

Abstract
Progranulin (PGRN) is an autocrine growth factor that exerts crucial roles within cartilage tissue; however, the molecular mechanisms underlying PGRN-mediated cartilage homeostasis remain elusive. In the present study, we investigated the role of PGRN in regulating chondrocyte homeostasis and its therapeutic potential for managing osteoarthritis (OA). We found that PGRN levels are significantly increased in human cartilage in mild OA and that its expression is decreased in the cartilage in severe OA. In vitro, treatment of primary rat chondrocytes with recombinant PGRN significantly enhanced the levels of collagen type II α 1 chain (COL2A1) and aggrecan, and attenuated TNFα-induced up-regulation of matrix metallopeptidase 13 (MMP13) and ADAM metallopeptidase with thrombospondin type 1 motif 5 (ADAMTS5) in chondrocytes. These effects were abrogated in SIRT1−/− cells, indicating a causative role of SIRT1 in the effects of PGRN on protein expression in chondrocytes. Mechanistically, PGRN increased SIRT1 expression and activity, which reduced the acetylation levels of SRY-box transcription factor (SOX9) and transcription factor P65 (P65) and thereby promoted nuclear translocation of SOX9 and inhibited TNFα-induced P65 nuclear accumulation to maintain chondrocyte homeostasis. In conclusion, our findings reveal a mechanism of action for PGRN that maintains cartilage homeostasis and supports the notion that PGRN up-regulation may be a promising strategy for managing OA.
Keywords: chondrocytes, osteoarthritis, progranulin, PGRN, sirtuin 1, SIRT1, deacetylation, growth factor, cartilage, inflammation, joint disease
Osteoarthritis (OA) is the most prevalent disorder in joint disease, characterized by progressive loss of articular cartilage, synovial inflammation, and osteophyte formation (1), causing joint pain and disability in older adults. A series of pathological changes are implicated in OA development, including loss of chondrocyte cellularity, an imbalance between anabolism and catabolism of chondrocytes (1, 2), increased apoptosis (3), and bone regeneration of subchondral bone. Although it is well established that multiple risk factors have been involved in OA development, including age, sex, joint injury, and mechanical and genetic factors(4), much remains to be explored for its underlying molecular mechanisms.
Progranulin (PGRN), also known as proepithelin, granulin/epithelin precursor, and PC cell-derived growth factor, is a 593 amino acid autocrine growth factor which is involved in a variety of physiological and disease processes such as inflammation (5, 6), wound healing (7), tumorigenesis (8), and bone regeneration (9). Interestingly, several previous studies indicated that PGRN could act as a cartilage growth factor and exert a crucial role in cartilage homeostasis. For instance, PGRN-deficient mice showed more severe degeneration of articular cartilage compared with control mice, whereas intra-articular injection of recombinant PGRN dramatically improved OA score and attenuated cartilage matrix loss in surgically induced OA mice (10). Consistently, it is reported that PGRN could inhibit the degradative processes that occur in OA and RA patients by disrupting the interaction between cartilage oligomeric matrix protein and ADAMTS7/ADAMTS12 (11). Additionally, several clinical investigations indicated that both serum and protein levels of PGRN were significantly higher in patients with OA and rheumatoid arthritis (RA) (12–14). Collectively, these observations support an important regulatory role for PGRN in cartilage homeostasis.
Although the regulatory role of PGRN in cartilage homeostasis has been identified, the intracellular events responsible for PGRN-mediated protective role against OA progression needed to be elucidated in more detail. It is reported that sirtuin-1 (SIRT1), an NAD+-dependent histone deacetylase, exerts protective roles in human chondrocytes through enhancing the expression of cartilage anabolic markers such as collagen type II α 1 chain (COL2A1) (15) and aggrecan (16) while inhibiting apoptosis (17). Furthermore, inhibition of SIRT1 in human chondrocytes leads to OA-like gene expression changes (18), and cartilage-specific SIRT1 knockout mice show accelerated OA progression (19). We recently showed that SIRT1 facilitates growth plate chondrogenesis via deacetylating PERK and attenuating the PERK-eIF-2α-CHOP axis of the unfolded protein response pathway (20). Of note, treatment with PGRN in podocytes has been shown to increase SIRT1 expression and activity, resulting in decreased level in acetylation of PGC-1α and FoxO1 (21). However, whether and how PGRN modulates SIRT1 expression and activity in the articular chondrocytes have not been elucidated yet.
