

Abstract
The Mdm4 (alias MdmX) oncoprotein, like its paralogue and interaction partner Mdm2, antagonizes the tumor suppressor p53. p53-independent roles of the Mdm-proteins are emerging, and we have reported the ability of Mdm2 to modify chromatin and to support DNA replication by suppressing the formation of R-loops (DNA/RNA-hybrids). We show here that the depletion of Mdm4 in p53-deficient cells compromises DNA replication fork progression as well. Among various deletion mutants, only full-length Mdm4 was able to support DNA replication fork progression. Co-depletion of Mdm4 and Mdm2 further impaired DNA replication, and the overexpression of each partially compensated for the other’s loss. Despite impairing replication, Mdm4 depletion only marginally hindered cell proliferation, likely due to compensation through increased firing of replication origins. However, depleting Mdm4 sensitized p53−/− cells to the nucleoside analogue gemcitabine, raising the future perspective of using Mdm4 inhibitors as chemosensitizers. Mechanistically, Mdm4 interacts with members of the Polycomb Repressor Complexes and supports the ubiquitination of H2A, thereby preventing the accumulation of DNA/RNA-hybrids. Thus, in analogy to previously reported activities of Mdm2, Mdm4 enables unperturbed DNA replication through the avoidance of R-loops.
Keywords: Mdm4, Mdmx, Mdm2, p53, DNA replication, DNA fiber assays
INTRODUCTION
Mutations of the tumor suppressor p53 are observed in more than 50% of human malignancies. Alternatively, elevated levels and activity of its negative regulators Mdm2 and Mdm4 (also known as Mdmx or Hdm4/Hdmx in humans) can drive oncogenic transformation. The negative feedback loop between Mdm2 and p53 controls p53 activity, but only the formation of a heterodimer between the two Mdm-proteins efficiently antagonizes p53 [1]. Increased levels of Mdm4 were reported for a variety of cancer entities such as carcinomas of the lung, breast and colon [2], retinoblastomas [3], cutaneous melanomas [4] and acute myeloid leukemia (AML) [5]. Elevated expression of such a critical negative regulator leads to pathological inactivation of p53.
Additionally, Mdm4 carries out p53-independent functions. The lack of Mdm4 promotes genome instability [6]. When expressed ectopically, Mdm4 inhibits DNA break repair by associating with Nbs1, a member of the MRN complex that also consists of Mre11 and Rad50 [7]. Moreover, Mdm4 negatively regulates E2F1 [8] but it can also mediate the degradation of the E2F1 antagonist RB [9]. And like Mdm2, Mdm4 is able to increase the proteasomal turnover of p21 [10].
The activities of Mdm2 are not limited to the degradation of p53 either [11]. In addition to its role in DNA repair [12,13], we reported previously that Mdm2 is necessary for maintaining processive DNA replication [14]. For this, it acts as a chromatin modifier to prevent conflicts between replication and transcription [15]. Mdm2 mediates histone 2A ubiquitination via its RING-domain, in cooperation with members of the Polycomb Repressor Complexes (PRCs). With this, it helps to avoid supraphysiological levels of DNA/RNA-hybrids (R-loops) [16]. Such DNA/RNA-hybrids, typically formed by hybridization of RNA to its template DNA co-transcriptionally, represent a common obstacle to DNA replication forks [17]. Thus, Mdm2 enables unperturbed DNA replication by preventing R-loop formation.
Mdm2 and Mdm4 share 90% of their amino acid sequence and show high similarity in their p53-binding and RING-domains [18]. Yet, little is known about the potential role of Mdm4 in DNA-replication. Based on the high level of structural homology and the cooperative effects of Mdm4 and Mdm2 described in the literature, we asked whether Mdm4 interferes with DNA replication as well.
In our hands, processive replication of DNA in tumor-derived cells, as well as in primary cells lacking wild-type p53, heavily relied on sufficient amounts of full-length Mdm4. In the absence of Mdm4, DNA replication was strongly compromised but was rescued by re-introducing Mdm4, RNF2 (RING finger protein 2, alias RING1B) or by eliminating R-loops. Thus, Mdm4 acts as a supporter of DNA replication, independent of p53.
MATERIALS AND METHODS
Detailed descriptions regarding the experimental procedures are provided in the Supplemental Materials and Methods.
Cell culture
H1299 (non-small cell lung carcinoma, TP53−/−; male), MIA PaCa-2 (pancreatic adenocarcinoma, mutp53 R248W; male), Panc-1 (pancreatic epithelial carcinoma, mutp53 R273H; male), U2OS (osteosarcoma, TP53+/+; female) cells and mouse embryonic fibroblasts derived from p53−/−, p53−/− Mdm4−/−, p53−/− Mdm2−/− as well as p53−/− Mdm2−/− Mdm4−/− animals were maintained in DMEM and routinely tested to exclude mycoplasma contamination.
DNA fiber assays
DNA fiber assays to analyze replication fork progression and processivity and the share of ORIs were carried out as described in [16]. During microscopy and measurement of the labeled tracks, the groups were blinded to ensure unbiased analysis. To reliably estimate fork progression a minimum of 100 fibers [19] was measured.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation was done according to a protocol previously described by our group [15] but modified as described in the supplemental information.
Dot blot analysis
Cells were washed in PBS and fixed with 1.1% paraformaldehyde in 0.1 M NaCl, 1 mM EDTA, 0.5 mM EGTA and 50 mM HEPES pH7 for 30 min. Glycine was added to a final concentration of 0.125 M to quench the reaction. Subsequently, the cells were lysed in 1% Triton X-100, 0.15 M NaCl, 1 mM EDTA, 0.3% SDS with protease inhibitors and sonicated for 10 cycles. Following proteinase K (2 mg/ml, ThermoFisher, Waltham, MA, USA) treatment for 1 h at 50°C, DNA was isolated using phenol-chloroform extraction. An equal amount of DNA was spotted onto pre-wet nitrocellulose membrane and cross-linked with UVC for 5 min. As a negative control, one half of the samples were also pre-treated with RNaseH (0.03 U/ng DNA, ThermoFisher) for 3 h at 37°C prior to spotting. The membrane was blocked in 5% BSA containing 0.25% Tween-20 and then incubated with S9.6 antibody (Kerafast, Boston, MA, USA, 1:300) overnight. DNA/RNA-hybrids were detected using Femto Maximum Sensitivity Substrate (ThermoFisher) following incubation with peroxidase-conjugated donkey anti-mouse IgG (Jackson Immunoresearch, Cambridgeshire, UK, 1:10000). To ensure equal loading, the membrane was incubated with antibody to single-stranded DNA (Millipore, Saint-Louis, MO, USA, 1:1000) following treatment with 2.5 M HCl for 15 min to denature the DNA. Similarly, detection of ssDNA was performed following exposure to the secondary antibody.
Non-denaturing CldU Assay for detection of single-stranded DNA
Twenty-four hours after transfection of H1299 cells, medium was changed and replaced with DMEM containing 25 μM CldU. Following a further 24 h incubation, the cells were harvested according to the immunofluorescence protocol and incubated with Anti-BrdU antibody (ab6326, Abcam, Cambridge, UK), DAPI and Alexa Fluor 555 (A-21434, ThermoFisher). The primary antibody detects the incorporated CldU (by cross-reactivity), but only when present as single-stranded DNA.
Quantification and statistical analysis
Fiber images were acquired by fluorescence microscopy and analyzed manually using Image J. The number of fibers measured for each condition is indicated in the graphs and biological replicates shown in corresponding supplemental figures. Statistical testing was performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). For normally distributed samples, ANOVA or a 2-sided unpaired t-test were used to calculate the significance. In the remaining cases, Kruskal-Wallis test or Mann-Whitney test were calculated with an assumed significance for p-values ≤ 5%. For multiple comparisons, the adjusted p-values are shown.
RESULTS
Loss of Mdm4 impairs DNA replication fork progression
Our previously published results suggested that the chromatin modifiers Mdm2 and RNF2 support DNA replication through a mechanism that is mediated via their RING domains [14,16]. To address the question whether Mdm4, a structural homologue of Mdm2, is also able to support DNA replication, we depleted Mdm4 in various cancer cell lines and murine embryonic fibroblasts (MEFs) to carry out DNA fiber assays after confirming Mdm4 depletion using immunoblot analysis and immunoprecipitation (Fig. 1 A–B, Suppl. Fig. 1 A). Fiber assays allowed us to visualize the progression of single DNA replication forks and quantify the length of the DNA that was synthesized in each fork during the labeling time. Similar to Mdm2, depletion of Mdm4 in the TP53−/− lung large cell carcinoma-derived cell line H1299 [20] impaired the progression of DNA replication forks (Fig. 1 C–E, Suppl. Fig. 1 B–C). Decreased fork progression was not only observed after Mdm4 depletion in p53-null cells but also in two pancreatic adenocarcinoma cell lines with mutant p53 status. MIA PaCa-2 cells carrying the p53 R248W mutation as well as Panc-1 cells with the p53 R273H substitution [21] displayed a reduced fork rate after Mdm4 depletion (Fig. 1 F–I, Suppl. Fig. D–G). Surprisingly, even in the osteosarcoma cell line U2OS harboring wild-type p53, the depletion of Mdm4 led to a strong decrease in replication fork progression despite p53 accumulation (Fig. 1 J–K, Suppl. Fig. 1 H–I). Due to the importance of Mdm4 during embryonic development [22], we additionally tested the role of Mdm4 in supporting DNA replication in MEFs of mice lacking Mdm4 in a p53-null background. Strikingly, the loss of Mdm4 reduced replication fork progression compared to a control cell preparation lacking only p53 (Fig. 1 L–N, Suppl. Fig. 1 J–K). Taken together, Mdm4 depletion impairs replication fork progression independent of p53.
