annu. Rev. Immunol 23: 549–600. [DOI] [PubMed] [Google Scholar]
- 2.Andreotti AH, Schwartzberg PL, Joseph RE, and Berg LJ. 2010. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb. Perspect. Biol DOI: 10.1101/cshperspect.a002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andreotti AH, Joseph RE, Conley JM, Iwasa J, and Berg LJ. 2018. Multidomain control over TEC kinase activation state tunes the T cell response. Annu. Rev. Immunol 36: 549–578. [DOI] [PubMed] [Google Scholar]
- 4.Fowell DJ, Shinkai K, Liao XC, Beebe AM, Coffman RL, Littman DR, and Locksley RM. 1999. Impaired NFATc trans-location and failure of Th2 development in Itk-deficient CD4+ T cells. Immunity 11: 399–409. [DOI] [PubMed] [Google Scholar]
- 5.Miller AT, Wilcox HM, Lai Z, and Berg LJ. 2004. Signaling through Itk promotes T helper 2 differentiation via negative regulation of T-bet. Immunity 21: 67–80. [DOI] [PubMed] [Google Scholar]
- 6.Schwartzberg PL, Finkelstein LD, and Readinger JA. 2005. TEC-family kinases: regulators of T-helper-cell differentiation. Nat. Rev. Immunol 5: 284–295. [DOI] [PubMed] [Google Scholar]
- 7.Gomez-Rodriguez J, Sahu N, Handon R, Davidson TS, Anderson SM, Kirby MR, August A, and Schwartzberg PL. 2009. Differential expression of interleukin-17A and -17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity 31: 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomez-Rodriguez J, Meylan F, Handon R, Hayes ET, Anderson SM, Kirby MR, Siegel RM, and Schwartzberg PL. 2016. Itk is required for Th9 differentiation via TCR-mediated induction of IL-2 and IRF4. Nat. Commun DOI: 10.1038/ncomms10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain N, Miu B, Jiang J-K, McKinstry KK, Prince A, Swain SL, Greiner DL, Thomas CJ, Sanderson MJ, Berg LJ, and Kang J. 2013. CD28 and ITK signals regulate autoreactive T cell trafficking. Nat. Med 19: 1632–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mueller C, and August A. 2003. Attenuation of immunological symptoms of allergic asthma in mice lacking the tyrosine kinase ITK. J. Immunol 170: 5056–5063. [DOI] [PubMed] [Google Scholar]
- 11.Schaeffer EM, Yap GS, Lewis CM, Czar MJ, McVicar DW, Cheever AW, Sher A, and Schwartzberg PL. 2001. Mutation of Tec family kinases alters T helper cell differentiation. Nat. Immunol 2: 1183–1188. [DOI] [PubMed] [Google Scholar]
- 12.Cho H-S, Shin HM, Haberstock-Debic H, Xing Y, Owens TD, Funk JO, Hill RJ, Bradshaw JM, and Berg LJ. 2015. A small molecule inhibitor of ITK and RLK impairs Th1 differentiation and prevents colitis disease progression. J. Immunol 195: 4822–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kannan AK, Kim D-G, August A, and Bynoe MS. 2015. Itk signals promote neuroinflammation by regulating CD4+ T-cell activation and trafficking. J. Neurosci 35: 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, et al. 2010. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med 207: 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller SN, and Mackay LK. 2016. Tissue-resident memory T cells: local specialists in immune defence. Nat. Rev. Immunol 16: 79–89. [DOI] [PubMed] [Google Scholar]
- 16.Shin H, and Iwasaki A. 2013. Tissue-resident memory T cells. Immunol. Rev 255: 165–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johansson-Lindbom B, Svensson M, Wurbel M-A, Malissen B, Márquez G, and Agace W. 2003. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J. Exp. Med 198: 963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Förster R, and Agace WW. 2005. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J. Exp. Med 202: 1063–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, and Von Andrian UH. 2003. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 424: 88–93. [DOI] [PubMed] [Google Scholar]
- 20.Mora JR, Cheng G, Picarella D, Briskin M, Buchanan N, and von Andrian UH. 2005. Reciprocal and dynamic control of CD8 T cell homing by dendritic cells from skin- and gut-associated lymphoid tissues. J. Exp. Med 201: 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon M-L, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, et al. 2013. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol 14: 1294–1301. [DOI] [PubMed] [Google Scholar]