Based on all these findings, we hypothesized that PGRN facilitates cartilage homeostasis by up-regulating SIRT1 expression and activity. In the current study, PGRN promoted SIRT1 expression and activity and then initiated a cascade of target genes involved in anabolism and catabolism of chondrocytes, which clearly demonstrated the relationship between PGRN and SIRT1 in chondrocytes. To the best of our knowledge, this is the first study to demonstrate that induction of SIRT1 expression and activity in articular chondrocytes is critical in PGRN-mediated cartilage homeostasis.
Results
PGRN expression in human articular cartilage of OA
To gain insight into the role of PGRN during OA development, we obtained normal cartilage of the femoral head from patients with femoral neck fracture without joint disease, the lateral femoral condyle with mild OA, and the medial femoral condyle with severe OA. Safranin-O staining of cartilage was weaker in the cartilage of lateral femoral condyle (mild OA group) than in the normal group, and the cartilage of medial femoral condyle (severe OA group) was barely stained with safranin-O and was severely degenerated (Fig. 1a). Immunohistochemistry analysis showed that PGRN levels were increased in human cartilage in mild OA, with PGRN-positive cells mainly observed in the superficial zone, while its expression was decreased in severe OA (Fig. 1b). Moreover, the levels of PGRN in human articular cartilage were further confirmed by Western blotting and real-time PCR (Fig. 1, c and d). Similarly, the expression of PGRN receptor EphA2 was increased in mild OA cartilage but decreased in severe OA cartilage (Fig. S1a).
Figure 1.
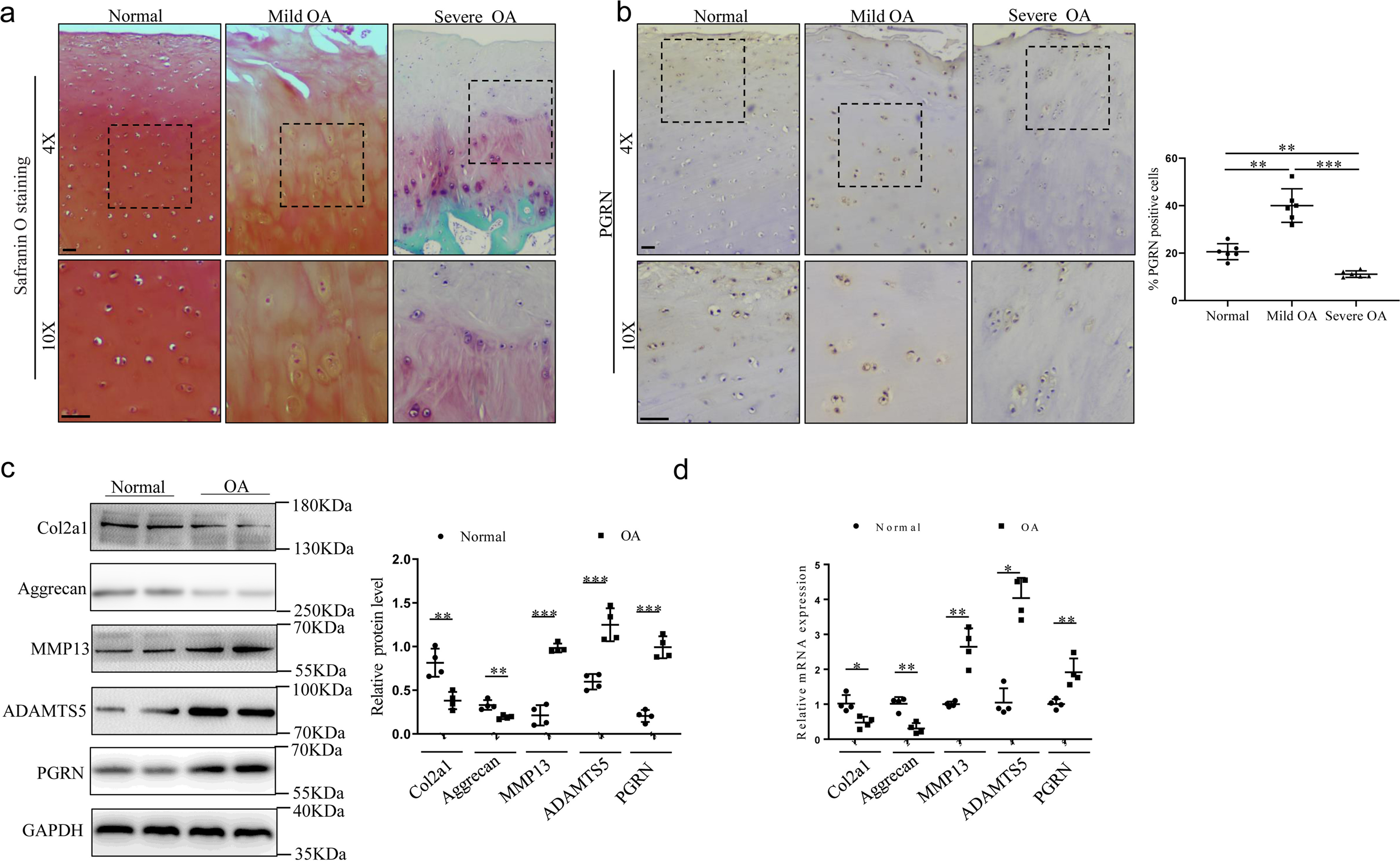
Expression of PGRN in human articular cartilage tissue in OA patients. a, safranin-O staining was performed on articular cartilage (n = 6/group). Mild OA indicates the articular cartilage of lateral femoral condyle from OA patients. Severe OA indicates the articular cartilage of medial femoral condyle from OA patients. Scale bar, 50 μm. b, immunohistochemistry and statistical analyses of PGRN expression in articular cartilage from normal humans and OA patients (n = 6/group). Scale bar, 50 μm. Brown-Forsythe and Welch ANOVA test with Dunnett's T3 multiple comparison