FIGURE 1. Loss of Mdm4 impairs DNA replication fork progression.
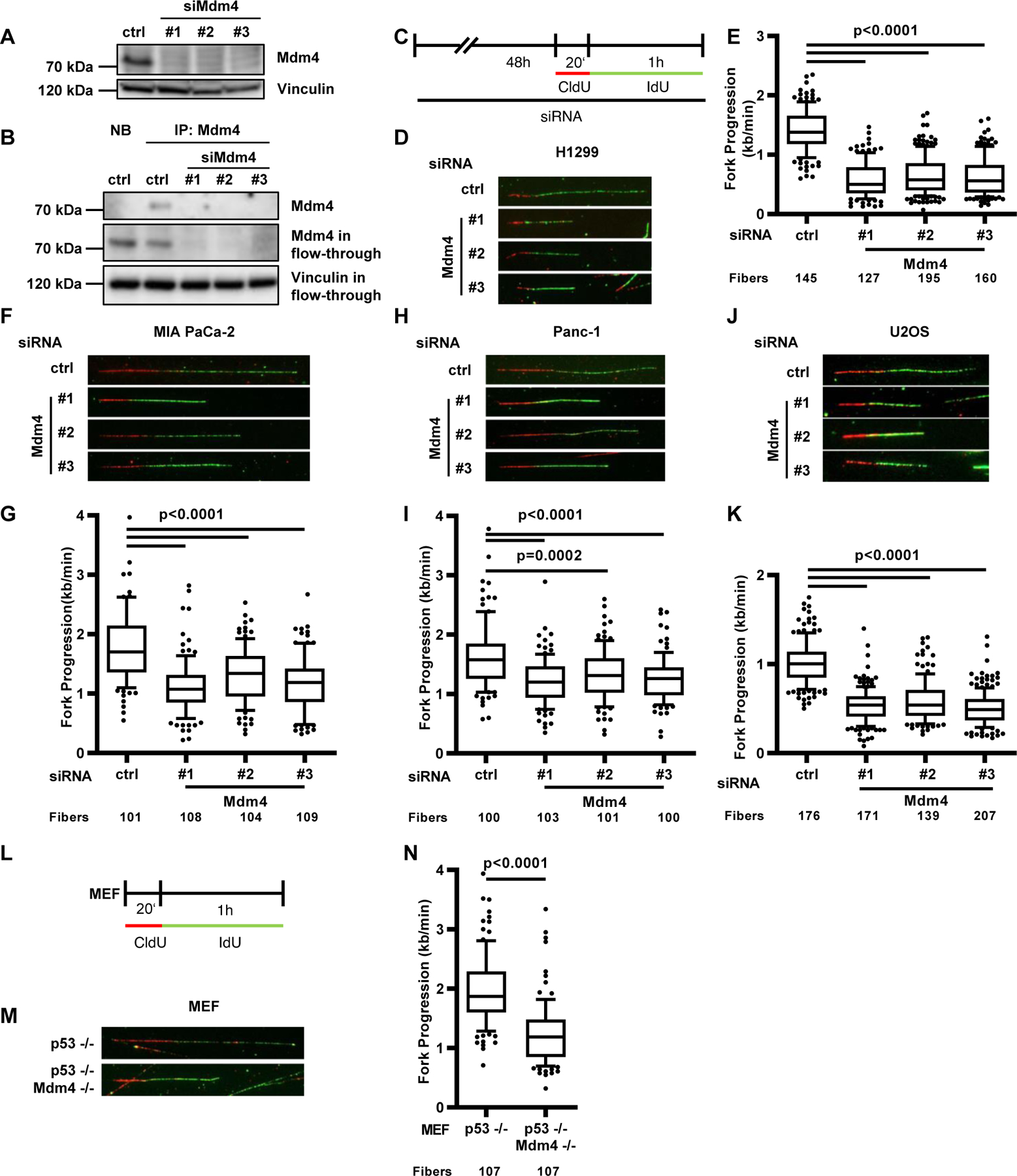
(A) Immunoblot to confirm the depletion of Mdm4 in H1299 cells 48 h after siRNA transfection. A biological replicate is shown in Suppl. Fig. 1A.
(B) Immunoprecipitation of Mdm4 in H1299 after transfection with control siRNA and three different siRNAs targeting Mdm4. Precipitation with non-binding beads (NB) and Mdm4 staining in the flow-through served as controls. Vinculin in the flow-through was detected as an input control.
(C) H1299, MIA PaCa-2, Panc-1 and U2OS cells were depleted of Mdm4 by siRNA transfection for 48 h and labeled with 5’-chloro-2’-deoxyuridine (CldU, 25 μM) for 20 min and iododeoxyuridine (IdU, 25 μM) for 60 min, followed by staining and microscopy. Analysis of the newly synthesized DNA, visualized by fluorescent tracks, allows for calculation of DNA replication fork progression within the labeling period.
(D) Representative images of labeled tracks after immunostaining of CldU (red) and IdU (green) in H1299 cells after Mdm4 depletion.
(E) DNA fork progression in H1299 cells was determined from the track length of the second label (IdU; kilobases per minute) and displayed in a boxplot with 10–90 percentile whiskers. For this analysis, more than 100 tracks per sample were measured. Two biological replicates of experiments carried out with the same protocol but different passage of cells are shown in Suppl. Fig. 1 B–C, confirming the results.
(F) Representative images of tracks of newly synthesized DNA, visualized by immunostaining of CldU (red) and IdU (green) in MIA PaCa-2 cells.
(G) Boxplot analysis of IdU-labeled tracks (indicating DNA replication fork progression) upon Mdm4 depletion using three siRNAs with 10–90 percentile whiskers in MIA PaCa-2 cells. Two biological replicates are shown in Suppl. Fig. 1 D–E.
(H) Immunostained tracks of CldU (red) and IdU (green) in representative images.
(I) IdU-stained DNA fibers in Panc-1 cells displayed as boxplots with 10–90 percentile whiskers. Two biological replicates are shown in Suppl. Fig. 1 F–G.
(J) Representative images of labeled tracks in U2OS cells.
(K) Boxplot analysis of IdU-labeled tracks upon Mdm4 depletion, with 10–90 percentile whiskers. A biological replicate is shown in Suppl. Fig. 1 H.
(L) Mouse embryonic fibroblasts lacking p53, alone or in combination with a deletion of Mdm4, were labeled with CldU (20 min) and IdU (60 min).
(M) Labeled tracks were immunostained in red (CldU) and green (IdU).
(N) Fork progression in MEFs is displayed in a boxplot with 10–90 percentile whiskers. Two biological replicates are shown in Suppl. Fig. 1 J–K.
Mdm4 and Mdm2 act through partially distinct mechanisms
Mdm2 and Mdm4 form heterodimers via their respective RING domains to ubiquitinate p53 efficiently [1]. In order to investigate whether Mdm2 and Mdm4 act cooperatively to support DNA replication, we depleted H1299 cells of both Mdm2 and Mdm4 with siRNAs and used MEFs from mice lacking Mdm2 as well as Mdm4 in a p53-null background for replication analysis. In both cases, co-depletion of Mdm2 and Mdm4 led to a further decrease in replication fork progression compared to single depletions (Fig. 2 A–F, Suppl. Fig. 2 A–D). These results argue that the two proteins are not entirely dependent on each other for their impact on DNA replication. To explore whether Mdm2 and Mdm4 can substitute for each other in supporting replication, we performed cross-rescue experiments. A control experiment showed that replication upon Mdm4 depletion can indeed be rescued by ectopic expression of Mdm4, but replication did not further benefit from higher than endogenous Mdm4 levels (Fig. 2 G–I, Suppl. Fig. 2 E–F). Only partial cross-rescue was observed upon Mdm2 depletion and Mdm4 ectopic expression (Fig. 2 J–K, Suppl. Fig. 2 G–H). Analogous results were obtained when using MEFs (Fig. 2 L–N, Suppl. Fig. I–J). In the same way, the ectopic expression of Mdm4 in cells lacking p53 and Mdm4 could completely restore DNA replication fork progression. On the other hand, only a partial rescue in replication was observed when Mdm4 was overexpressed in the p53−/− Mdm2−/− and p53−/− Mdm4−/− Mdm2−/− MEFs. Similar results were found upon ectopic expression of wild-type Mdm2 in Mdm4-depleted H1299 cells (Fig. 2 O–P, Suppl. Fig. 2 K–L). A mutant version of Mdm2 carrying a point mutation in its RING domain that inactivates its E3 ubiquitin ligase activity was not able to rescue replication after Mdm4 depletion (Fig. 2 O–P). This leads to the conclusion that Mdm2 and Mdm4 can partially compensate for each other in supporting fork progression when present in excess, although their simultaneous depletion is most deleterious to DNA replication. This argues that there are overlapping but not entirely redundant activities of both proteins in supporting DNA replication.