- 22.Skon CN, Lee J-Y, Anderson KG, Masopust D, Hogquist KA, and Jameson SC. 2013. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol 14: 1285–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang N, and Bevan MJ. 2013. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39: 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, Pellegrini M, Belz GT, Smyth GK, Febbraio MA, et al. 2013. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. [Published erratum appears in 2014 Nat. Immunol 15: 894.] Nat. Immunol. 14: 1155–1165. [DOI] [PubMed] [Google Scholar]
- 25.Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt F, Febbraio MA, et al. 2017. Transcription factor IRF4 promotes CD8+ T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity 47: 1129–1141.e5. [DOI] [PubMed] [Google Scholar]
- 26.Yao S, Buzo BF, Pham D, Jiang L, Taparowsky EJ, Kaplan MH, and Sun J. 2013. Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity 39: 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwata A, Durai V, Tussiwand R, Briseño CG, Wu X, Grajales-Reyes GE, Egawa T, Murphy TL, and Murphy KM. 2017. Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat. Immunol 18: 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nayar R, Enos M, Prince A, Shin H, Hemmers S, Jiang J-K, Klein U, Thomas CJ, and Berg LJ. 2012. TCR signaling via Tec kinase ITK and interferon regulatory factor 4 (IRF4) regulates CD8+ T-cell differentiation. Proc. Natl. Acad. Sci. USA 109: E2794–E2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho S-H, Lerner H, et al. 2017. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 20: 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villadangos J, Smyth G, and Bevan MJ. 2012. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol 189: 3462–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guérin A, Kerner G, Marr N, Markle JG, Fenollar F, Wong N, Boughorbel S, Avery DT, Ma CS, Bougarn S, et al. 2018. IRF4 haploinsufficiency in a family with Whipple’s disease. Elife. DOI: 10.7554/eLife.32340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huck K, Feyen O, Niehues T, Rüschendorf F, Hübner N, Laws H-J, Telieps T, Knapp S, Wacker H-H, Meindl A, et al. 2009. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J. Clin. Invest 119: 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linka RM, Risse SL, Bienemann K, Werner M, Linka Y, Krux F, Synaeve C, Deenen R, Ginzel S, Dvorsky R, et al. 2012. Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia 26: 963–971. [DOI] [PubMed] [Google Scholar]
- 34.Mansouri D, Mahdaviani SA, Khalilzadeh S, Mohajerani SA, Hasanzad M, Sadr S, Nadji SA, Karimi S, Droodinia A, Rezaei N, et al. 2012. IL-2-inducible T-cell kinase deficiency with pulmonary manifestations due to disseminated Epstein-Barr virus infection. Int. Arch. Allergy Immunol 158: 418–422. [DOI] [PubMed] [Google Scholar]
- 35.Stepensky P, Weintraub M, Yanir A, Revel-Vilk S, Krux F, Huck K, Linka RM, Shaag A, Elpeleg O, Borkhardt A, and Resnick IB. 2011. IL-2-inducible T-cell kinase deficiency: clinical presentation and therapeutic approach. Haematologica 96: 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atherly LO, Lucas JA, Felices M, Yin CC, Reiner SL, and Berg LJ. 2006. The Tec family tyrosine kinases Itk and Rlk regulate the development of conventional CD8+ T cells. Immunity 25: 79–91. [DOI] [PubMed] [Google Scholar]
- 37.Bachmann MF, Littman DR, and Liao XC. 1997. Antiviral immune responses in Itk-deficient mice. J. Virol 71: 7253–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho H-S, Reboldi A, Hall JA, and Berg LJ. 2019. The Tec kinase ITK is essential for ILC2 survival and epithelial integrity in the intestine. Nat. Commun DOI: 10.1038/s41467-019-08699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith CM, Rosa GTL, May JS, Bennett NJ, Mount AM, Belz GT, and Stevenson PG. 2006. CD4+ T cells specific for a model latency-associated antigen fail to control a gammaherpesvirus in vivo. Eur. J. Immunol 36: 3186–3197. [DOI] [PubMed] [Google Scholar]