test. c, Western blot analysis of expression of COL2A1, aggrecan, MMP13, ADAMTS5, and PGRN in articular cartilage, and the relative quantity was analyzed using ImageJ software. Unpaired two-tailed Student's t tests. d, mRNA levels of col2a1, aggrecan, MMP13, ADAMTS5, and PGRN were examined by real-time PCR in articular cartilage. Results were presented as gene expression levels in all groups normalized to controls. Unpaired two-tailed Student's t tests. Data were expressed as mean ± S.D. in each scatterplot. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
PGRN positively regulates cartilage gene expression and up-regulates SIRT1 in cultured chondrocytes
Because PGRN is known as a growth factor that promotes cell cycle progression in many cellular systems (22), we sought to determine whether PGRN affects cartilage-specific gene expression in cultured chondrocytes. Therefore, we tested the expression of cartilage genes markers, including COL2A1 and aggrecan, in chondrocytes isolated from rat articular cartilage in the presence of recombinant PGRN (0–200 ng/ml). As expected, graded concentrations of PGRN stimulated COL2A1 and aggrecan expression and secretion in a dose-dependent manner, with the lowest stimulated concentration of 100 ng/ml (Fig. 2, a–e and Fig. S2a). In addition, 50 ng/ml and 100 ng/ml recombinant PGRN increased the level of PGRN receptor EphA2 in chondrocytes (Fig. S1b). It is well accepted that overexpression or activation of SIRT1 promoted cartilage gene expression in human chondrocytes (15), together with recent findings that PGRN regulated SIRT1-PGC-1α/FoxO1 signaling in podocytes (21), led us to explore whether SIRT1 was involved in PGRN-mediated anabolism of chondrocytes. PGRN enhanced SIRT1 expression and activity in a dose-dependent manner, with higher concentrations (100 and 200 ng/ml) causing a statistically significant up-regulation (Fig. 2, f–h and Fig. S2b). Interestingly, inhibition of SIRT1 by SIRT1 siRNA (the efficiency was validated by real-time PCR and Western blotting) (Fig. 2, i and j and Fig. S2c) dramatically repressed PGRN-mediated up-regulation of cartilage genes (Fig. 2, k–o and Fig. S2d), indicating that PGRN may promote COL2A1 and aggrecan expression and secretion via up-regulation of SIRT1.
Figure 2.