FIGURE 2. Mdm4 and Mdm2 act through partially distinct mechanisms.

(A) H1299 cells were transfected with siRNA for 48 h to knockdown Mdm4, Mdm2, or both, and labeled with IdU and CldU as indicated to detect single replication forks.
(B) Representative images of tracks of newly synthesized DNA were visualized by immunofluorescence, staining CldU (red) and IdU (green).
(C) Analysis of IdU-labeled tracks upon depletion of Mdm4 or Mdm2 or Mdm4 and Mdm2 with 10–90 percentile whiskers. Two biological replicates are shown in Suppl. Fig. 2 A–B.
(D) MEFs with four different knockouts (p53−/− , p53−/− Mdm2−/− , p53−/− Mdm4−/− , p53−/− Mdm2−/− Mdm4−/−) were labeled with CldU for 20 min and IdU for 60 min.
(E) Immunostained tracks of CldU (red) and IdU (green) in representative images.
(F) Fork progression in MEFs displayed as a boxplot. A biological replicate is shown in Suppl. Fig. 2 D.
(G) H1299 cells were first transfected with siRNAs to deplete Mdm2 or Mdm4, followed by plasmid transfection to overexpress Mdm2 or Mdm4 after 24 h. After another 30 h, samples were subjected to fiber assay labeling with 25 μM CldU (20 min) and 25 μM IdU (60 min).
(H) Labeled tracks of CldU and IdU stained in red and green, respectively.
(I) Fluorescently labeled tracks displayed as boxplots with 10–90 percentile whiskers. A biological replicate is shown in Suppl. Fig. 2 E.
(J) Labeled tracks were immunostained in red (CldU) and green (IdU).
(K) The fork rate of replication during the IdU label is shown in boxplots. A biological replicate is shown in Suppl. Fig. 2 G.
(L) Mdm4 was ectopically expressed in MEFs for 30 h before fiber assays were conducted.
(M) Representative images of replication tracks from MEFs with single p53 knockouts, p53/Mdm4 and p53/Mdm2 double knockouts as well as triple knockouts, transfected with an expression plasmid for wildtype Mdm4.
.(N) IdU track lengths representing fork progression in MEFs transfected with Mdm4 as in M, displayed in boxplots with 10–90 percentile whiskers. A biological replicate is shown in Suppl. Fig. 2 J.
(O) Fluorescently labeled tracks in red (CldU) and green (IdU).
(P) Analysis of fork progression in the IdU label after Mdm4 depletion and ectopic expression of wildtype Mdm2 or Mdm2 with the mutation C462A, lacking its E3 ubiquitin ligase function. A biological replicate is shown in Suppl. Fig. 2 K.
Full-length Mdm4 is required for supporting DNA replication
Between the structural homologues Mdm4 and Mdm2, the p53-binding domains (p53BD) and the RING domains are both particularly well conserved [18]. We set out to study whether these domains are required for the function of Mdm4 in DNA replication by first depleting cells of Mdm4 and then ectopically expressing full-length Mdm4 protein (wildtype) or specific Mdm4 fragments (Fig. 3 A). First, we investigated the N-terminal p53BD and used a Mdm4 fragment lacking the first 200 amino acids or a fragment comprising the p53BD only. Using DNA fiber assays, we observed a rescue of DNA replication fork progression only after overexpression of full-length, wildtype Mdm4 (Fig. 3 B–D, Suppl. Fig. 3 A–B). The role of Mdm2 as a chromatin modifier depends on its RING domain that carries E3 ubiquitin ligase activity [16]. Mdm4 contains a RING domain as well but so far no associated E3 ubiquitin ligase activity has been described [23]. We therefore asked whether the RING domain of Mdm4 is essential for supporting DNA replication. We conducted fiber assay experiments overexpressing either the C-terminal RING domain or a fragment lacking the most carboxyterminal 145 amino acids of Mdm4, which comprise the RING finger. Again, only full-length Mdm4 was able to restore DNA replication after depletion of endogenous Mdm4, whereas the ΔRING (Fig. 3 E–F, Suppl. Fig. 3 C–E) and RING fragments could not do so (Fig. 3 G–H, Suppl. Fig. 3 F–H). Hence, only full-length Mdm4 is capable of supporting DNA replication.
FIGURE 3. Full-length Mdm4 is required for supporting DNA replication.

(A) Schematic diagram of five plasmid constructs expressing either full-length Mdm4, Mdm4 carrying a deletion of its p53-binding and RING domains, or the p53-binding and RING domain alone.
(B) H1299 cells were depleted of endogenous Mdm4 by siRNA transfection for 48 h, transfected with plasmid DNA to overexpress Mdm4 or fragments of it for 30 h, and labeled with CldU (20 min) and IdU (60 min) for fiber analysis.
(C) Replication fork progression upon overexpression of Mdm4 fragments while depleting endogenous Mdm4. Immunostained tracks of CldU (red) and IdU (green) in representative images.
(D) Boxplot analysis of IdU-labeled tracks upon Mdm4 depletion and overexpression of wt-Mdm4 as well as Mdm4 (aa 201–489) or the p53 binding domain alone (aa 1–198) with 10–90 percentile whiskers. A biological replicate is shown in Suppl. Fig. 3 B.
(E) Replication tracks after immunostaining of CldU (red) and IdU (green).
(F) Boxplot analysis of IdU-labeled tracks upon Mdm4 depletion and overexpression of wt-Mdm4 and Mdm4 (aa 1–344) with 10–90 percentile whiskers. Two biological replicates are shown in Suppl. Fig. 3 D–E.
(G) Replication tracks.
(H) Analysis of fork progression upon Mdm4 depletion and overexpression of wt-Mdm4 or the Mdm4 RING (aa 345–489) domain alone. Two biological replicates are shown in Suppl. Fig. 3 G–H.
Mdm4 depletion increases replicative stress, but overall DNA synthesis is maintained
To understand whether cells experience replicative stress upon loss of Mdm4, we studied different indicators of this phenomenon. First, we asked whether the velocity or the processivity of the DNA replication forks change upon Mdm4 knockdown. To distinguish this, we used a modified fiber assay protocol previously described by our group [14], labeling the DNA of ongoing forks with seven brief exposures to nucleoside analogues. In response to Mdm4 depletion, we observed an increase in replication fork stalling, indicated by the incorporation of fewer than 7 labels (Fig. 4 A–C). Fork velocity, assessed by the progression of labels 2 to 5, was also significantly reduced (Fig. 4 D). Thus, Mdm4 depletion impairs both the velocity and the processivity of DNA replication forks. Next, we assessed the levels of single-stranded nuclear DNA using immunofluorescence staining for CldU without previous DNA denaturation. The signal derived from incorporated CldU nucleotides that were accessible in single-stranded portions of DNA increased significantly after Mdm4 depletion (Fig. 4 E–F, Suppl. Fig. 4 A–B). Single-stranded DNA is a hallmark of replicative stress and is often accompanied by activation of the DNA damage signaling cascade [24]. One readout for this cascade is the phosphorylation of the histone variant H2A.X at Ser139, also referred to as γH2AX. We detected elevated levels of γH2AX upon Mdm4 depletion (Fig. 4 G–I, Suppl. Fig. 4 C–E). Thus, depletion of Mdm4 increases replicative stress, indicated by fork stalling, high levels of ssDNA, and DNA damage signaling. Surprisingly however, we observed that long-term proliferation of cells after Mdm4 depletion was hardly affected (Fig. 4 J, Suppl. Fig. 4 F). One way of compensating for high levels of replicative stress in cells is to induce dormant origin firing [25]. We determined firing of bidirectional origins during the first labeling pulse to be increased by 2-fold in samples lacking Mdm4 (Fig. 4 K, Suppl. Fig. 4 G–H), seen as structures labeled green-red-green [26]. Accordingly, overall EdU incorporation per cell remained largely unchanged (Fig. 4 L, Suppl. Fig. 4 I), and cell cycle progression was only marginally altered (Fig. 4 M, Suppl. Fig. 4 J–M), further arguing in favor of a compensatory effect of dormant origin firing to maintain DNA synthesis and cell proliferation upon Mdm4 depletion.
FIGURE 4. Mdm4 depletion increases replicative stress, but overall DNA synthesis is maintained.