- 40.Sunil-Chandra NP, Efstathiou S, Arno J, and Nash AA. 1992. Virological and pathological features of mice infected with murine gamma-herpesvirus 68. J. Gen. Virol 73: 2347–2356. [DOI] [PubMed] [Google Scholar]
- 41.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP. 2011. Integrative genomics viewer. Nat. Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mokrani M, Klibi J, Bluteau D, Bismuth G, and Mami-Chouaib F. 2014. Smad and NFAT pathways cooperate to induce CD103 expression in human CD8 T lymphocytes. J. Immunol 192: 2471–2479. [DOI] [PubMed] [Google Scholar]
- 43.Gredmark-Russ S, Cheung EJ, Isaacson MK, Ploegh HL, and Grotenbreg GM. 2008. The CD8 T-cell response against murine gammaherpesvirus 68 is directed toward a broad repertoire of epitopes from both early and late antigens. J. Virol 82: 12205–12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang C, Thangamani S, Kim M, Gu B-H, Lee JH, Taparowsky EJ, and Kim CH. 2013. BATF is required for normal expression of gut-homing receptors by T helper cells in response to retinoic acid. J. Exp. Med 210: 475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nayar R, Schutten E, Bautista B, Daniels K, Prince AL, Enos M, Brehm MA, Swain SL, Welsh RM, and Berg LJ. 2014. Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J. Immunol 192: 5881–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu T-T, Corcoran L, Treuting P, Klein U, and Rudensky AY. 2009. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458: 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khanna KM, Bonneau RH, Kinchington PR, and Hendricks RL. 2003. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 18: 593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, and Hendricks RL. 2008. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322: 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu T, Khanna KM, Carriere BN, and Hendricks RL. 2001. Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J. Virol 75: 11178–11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheridan BS, Pham Q-M, Lee Y-T, Cauley LS, Puddington L, and Lefrançois L. 2014. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity 40: 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, et al. 2012. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol 188: 4866–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bein A, Zilbershtein A, Golosovsky M, Davidov D, and Schwartz B. 2017. LPS induces hyper-permeability of intestinal epithelial cells. J. Cell. Physiol 232: 381–390. [DOI] [PubMed] [Google Scholar]
- 53.Guo S, Nighot M, Al-Sadi R, Alhmoud T, Nighot P, and Ma TY. 2015. Lipopolysaccharide regulation of intestinal tight junction permeability is mediated by TLR4 signal transduction pathway activation of FAK and MyD88. J. Immunol 195: 4999–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Onyeagocha C, Hossain MS, Kumar A, Jones RM, Roback J, and Gewirtz AT. 2009. Latent cytomegalovirus infection exacerbates experimental colitis. Am. J. Pathol 175: 2034–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang W, Solouki S, Koylass N, Zheng S-G, and August A. 2017. ITK signalling via the Ras/IRF4 pathway regulates the development and function of Tr1 cells. Nat. Commun 8: 15871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Landrith TA, Sureshchandra S, Rivera A, Jang JC, Rais M, Nair MG, Messaoudi I, and Wilson EH. 2017. CD103+ CD8 T cells in the Toxoplasma-infected brain exhibit a tissue-resident memory transcriptional profile. Front. Immunol DOI: 10.1038/ncomms15871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, Fagarasan S, Mielke LA, Afshar-Sterle S, Masters SL, et al. 2015. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. [Published erratum appears in 2015 Nat. Immunol 16: 544.] Nat. Immunol. 16: 276–285. [DOI] [PubMed] [Google Scholar]
- 58.Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, Yates K, Godec J, Klatt MG, Regev A, et al. 2014. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol 15: 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li P, Spolski R, Liao W, Wang L, Murphy TL, Murphy KM, and Leonard WJ. 2012. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 490: 543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.