Effects of recombinant PGRN on chondrocytes anabolism and SIRT1 expression. Chondrocytes isolated from newborn Sprague-Dawley rat articular cartilage were cultured in the presence of graded concentrations of recombinant PGRN (0–200ng/ml) for 48 h. a, at the end of the culture period, col2a1 mRNA level was examined by real-time PCR. One-way ANOVA and Dunnett's multiple comparison test. b, quantification of secreted COL2A1 in the conditioned medium of chondrocytes treated with PGRN. One-way ANOVA and Dunnett's multiple comparison test. c and d, aggrecan expression and secretion were detected by real-time PCR and ELISA, respectively. One-way ANOVA and Dunnett's multiple comparison test. e, COL2A1 and aggrecan protein expression were detected by Western blotting. f–h, the expression and activity of SIRT1 in chondrocytes treated with PGRN. One-way ANOVA and Dunnett's multiple comparison test. Chondrocytes transfected with control siRNA or SIRT1 siRNA cultured in the absence or presence of 100 ng/ml PGRN. SIRT1i-1, SIRT1i-2, and SIRT1i-3 indicate three independent siRNAs against SIRT1. i–j, the protein and mRNA level of SIRT1 were detected by Western blotting and real-time PCR. One-way ANOVA and Tukey's multiple comparison test. k and l, COL2A1 expression and secretion were detected by real-time PCR and soluble collagen quantification assay. One-way ANOVA and Tukey's multiple comparison test. m and n, aggrecan expression and secretion were detected by real-time PCR and ELISA. One-way ANOVA and Tukey's multiple comparison test. o, the protein levels of COL2A1 and aggrecan were analyzed by Western blotting. Data were expressed as mean ± S.D. in each scatterplot. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
PGRN decreases the acetylation level of SOX9 and promotes SOX9 nuclear transportation
Because several studies reported that SIRT1 promotes cartilage-specific gene expression through deacetylation of SOX9 (15, 16), we hypothesized that PGRN increased the expression of COL2A1 and aggrecan through SIRT1-mediated SOX9 deacetylation. Co-immunoprecipitation assays showed that PGRN could significantly reduce the acetylation level of SOX9 (Fig. 3a), whereas such effect was abrogated in SIRT1−/− chondrocytes (Fig. 3b). Given that SOX9 acetylation state may affect SOX9 nuclear entry (16), we postulated that PGRN increased SIRT1 expression and activity to reduce the acetylation level of SOX9 and thereby promoted nuclear translocation of SOX9. As expected, PGRN significantly increased nuclear localization of SOX9, whereas co-treatment with SIRT1 siRNA reversed such stimulatory effect of PGRN (Fig. 3, c and d).