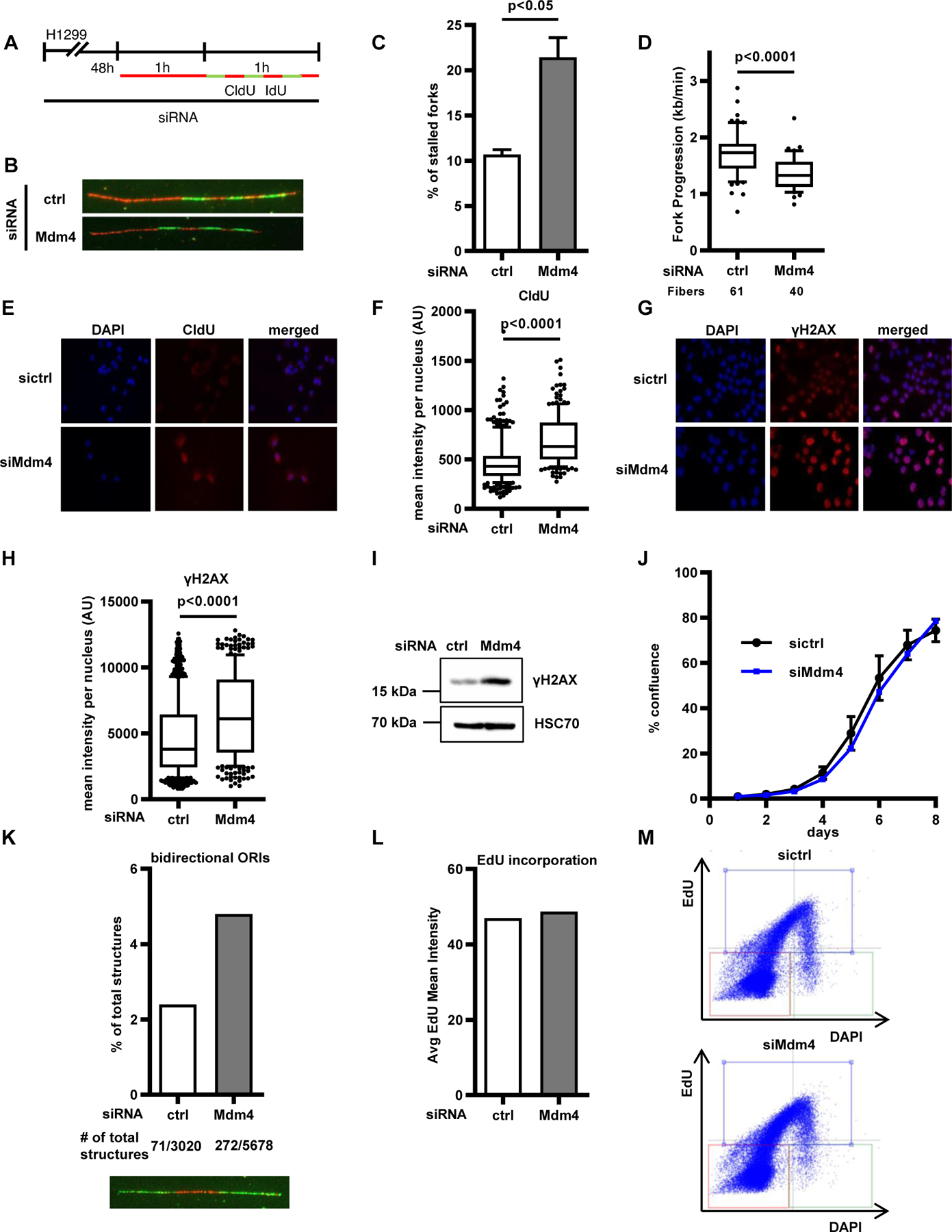
(A) H1299 cells were transfected with siRNAs targeting Mdm4 for 48 h and then labeled with CldU for 1 h, followed by short (10 min), alternating pulses of IdU and CldU for a total of seven labels.
(B) Representative images of replicated stretches that have incorporated all seven labels. Seven labels reflect the full progression of the fork throughout the entire labeling time. Numbers lower than 7 indicate premature termination during the labeling time.
(C) The percentages of forks with fewer than seven labels indicate that depletion of Mdm4 causes replication to run in a less processive manner than cells transfected with scrambled (control) siRNA. The bar chart shows mean + SD from two biological replicates.
(D) The length of labels 2–5 was used for fork progression analysis to reveal that fork velocity was also diminished by Mdm4 depletion.
(E) Representative images of immunostaining after incubating the cells with 25 μM CldU after 48 h of Mdm4 depletion. Fluorescence represents single-stranded DNA.
(F) Fluorescence intensity corresponding to single-stranded DNA was quantified by Fiji software and displayed in a boxplot, indicating that Mdm4 depletion causes accumulation of single-stranded DNA. Two biological replicates are shown in Suppl. Fig. 4 A–B.
(G) Staining of the DNA damage marker γH2AX in cells depleted of Mdm4.
(H) Fluorescence intensity per nucleus is shown in a boxplot with 10–90 percentile whiskers. Two biological replicates are shown in Suppl. Fig. 4 C–D.
(I) Immunoblot analysis to show increases in γH2AX levels after Mdm4 knockdown in H1299 cells. A biological replicate is shown in Suppl. Fig. 4 E.
(J) To assay for cell proliferation, H1299 cells were transfected with siRNA against Mdm4. Confluence of transfected cells was analyzed every 24 h by translucent, automated microscopy. A biological replicate is shown in Suppl. Fig. 4 F.
(K) After labeling as described in the legend to Fig. 1 C, all visible fiber track structures were counted (in total 8,698). The share of bidirectional origins of replication (green-red-green structures as shown in the representative image below) firing during the first labeling pulse, in relation to all track structures, increased after Mdm4 depletion, indicating enhanced origin firing.
(L) To assess the overall DNA synthesis, cells were labeled with 10 μM EdU for 4 h and the average EdU mean intensity was plotted as bar graphs. No change in DNA synthesis was detected after Mdm4 depletion. A biological replicate is shown in Suppl. Fig. 4I.
(M) Dual parameter plot of DNA content (DAPI) vs. EdU intensity, for both ctrl and Mdm4-depleted cells. Three populations of cells are visible, G1 (orange square), S-phase (blue), G2/M (green). A biological replicate is shown in Suppl. Fig. 4J. For a detailed quantification see Suppl. Fig. 4 K.
Mdm4 depletion sensitizes cells to gemcitabine
Although cells were able to proliferate despite the depletion of Mdm4, this situation still led to replicative stress (Figure 4). Therefore, we asked whether Mdm4 depletion could exacerbate the effect of a nucleoside analogue on DNA replication. To address this, we performed fiber assays on H1299 cells treated with gemcitabine, a drug that induces replication stress by inhibiting ribonucleotide reductase and also by misincorporation into nascent DNA [27]. As expected, a drop in DNA fork progression was observed following gemcitabine treatment. Importantly however, Mdm4 depletion impaired fork progression even further when combined with gemcitabine treatment (Fig. 5 A–C, Suppl. Fig. 5 A). The detrimental effect on DNA replication was accompanied by further accumulation of phosphorylated checkpoint kinase 1 (pChk1), an intermediate in DNA damage signaling, as determined by immunoblot analysis (Fig. 5 D, Suppl. Fig. 5 B) in Mdm4-depleted cells co-treated with gemcitabine. In addition, cell proliferation was compromised to a greater extent upon a combination of gemcitabine treatment and Mdm4 depletion compared to cells that only underwent treatment with the drug (Fig. 5 E, Suppl. Fig. 5 C). Moreover, we conducted cell viability assays in Mdm4-depleted cells treated with doses of gemcitabine ranging from 5 nM to 80 nM. Similar to the proliferation assay, a lack of Mdm4 alone did not significantly affect the viability of cells. However, when these Mdm4-depleted cells were treated with 5 to 20 nM gemcitabine, their viability was strongly impaired compared to their respective controls (Fig. 5 F). Hence, the depletion of Mdm4 sensitizes cells towards a nucleoside analogue used in cancer chemotherapy.
FIGURE 5. Mdm4 depletion sensitizes cells to gemcitabine.