Figure 3.
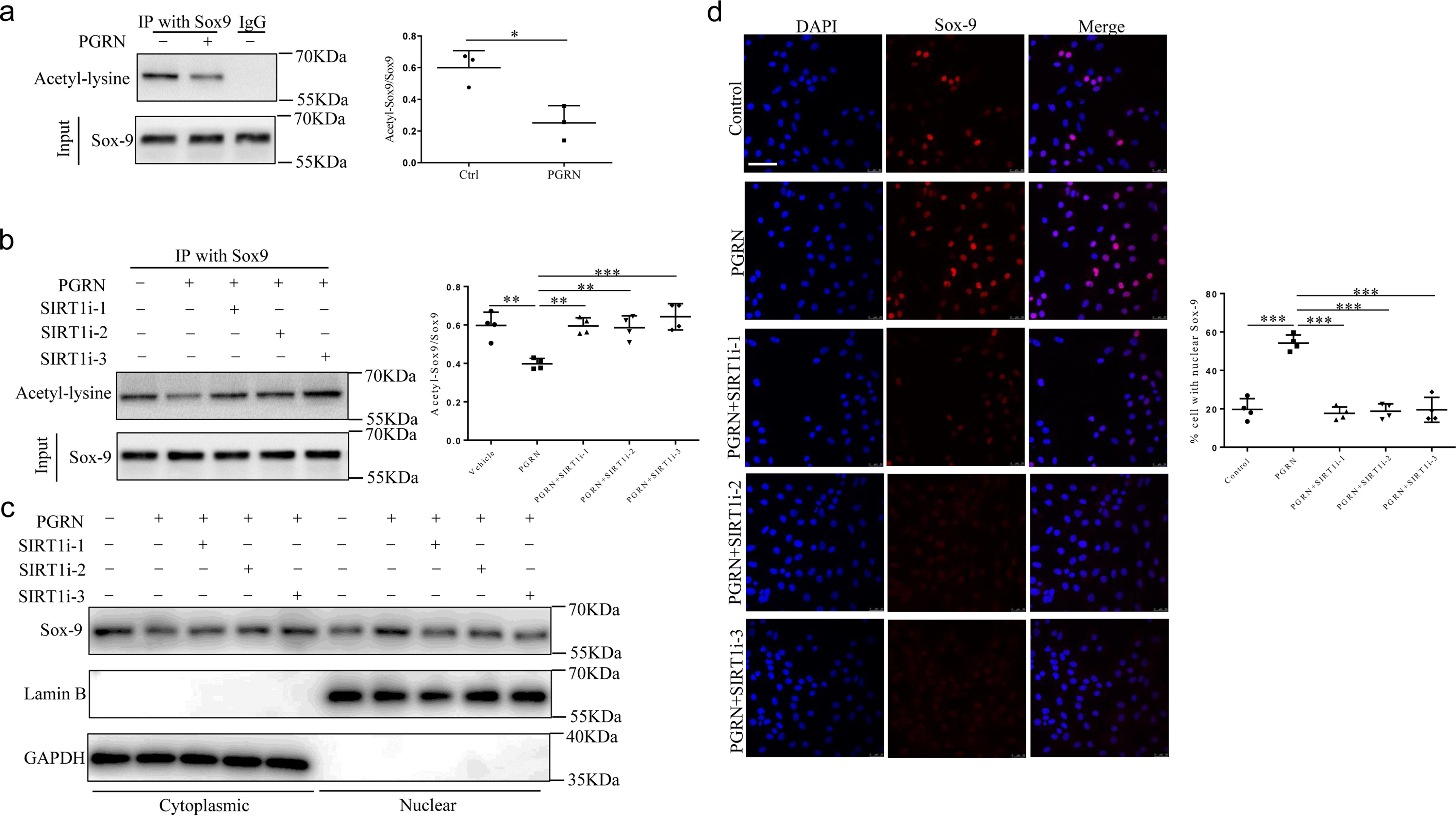
Effects of recombinant PGRN on SOX9 acetylation and nuclear translocation. a, Chondrocytes were treated with or without 100 ng/ml PGRN, and the level of acetylation of SOX9 was analyzed by IP–Western blot analysis. Unpaired two-tailed Student's t tests. b, chondrocytes transfected with control siRNA or SIRT1 siRNA cultured in the absence or presence of 100 ng/ml PGRN. The level of acetylation of SOX9 was analyzed by IP–Western blot analysis. One-way ANOVA and Tukey's multiple comparison test. c, subcellular localization of SOX9 was analyzed by Western blotting. d, immunofluorescence staining and statistical analyses of SOX9 nuclear translocation in chondrocytes. One-way ANOVA and Tukey's multiple comparison test. Scale bar, 50 μm. Data were expressed as mean ± S.D. in each scatterplot. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
PGRN inhibited TNFα-induced MMP13 and ADAMTS5 expression through SIRT1-mediated deacetylation of P65
To confirm whether PGRN affects catabolism of chondrocytes, we examined the effect of PGRN on expression of matrix metallopeptidase 13 (MMP13) and ADAM metallopeptidase with thrombospondin type 1 motif 5 (ADAMTS5). However, graded concentration of PGRN (0–200 ng/ml) had a minor effect on the expression of MMP13 and ADAMTS5 both at protein and mRNA levels (Fig. 4, a and b and Fig. S3a). To explore the role of PGRN in cellular models of OA, chondrocytes were treated with 10 ng/ml TNFα for 24 h. However, no changes were observed in either PGRN expression or secretion upon stimulation of TNFα as detected by real-time PCR and ELISA (Fig. 4, c and d). In addition, TNFα inhibited the activity of SIRT1, whereas a combination of PGRN with TNFα partially abrogated this inhibition (Fig. 4e). The stimulation with TNFα significantly induced MMP13 and ADAMTS5 expression, while the addition of PGRN abolished the chondrocyte catabolism induced by TNFα (Fig. 4, f and g and Fig. S3b). Considering the protective role of SIRT1 against OA-related catabolic stimuli in cultured chondrocytes (23), we reasoned that PGRN may suppress TNFα-induced chondrocyte catabolism through up-regulation of SIRT1. Indeed, under the stimulation with TNFα, PGRN significantly inhibited MMP13 and ADAMTS5 expression, whereas this suppression was largely nullified in SIRT1−/− chondrocytes (Fig. 4, h and i and Fig. S3c).
Figure 4.

Effects of recombinant PGRN on chondrocytes catabolism induced by TNFα. a and b, chondrocytes were cultured in the presence of graded concentrations of recombinant PGRN (0–200 ng/ml) for 48 h. At the end of the culture period, the expression of MMP13 and ADAMTS5 were analyzed by Western blotting and real-time PCR. One-way ANOVA and Tukey's multiple comparison test. c and d, chondrocytes were treated with or without 10 ng/ml TNFα for 24 h, PGRN expression and secretion were detected by real-time PCR and ELISA. Unpaired two-tailed Student's t tests. e, the activity of SIRT1 was detected in chondrocytes treated with or without 10 ng/ml TNFα for 48 h. One-way ANOVA and Tukey's multiple comparison test. f and g, chondrocytes were cultured in the absence or presence of 100 ng/ml PGRN and combination with or without 10 ng/ml TNFα for 48 h, levels of MMP13 and ADAMTS5 were analyzed by Western blotting and real-time PCR. Brown-Forsythe and Welch ANOVA test with Dunnett's T3 multiple comparison test. h and i, chondrocytes were treated with 10 ng/ml TNFα, transfected with control siRNA or SIRT1 siRNA cultured in the absence or presence of 100 ng/ml PGRN. The expression of MMP13 and ADAMTS5 was detected by Western blotting and real-time PCR. One-way ANOVA and Tukey's multiple comparison test. Data were expressed as mean ± S.D. in each scatter plot. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
It is well accepted that TNFα can induce NF-κB–P65 activation and up-regulate the levels of various MMPs and ADAMTS (24) in chondrocytes. Therefore, to explore the downstream mechanism that regulates chondrocytes catabolism, we assessed the acetylation level P65 at Lys-310 in cultured chondrocytes with or without SIRT1 siRNA. TNFα increased both the total P65 and acetylated P65 expression in chondrocytes (Fig. 5a). PGRN reduced the TNFα-induced acetylation level of P65, whereas a combination of PGRN with SIRT1 siRNA abolished this inhibition induced by PGRN (Fig. 5a). Simultaneously, PGRN inhibited TNFα-induced nuclear translocation of P65, whereas such effect was largely lost in SIRT1−/− chondrocytes (Fig. 5, b–d). The previous study reported that SIRT1 inhibited MMP13 expression by reducing LEF1 transcriptional activity, and LEF-1 was known to