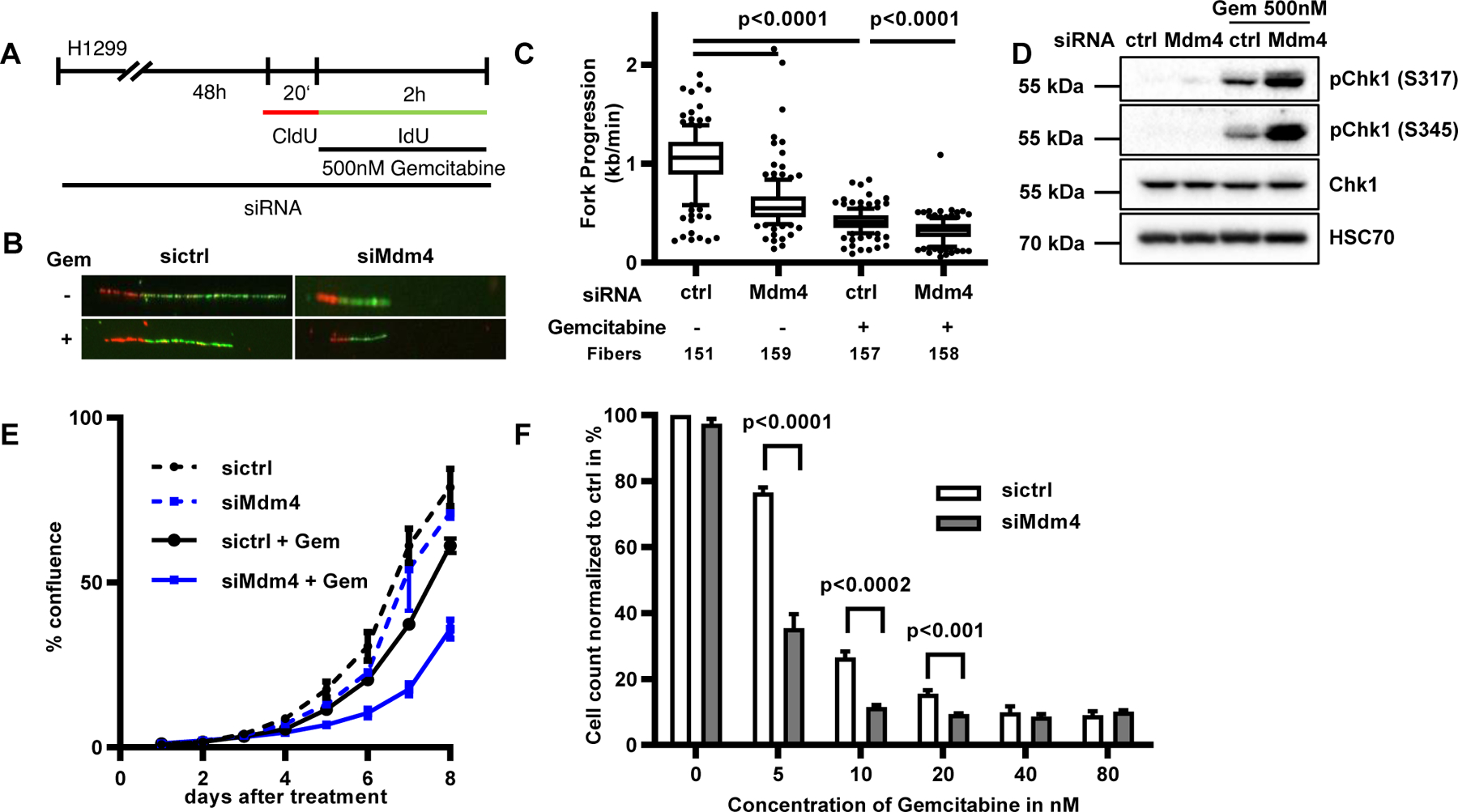
(A) H1299 cells were transfected with siRNA for 48 h and labeled with IdU and CldU as indicated. During the 120 min of IdU labeling the cells were treated with 500 nM gemcitabine.
(B) Representative images of labeled tracks after immunostaining.
(C) IdU stained DNA fibers in H1299 cells displayed as boxplots with 10–90 percentile whiskers. A biological replicate is shown in Suppl. Fig. 5 A.
(D) Cells were treated as in A and subjected to immunoblot analysis, revealing enhanced Chk1 phosphorylation when Mdm4 depletion was combined with gemcitabine treatment. A biological replicate is shown in Suppl. Fig. 5 B.
(E) H1299 cells were transfected with siRNA against Mdm4 and treated with 10 nM gemcitabine. Confluence of transfected cells was analyzed every 24 h to document decreased proliferation of Mdm4-depleted, gemcitabine-treated cells. Variability within triplicates is shown as standard deviation. A biological replicate is shown in Suppl. Fig. 5 C.
(F) H1299 cells were transfected with siRNA to knock down Mdm4 and treated with different concentrations of gemcitabine for 72 h. Cell viability was assessed by measuring ATP levels. Mean and SD were calculated from three biological replicates.
Mdm4 cooperates with Polycomb Repressor Complex members and regulates H2A ubiquitination
We and others have described that Mdm2 interacts with members of the PRC 1 [28] and 2 [15,29]. Importantly, Mdm2 cooperates with RNF2 to mediate chromatin compaction and maintain proper DNA replication. Therefore, we asked whether Mdm4 interacts with components of the PRCs. Co-immunoprecipitation followed by immunoblot analysis revealed that Mdm4 can bind to the PRC1 member RNF2 alias Ring1b (Fig. 6 A). Moreover, Mdm4 as well as Mdm2 co-precipitated with the PRC2 member EZH2 (Fig. 6 B, Suppl. Fig. 6 A). In line with these results, the overexpression of RNF2 restored DNA replication fork progression in Mdm4-depleted cells (Fig. 6 C–D, Suppl. Fig. 6 B–D), suggesting that the role of Mdm4 in replication overlaps with that of RNF2. Although Mdm4 lacks an intrinsic E3 ubiquitin ligase activity, it is able to associate with Mdm2 and the PRCs, leading us to further speculate that Mdm4 might participate in chromatin modification. And indeed, co-depletion of Mdm4 and RNF2 led to further reduction of global H2AK119ub1 levels compared to the single knockdowns, as seen in immunoblots (Fig. 6 E, Suppl. Fig. E). Additionally, chromatin immunoprecipitation using an antibody to H2AK119ub1, in cells depleted of Mdm4 or Mdm2, showed a marked reduction of H2AK119ub1 occupancy at known PRC-regulated target sites (Fig. 6 F). To investigate if this decrease in H2AK119ub1 was responsible for attenuating DNA replication in response to Mdm4 depletion, we examined whether the overexpression of histone 2A could rescue fork progression after Mdm4 knockdown. Indeed, fork progression was fully restored upon ectopic expression of wild-type H2A in H1299, but not in cells which were transfected with mutant H2A bearing point mutations at its PRC-responsive ubiquitination sites (K118R/K119R) (Fig. 6 G–H, Suppl. Fig. 6 F–H). Moreover, the overexpression of wild-type H2A rescued the fork progression in p53−/− Mdm4−/− and p53−/− Mdm2−/− MEFs as well (Fig. 6 I–J, Suppl. Fig. I–J). Interestingly, H2A overexpression in triple knockout MEFs (p53−/− Mdm4−/− Mdm2−/−) only led to a partial restoration of replication, suggesting that the increased supply of the substrate H2A is not sufficient to fully compensate for the absence of both Mdm-proteins. Taken together, these findings indicate a novel function of Mdm4 in regulating histone modifications by cooperating with RNF2, and they provide mechanistic insight into the p53-independent role of Mdm4 in supporting DNA replication.
FIGURE 6. Mdm4 cooperates with Polycomb Repressor Complex members and regulates H2A ubiquitination.

(A) Co-immunoprecipitation of Mdm4 and RNF4. Expression plasmids for Flag-tagged Mdm4 and/or non-tagged RNF2, or an empty control plasmid, were transfected into H1299 cells. Cell lysates (input) and the immunoprecipitated (IP) material were analyzed by immunoblotting (IB) with antibodies detecting the Flag-tag and RNF2, respectively. Immunoprecipitation was conducted using antibodies against RNF2 (rabbit antibody), and pre-immune IgG (rabbit antibody) was used as a negative control.
* indicates a background band, presumably dimerized IgH; ** indicates monomeric IgH.
(B) Co-immunoprecipitation of Mdm4 and EZH2. H1299 cells were transfected with expression plasmids for Flag-tagged Mdm4, non-tagged EZH2, and/or a control plasmid, for 30 h. Cell lysates were immunoprecipitated using Mdm4 (mouse antibody) and βGal (mouse antibody, negative control) antibodies and subjected to immunoblot analysis with antibodies detecting the Flag-tag and EZH2. An additional Co-IP upon ectopic expression of Mdm4 and/or Mdm2 and EZH2 is shown in Suppl. Fig. 6 A.
(C) DNA replication upon Mdm4 depletion and overexpression of RNF2. Representative images of immunostained tracks of CldU (red) and IdU (green) of H1299 cells upon transfection with Mdm4 siRNA and overexpression of RNF2.
(D) Boxplot analysis of fork progression in the IdU label (green) with 10–90 percentile whiskers of the experiment described in (C). Two biological replicates are shown in Suppl. Fig. 6 C–D.
(E) Immunoblot analysis of cells depleted of Mdm4, RNF2 or both. Samples stained for RNF2 as well as H2AK119ub1 and HSC70 and total H2A. A biological replicate is shown in Suppl. Fig. 6 E.
(F) Chromatin immunoprecipitation (ChIP) analysis of H2AK119ub1 on PRC1 target gene promotors and C-FOS as negative control after H1299 cells were transfected with siRNA against Mdm4 or Mdm2. (Enrichment normalized to input and total H2A shown as mean + SEM from three biological replicates).
(G) Overexpressed H2A rescuing DNA replication in Mdm4-depleted cells. Representative images of tracks of newly synthesized DNA that were visualized using immunofluorescence.
(H) Fork progression after overexpression of wild-type H2A (pcDNA3.1-Flag-H2A) or a mutant lacking the lysine residue that is known to be ubiquitinated by Mdm2 and RNF2 (pcDNA3.1-Flag-H2A-K118–119R), upon Mdm4 depletion. H2A but not the mutant rescued DNA replication. Two biological replicates are shown in Suppl. Fig. 6 F–G.
(I) Overexpressed H2A rescuing DNA replication in MEFs lacking p53 and the respective Mdm proteins. Representative images of immunostained tracks of CldU (red) and IdU (green) of a fiber assay with mouse embryonic fibroblasts (MEFs).
(J) MEFs with targeted deletions of p53 alone, p53 in combination with Mdm4 or Mdm2, as well as a triple knockout were transfected with a plasmid carrying wildtype H2A or an empty plasmid as a control. Fork progression analysis of these cells is displayed in a boxplot with 10–90 percentile whiskers. A biological replicate is shown in Suppl. Fig. 6 I.
Removal of accumulated DNA/RNA-hybrids allows replication fork progression despite the loss of Mdm4
The formation of DNA/RNA-hybrids presents a major obstacle to moving replication forks [30]. We previously described a novel role of Mdm2 in maintaining dynamic chromatin compaction to prevent the formation of persistent hybrids [16]. To assess if these structures are also present after the loss of Mdm4, we performed dot blot analyses of cellular DNA using an antibody (S9.6) that detects DNA/RNA-hybrids [31,32]. Indeed, loss of Mdm4 resulted in a strong induction of R-loops in both H1299 cells and the corresponding knockout MEFs (Fig. 7 A–D, Suppl. Fig. 7 A–D). We next asked whether replicative stress caused by Mdm4 depletion can also be relieved by the overexpression of RNaseH1, an enzyme resolving DNA/RNA-hybrids. Strikingly, ectopic expression of wild-type RNaseH1, but not its catalytically inactive mutant, rescued replication fork impairment in Mdm4-depleted H1299 cells (Fig. 7 E–G, Suppl. Fig. 7 E–F). Analogous observations were made in p53−/− MEFs with or without additional deletions of Mdm4 and/or Mdm2 (Fig. 7 H–I, Suppl. Fig. 7 G–H). This indicates that changes in the chromatin upon depletion of Mdm4 lead to the formation of DNA/RNA-hybrids that act as obstacles to DNA replication forks.
FIGURE 7. Removal of accumulated DNA/RNA-hybrids allows replication fork progression despite the loss of Mdm4.
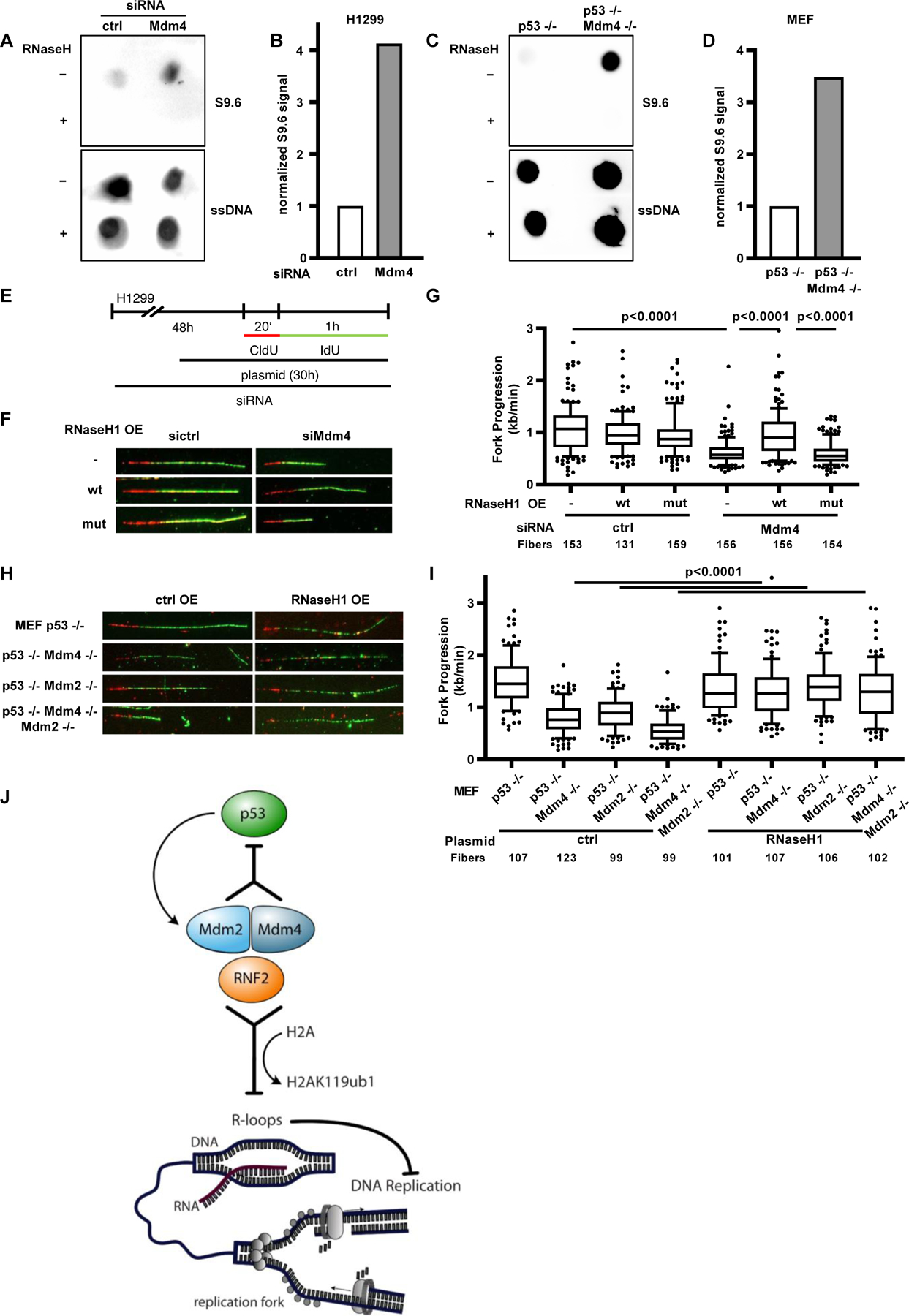
(A) Accumulation of DNA/RNA-hybrids (R-loops) upon depletion of Mdm4 in H1299 cells. Dot blot analysis of DNA from cells treated with siRNA targeting Mdm4 for 48 h to investigate the formation of R-loops. Incubation with RNaseH was carried out as negative control before staining with S9.6 antibody to detect DNA/RNA-hybrids. Staining of single-stranded DNA (ssDNA) after denaturation was used as a loading control. A biological replicate is shown in Suppl. Fig. 7 A–B.
(B) Bar chart of the S9.6 signal normalized to ssDNA.
(C) Dot blot analysis as described in (A) of MEFs with p53 or p53/Mdm4 knockouts. A biological replicate is shown in Suppl. Fig. 7 C–D.
(D) Quantification of the normalized S9.6 signal indicating the increase in DNA/RNA-hybrids due to deletion of Mdm4.
(E) H1299 cells were subjected to Mdm4 siRNA transfection for 48 h followed by a plasmid transfection for the last 30 h before fiber assay analysis.
(F) Immunostained tracks of CldU (red) and IdU (green) in representative images.
(G) A boxplot displaying the fork progression of Mdm4-depleted H1299 cells transfected with plasmids to express wild-type RNaseH1 (pICE-RNaseH1-NLS-mCherry) or a catalytically inactive mutant (pICE-RNaseH1-D10-E48R-NLS-mCherry), showing that RNaseH1 can rescue the defective DNA replication upon Mdm4 depletion. Two biological replicates are shown in Suppl. Fig. 7 E–F.
H) Representative images of CldU (red) and IdU (green) stained tracks of DNA.
(I) MEFs with a single p53 knockout, p53 in combination with Mdm4 or Mdm2 as well as the triple knockout were transfected with a plasmid carrying wildtype RNaseH as well as an empty control. Fork progression analysis of these cells is displayed in a boxplot with 10–90 percentile whiskers. A biological replicate is shown in Suppl. Fig. 7 H.
(J) Graphical abstract: Mdm4 as well as Mdm2 prevent the occurrence of replicative stress by cooperation with RNF2. As for p53 inhibition, Mdm2 and Mdm4 are most effective in supporting DNA replication when present together, although the single components can still perform a similar function and avoid the formation of R-loops.
DISCUSSION
Our results strongly suggest that Mdm4, much like its paralogue Mdm2, prevents the occurrence of replicative stress by cooperation with RNF2 and is therefore required for an unperturbed progression of DNA replication forks. As for p53 inhibition, Mdm4 and Mdm2 are most effective in supporting DNA replication when present together, although the single components can still perform a similar function and avoid the formation of R-loops which we identified as a cause of replicative stress (Fig. 7 J). This effect is observed in p53-deficient cells, excluding p53-binding as an underlying mechanism.