bind to the MMP13 promoter and transactivate its expression (25). Then, we determined whether the SIRT1/P65 axis was involved in the regulatory sequences of ADAMTS5 promoter by ChIP assay. As shown in Fig. 5e, PGRN inhibited TNFα-induced enrichment of P65 on ADAMTS5 promoter, with these inhibitions being neutralized by co-treatment with SIRT1 siRNA, indicating that PGRN may regulate the transcriptional activity of ADAMTS5 through SIRT1/P65 axis. Collectively, our data indicated that PGRN could inhibit chondrocytes catabolism induced by TNFα by deacetylation and suppressing P65 nuclear accumulation via enhancing the level of SIRT1.
Figure 5.
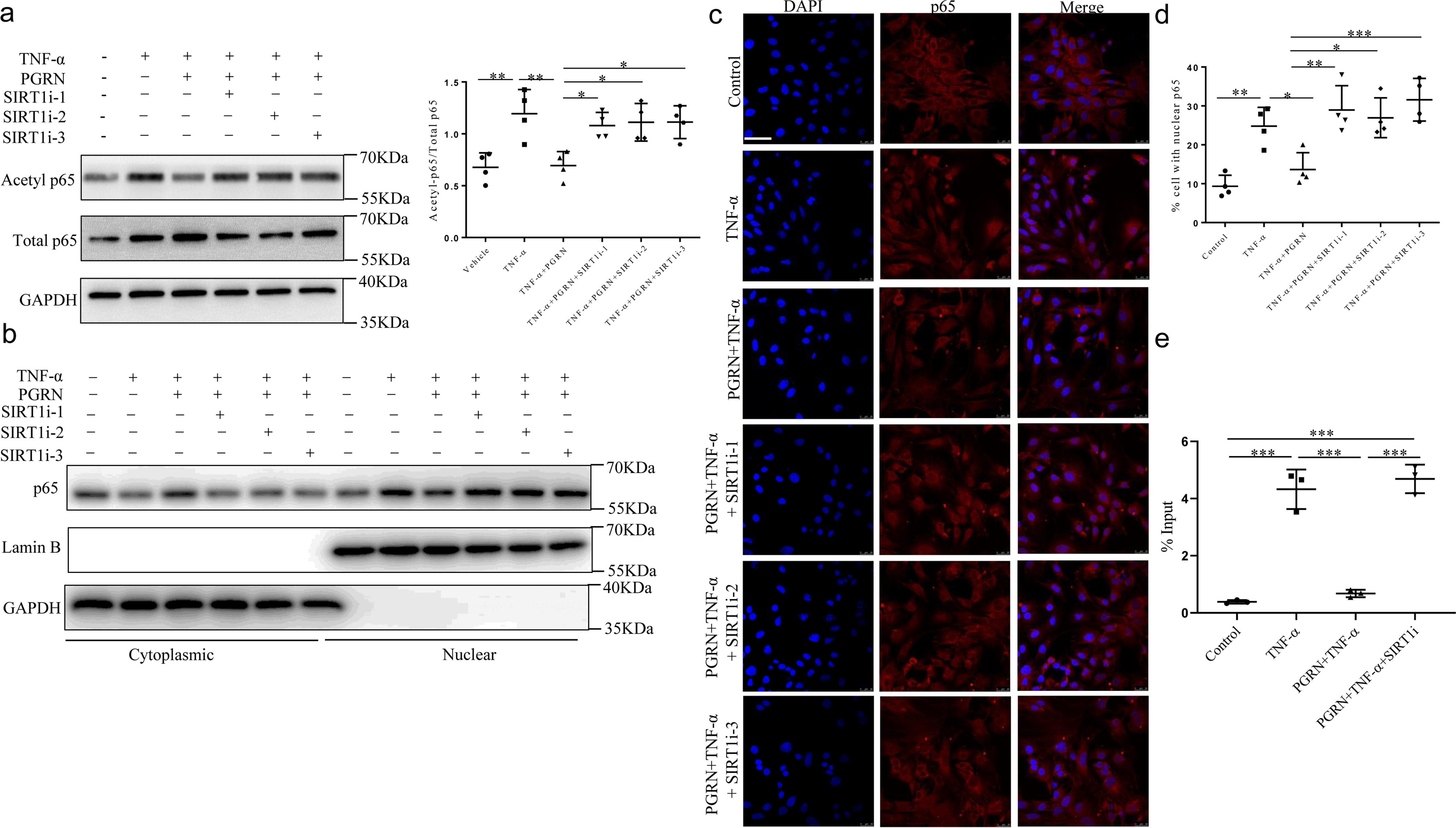
Effects of recombinant PGRN on P65 acetylation and nuclear translocation. Chondrocytes were treated with or without 10 ng/ml TNFα, transfected with control siRNA or SIRT1 siRNA cultured in the absence or presence of 100 ng/ml PGRN. a, chondrocytes were lysed and the level of acetylation of P65 was analyzed by Western blotting. One-way ANOVA and Tukey's multiple comparison test. b, subcellular localization of P65 was analyzed by Western blotting. c and d, immunofluorescence staining and statistical analyses of P65 nuclear translocation in chondrocytes. Scale bar, 50 μm. One-way ANOVA and Tukey's multiple comparison test. e, ChIP analysis for recruitment of P65 to ADAMTS5 promoter in C28/I2 human chondrocyte cell line. One-way ANOVA and Tukey's multiple comparison test. Data were expressed as mean ± S.D. in each scatterplot. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Effects of endogenous PGRN ablation on cartilage gene expression
To determine the effect of endogenous PGRN, we used siRNA for PGRN in cultured chondrocytes, and its validation was measured by the reduction of PGRN protein and mRNA expression by Western blotting and real-time PCR (Fig. 6, a and b and Fig. S4a). Meanwhile, PGRN siRNA also resulted in a considerable decrease in PGRN secretion in the culture medium (Fig. 6c). Knockdown of PGRN inhibited the anabolism of chondrocytes as evidenced by a decrease in COL2A1 and aggrecan expression and secretion (Fig. 6, d–g), whereas it did not affect levels of catabolic markers, including MMP13 and ADAMTS5 (Fig. 6h and Fig. S4b).
Figure 6.