The impact of Mdm4 on DNA replication resembles that of Mdm2 or p53. We have previously described p53 as a supporter of DNA replication, capable of enhancing the processivity of single replication forks [14]. On the other hand, we observed similar activities in the case of Mdm2, independent of p53 [16]. Furthermore, Mdm2 is the product of one of the most strongly p53-inducible genes [33]. This argues that p53 may carry out its supportive function for DNA replication through the induction of Mdm2.
Since Mdm4 resembles Mdm2 in its structure and most likely represents a paralogue of Mdm2 with a common ancestor gene [34], it seems plausible that the two proteins might act on DNA replication through similar mechanisms that may have been preserved through long periods of evolution. Both proteins bind to Polycomb Repressor Complexes, regulate chromatin modification and act, at least in part, by preventing DNA/RNA-hybrids (R-loops) thereby enabling smooth progression of DNA replication forks.
For counteracting p53 activity, Mdm2 teams up with Mdm4, and the heterodimer of both proteins appears most efficient for p53 antagonism. Since Mdm4 and Mdm2 form a specific complex through interaction of their RING finger domains, and since the RING finger of each protein is required for supporting DNA replication (this work, Fig. 3, and [16]), it would be conceivable to assume that this complex must be formed for supporting DNA replication. However, the observation that each of the two proteins can partially compensate for the depletion of the other suggests that they can support DNA replication individually.
Additionally, we provide evidence for partially exclusive roles of the two Mdm proteins. The effect of double knockout in murine embryonic fibroblast as well as co-depletion of Mdm4 and Mdm2 in cancer cell lines exceeds the impairment of DNA replication by depleting just one of them (Fig. 2). We propose that Mdm4 and Mdm2 act through only partially overlapping mechanisms when present at physiological levels and in addition have individual roles in DNA replication, e.g. through the individual interaction partners of each protein [35–37].
At least in the case of the Mdm4 binding partner p53, ubiquitination activity directly driven by Mdm4 has not been described [23]. Nevertheless, the RING finger domain of Mdm4 is required for supporting DNA replication. One might hypothesize that Mdm4 interacts with additional binding partners via this domain and therefore mediates ubiquitination of a substrate relevant to replication, such as H2A. Our observation that Mdm4 binds to members of the PRCs, consistent with recently published results on a role of Mdm4 in EZH2 ubiquitination [29], raises the possibility of Mdm4 acting on PRCs as shown for Mdm2 [38]. Interestingly, Mdm4 overexpression alone reduced DNA replication fork progression to some extent, although not as profoundly as the depletion of endogenous Mdm4. Supraphysiological levels of Mdm4 may alter the stoichiometry between Mdm4 and its binding partners. It is tempting to speculate that balanced levels of Mdm4 and Mdm2, on the one hand, and the PRCs on the other hand are essential for proper chromatin modification and thus an unperturbed DNA replication.
Mdm4 physically interacts with the MRN complex, consisting of the DNA repair mediators Mre11, Rad51, and Nbs1 [7]. Thereby, Mdm4 suppresses the repair of double-strand DNA breaks, and this occurs independent of Mdm2 and p53. This is in correspondence with the results reported here, regarding the impact of Mdm4 on DNA replication. Interestingly, the MRN complex, through the nuclease activity of Mre11, was found to be required for the activation of Chk1 in the context of replication stress, at least in an in vitro system [39]. Thus, Mdm4 might further support the progression of DNA replication forks by interacting with the MRN complex.
Since developing tissues are very proliferative and express high levels of Mdm4 [40], one would expect consequences of the impaired DNA replication in p53−/− Mdm4−/− mice. Different groups published conflicting observations on the consequences of Mdm4 loss in a p53−/− background in vivo. One study reported no obvious phenotypical differences or altered rate of tumor development in p53−/− Mdm4−/− mice, compared to those harboring a p53 deletion alone [22]. However, another group observed tumor onset in p53−/− Mdm4−/− mice five weeks earlier than in p53 null mice [41]. Mdm4 does not seem to be essential for proper development, at least not within laboratory mice. We suggest however, that Mdm4 and Mdm2 might provide a greater robustness of DNA replication under more natural conditions, which include stressors such as infections or inflammations. Furthermore, we describe a possible way how cells may at least partially compensate for the decreased DNA replication fork progression by inducing the firing of dormant replication origins. Only in the presence of an external stressor, replication stress led to a decrease in cell growth and cell viability. This mechanism, as well as a partial compensation by Mdm2, could prevent growth restrictions and tumor onset in Mdm4-deficient mice as well.
A recently proposed model by Marine and Jochemsen [42] regarding the role of different Mdm4 isoforms in human cancer considers a splicing switch from Mdm4-S (short) to Mdm4-FL (full-length) as crucial for the malignant transformation. In our hands, the supportive function of Mdm4 on DNA-replication required a full-length protein including the RING domain. This adds a new aspect of Mdm4 as a therapeutic target in human cancer: If Mdm4 enables some cancer cells to replicate their genome in a fast and processive manner, the rationale behind targeting Mdm4 would not only be the reactivation of wild-type p53 but also to impair the DNA replication in malignant cells and to sensitize these cells to other anti-tumor treatments. Since most differentiated adult tissues do not produce full-length Mdm4 due to unproductive splicing [40] and can therefore, tolerate the loss of Mdm4 [43,44], this argues for the possibility of inhibiting full-length Mdm4 in cancer cells with limited toxicity in healthy tissues.
Mdm4 has been suggested as a drug target under various circumstances [4,45–49]. The results presented in this work further encourage the search for Mdm4-inactivating small molecules. Although it is difficult to define precise target sites on Mdm4 regarding its impact on DNA replication, it is conceivable to eliminate Mdm4 by a drug that binds its p53-interacting domain [50] and couples it to a ubiquitin ligase, an approach termed PROTAC [51]. Such an approach is expected to increase replication stress, and, as observed in this study with Mdm4 depletion (Fig. 5), might cooperate with conventional drugs that interfere with DNA replication.
Supplementary Material
HIGHLIGHTS.
Mdm4, like Mdm2, is required for efficient DNA replication fork progression
Mdm4 binds to Polycomb repressors and supports H2A ubiquitination
Loss of Mdm4 induces R-loop formation, leading to replication stress
Mdm4 depletion sensitizes cancer cells towards gemcitabine treatment
ACKNOWLEDGEMENTS
We thank Guillermina Lozano for the MEFs with p53/Mdm4/Mdm2 deletions. pCMV-Flag-Mdm4 was a gift from Zhi-Min Yuan. pCMV-MDM2 was a gift from Bert Vogelstein (Addgene plasmid #16441), pCMV-MDM2(C464A) was provided by Tyler Jacks (Addgene plasmid #12086), pICE-RNaseHI-WT-NLS-mCherry (Addgene plasmid #60365) as well as pICE-RNaseHI-D10R-E48R-NLS-mCherry (Addgene plasmid #60367) were obtained from Patrick Calsou. H2A and EZH2 expression plasmids were from Titia Sixma (Addgene plasmids #63561 and #63564) and Kristian Helin (Addgene plasmid #24230), respectively. pLenti6/V5-DEST-RNF2 was a gift from Lynda Chin (Addgene plasmid #31216). This work was supported by the Deutsche Krebshilfe (to M.D. and K.W.), the Wilhelm Sander Stiftung, the Else Kröner Fresenius Stiftung, the Deutsche José Carreras Leukämie Stiftung, the Deutsche Forschungsgemeinschaft, the Boehringer Ingelheim Fonds (to I.K.) and the German Academic Scholarship Foundation (to K.W.). I.K., P.D. and V.M. were members of the IMPRS/MSc/PhD program Molecular Biology and I.K., V.M., C.G. and J.C. also of the Göttingen Graduate School GGNB Göttingen.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci. 2003; 100: 12009–12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R et al. Amplification of Mdmx (or Mdm4) Directly Contributes to Tumor Formation by Inhibiting p53 Tumor Suppressor Activity. Mol Cell Biol. 2004; 24: 5835–5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006; 444: 61–66. [DOI] [PubMed] [Google Scholar]
- 4.Gembarska A, Luciani F, Fedele C, Russell EA, Dewaele M, Villar S et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat Med. 2012; 18: 1239–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han X, Medeiros LJ, Zhang YH, You MJ, Andreeff M, Konopleva M et al. High Expression of Human Homologue of Murine Double Minute 4 and the Short Splicing Variant, HDM4-S, in Bone Marrow in Patients With Acute Myeloid Leukemia or Myelodysplastic Syndrome. Clin Lymphoma Myeloma Leuk. 2016; 16: S30–S38. [DOI] [PubMed] [Google Scholar]
- 6.Matijasevic Z, Krzywicka-Racka A, Sluder G, Jones SN. MdmX regulates transformation and chromosomal stability in p53-deficient cells. Cell Cycle. 2008; 7: 2967–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carillo Alexia M, Bouska Alzssa, Arrate Maria Pia ECM. NIH Public Access. Oncogene. 2015; 34: 846–856.24608433 [Google Scholar]
- 8.Strachan GD, Jordan-Sciutto KL, Rallapalli R, Tuan RS, Hall DJ. The E2F-1 transcription factor is negatively regulated by its interaction with the MDMX protein. J Cell Biochem. 2003; 88: 557–568. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H, Hu L, Qiu W, Deng T, Zhang Y, Bergholz J et al. MDMX exerts its oncogenic activity via suppression of retinoblastoma protein. Oncogene. 2015; 34: 5560–5569. [DOI] [PubMed] [Google Scholar]
- 10.Jin Y, Zeng SX, Sun X-X, Lee H, Blattner C, Xiao Z et al. MDMX Promotes Proteasomal Turnover of p21 at G1 and Early S Phases Independently of, but in Cooperation with, MDM2. Mol Cell Biol. 2008; 28: 1218–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bohlman S, Manfredi JJ. p53-independent effects of Mdm2. In: Sub-Cellular Biochemistry. 2014, pp 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alt JR, Bouska A, Fernandez MR, Cerny RL, Xiao H, Eischen CM. Mdm2 Binds to Nbs1 at Sites of DNA Damage and Regulates Double Strand Break Repair. J Biol Chem. 2005; 280: 18771–18781. [DOI] [PubMed] [Google Scholar]
- 13.Bouska A, Lushnikova T, Plaza S, Eischen CM. Mdm2 Promotes Genetic Instability and Transformation Independent of p53. Mol Cell Biol. 2008; 28: 4862–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klusmann I, Rodewald S, Müller L, Friedrich M, Wienken M, Li Y et al. p53 Activity Results in DNA Replication Fork Processivity. Cell Rep. 2016; 17: 1845–1857. [DOI] [PubMed] [Google Scholar]
- 15.Wienken M, Dickmanns A, Nemajerova A, Kramer D, Najafova Z, Weiss M et al. MDM2 Associates with Polycomb Repressor Complex 2 and Enhances Stemness-Promoting Chromatin Modifications Independent of p53. Mol Cell. 2016; 61: 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klusmann I, Wohlberedt K, Magerhans A, Teloni F, Korbel JO, Altmeyer M et al. Chromatin modifiers Mdm2 and RNF2 prevent RNA:DNA hybrids that impair DNA replication. Proc Natl Acad Sci. 2018; 115: E11311–E11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL et al. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011; 25: 2041–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shvarts A, Bazuine M, Dekker P, Ramos YFM, Steegenga WT, Merckx G et al. Isolation and identification of the human homolog of a new p53-binding protein, Mdmx. Genomics 1997; 43: 34–42. [DOI] [PubMed] [Google Scholar]
- 19.Técher H, Koundrioukoff S, Azar D, Wilhelm T, Carignon S, Brison O et al. Replication dynamics: Biases and robustness of DNA fiber analysis. J Mol Biol. 2013; 425: 4845–4855. [DOI] [PubMed] [Google Scholar]
- 20.Gazdar AF, Gao B, Minna JD. Lung cancer cell lines: Useless artifacts or invaluable tools for medical science? Lung Cancer. 2010; 68: 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gradiz R, Silva HC, Carvalho L, Botelho MF, Mota-Pinto A. MIA PaCa-2 and PANC-1 – pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci Rep. 2016; 6: 21648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001; 29: 92–95. [DOI] [PubMed] [Google Scholar]
- 23.Meulmeester E, Frenk R, Stad R, de Graaf P, Marine J-C, Vousden KH et al. Critical role for a central part of Mdm2 in the ubiquitylation of p53. Mol Cell Biol. 2003; 23: 4929–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobbelstein M, Sørensen CS. Exploiting replicative stress to treat cancer. Nat Rev Drug Discov. 2015; 14: 405–423. [DOI] [PubMed] [Google Scholar]
- 25.Blow JJ, Ge XQ, Jackson DA. How dormant origins promote complete genome replication. Trends Biochem Sci. 2011; 36: 405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quinet A, Carvajal-Maldonado D, Lemacon D, Vindigni A. DNA Fiber Analysis: Mind the Gap! In: Methods in enzymology. 2017, pp 55–82. [DOI] [PubMed] [Google Scholar]
- 27.Plunkett W, Huang P, Searcy CE, Gandhi V. Gemcitabine: preclinical pharmacology and mechanisms of action. Semin Oncol. 1996; 23: 3–15. [PubMed] [Google Scholar]
- 28.Wen W, Peng C, Kim MO, Ho Jeong C, Zhu F, Yao K et al. Knockdown of RNF2 induces apoptosis by regulating MDM2 and p53 stability. Oncogene. 2014; 33: 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuser-Abali G, Gong L, Yan J, Liu Q, Zeng W, Williamson A et al. An EZH2-mediated epigenetic mechanism behind p53-dependent tissue sensitivity to DNA damage. Proc Natl Acad Sci. 2018; 115: 201719532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguilera A, García-Muse T. Loops R: From Transcription Byproducts to Threats to Genome Stability. Mol Cell. 2012; 46: 115–124. [DOI] [PubMed] [Google Scholar]
- 31.Boguslawski SJ, Smith DE, Michalak MA, Mickelson KE, Yehle CO, Patterson WL et al. Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J Immunol Methods. 1986; 89: 123–30. [DOI] [PubMed] [Google Scholar]
- 32.El Hage A, French SL, Beyer AL, Tollervey D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010; 24: 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischer M, Steiner L, Engeland K. The transcription factor p53: not a repressor, solely an activator. Cell Cycle. 2014; 13: 3037–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Momand J, Villegas A, Belyi VA. The evolution of MDM2 family genes. Gene. 2011; 486: 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fåhraeus R, Olivares-Illana V. MDM2’s social network. Oncogene. 2014; 33: 4365–4376. [DOI] [PubMed] [Google Scholar]
- 36.Riley MF, Lozano G. The Many Faces of MDM2 Binding Partners. Genes Cancer. 2012; 3: 226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haupt S, Mejía-Hernández JO, Vijayakumaran R, Keam SP, Haupt Y. The long and the short of it: the MDM4 tail so far. J Mol Cell Biol. 2019; 11: 231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stad R, Little NA, Xirodimas DP, Frenk R, Eb AJ van der, Lane DP et al. Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep. 2001; 2: 1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee J, Dunphy WG. The Mre11-Rad50-Nbs1 (MRN) complex has a specific role in the activation of Chk1 in response to stalled replication forks. Mol Biol Cell. 2013; 24: 1343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dewaele M, Tabaglio T, Willekens K, Bezzi M, Teo SX, Low DHP et al. Antisense oligonucleotide–mediated MDM4 exon 6 skipping impairs tumor growth. J Clin Invest. 2015; 126: 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matijasevic Z, Steinman HA, Hoover K, Jones SN. MdmX Promotes Bipolar Mitosis To Suppress Transformation and Tumorigenesis in p53-Deficient Cells and Mice. Mol Cell Biol. 2008; 28: 1265–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marine J-C, Jochemsen AG. MDMX (MDM4), a Promising Target for p53 Reactivation Therapy and Beyond. Cold Spring Harb Perspect Med. 2016; 6. doi: 10.1101/cshperspect.a026237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valentin-Vega YA, Box N, Terzian T, Lozano G. Mdm4 loss in the intestinal epithelium leads to compartmentalized cell death but no tissue abnormalities. Differentiation. 2009; 77: 442–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia D, Warr MR, Martins CP, Brown Swigart L, Passegué E, Evan GI. Validation of MdmX as a therapeutic target for reactivating p53 in tumors. Genes Dev. 2011; 25: 1746–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heijkants RC, Nieveen M, Hart KC ‘t, Teunisse AFAS, Jochemsen AG. Targeting MDMX and PKCδ to improve current uveal melanoma therapeutic strategies. Oncogenesis. 2018; 7: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miranda PJ, Buckley D, Raghu D, Pang J-MB, Takano EA, Vijayakumaran R et al. MDM4 is a rational target for treating breast cancers with mutant p53. J Pathol. 2017; 241: 661–670. [DOI] [PubMed] [Google Scholar]
- 47.Park DE, Cheng J, Berrios C, Montero J, Cortés-Cros M, Ferretti S et al. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc Natl Acad Sci. 2019; 116: 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carvajal LA, Neriah D Ben, Senecal A, Benard L, Thiruthuvanathan V, Yatsenko T et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Transl Med. 2018; 10: eaao3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pellegrino M, Mancini F, Lucà R, Coletti A, Giacchè N, Manni I et al. Targeting the MDM2/MDM4 Interaction Interface as a Promising Approach for p53 Reactivation Therapy. Cancer Res. 2015; 75: 4560–4572. [DOI] [PubMed] [Google Scholar]
- 50.Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL et al. A Stapled p53 Helix Overcomes HDMX-Mediated Suppression of p53. Cancer Cell. 2010; 18: 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y, Yang J, Aguilar A, McEachern D, Przybranowski S, Liu L et al. Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J Med Chem. 2019; 62: 448–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.