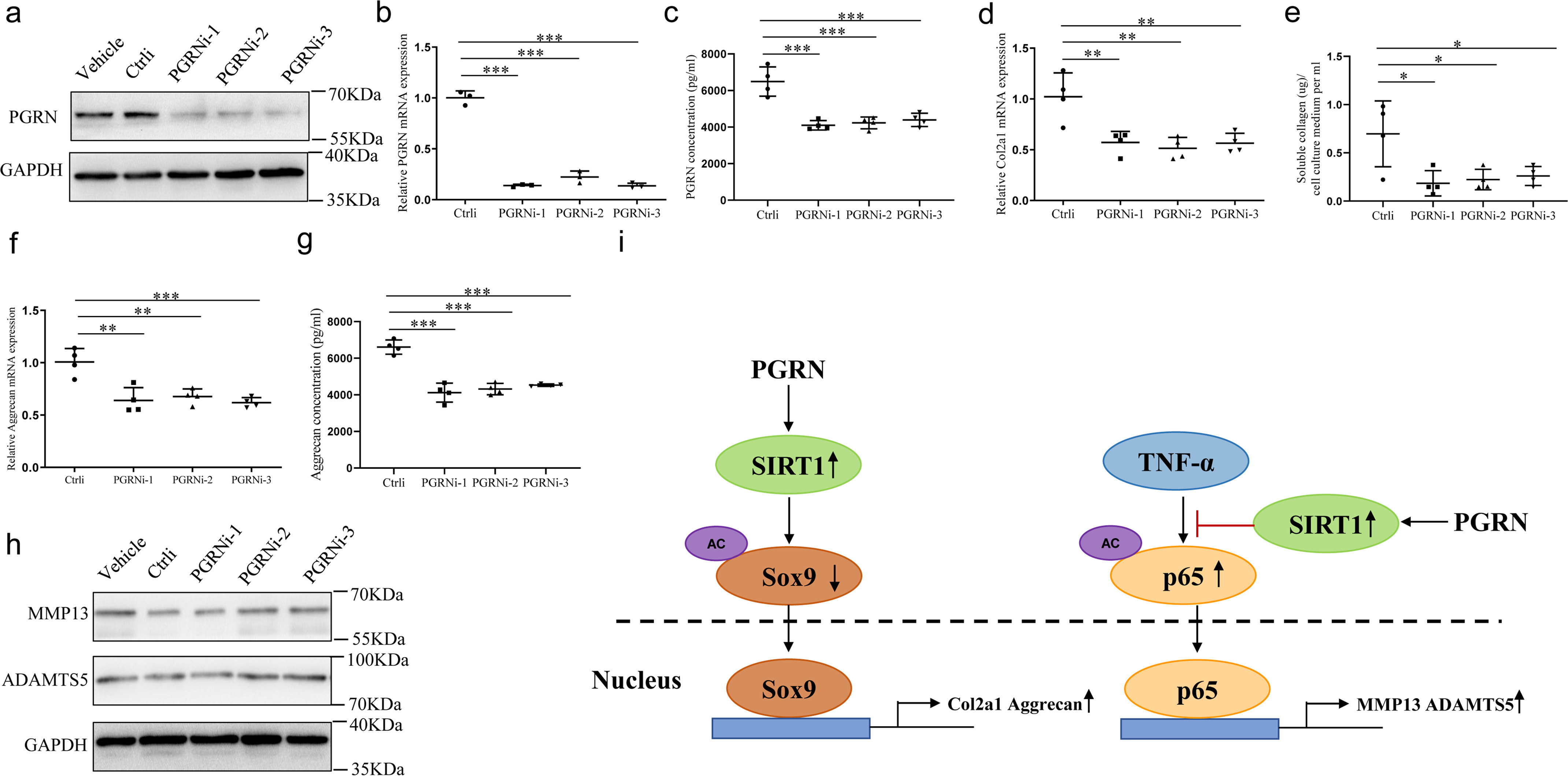
Effects of endogenous PGRN on cartilage gene expression. Chondrocytes were transfected with control siRNA or PGRN siRNA for 48 h. a and b, the protein and mRNA levels of PGRN were measured. PGRNi-1, PGRNi-2, and PGRNi-3 indicate three independent siRNAs against PGRN. One-way ANOVA and Tukey's multiple comparison test. c, PGRN secretion was detected by ELISA in the conditioned medium of chondrocytes. One-way ANOVA and Tukey's multiple comparison test. d and e, COL2A1 expression and secretion were detected by real-time PCR and soluble collagen quantification assay. One-way ANOVA and Tukey's multiple comparison test. f and g, aggrecan expression and secretion were detected by real-time PCR and ELISA. One-way ANOVA and Tukey's multiple comparison test. h, levels of MMP13 and ADAMTS5 were analyzed by Western blotting. i, model depicting the molecular mechanism that PGRN plays an important role in maintaining chondrocyte homeostasis. Data were expressed as mean ± S.D. in each scatterplot. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
PGRN has been purified and identified as a growth factor from conditioned tissue culture media by several independent laboratories (26–28), suggesting it may act as an autocrine manner. Additionally, PGRN is expressed in both growth plate chondrocytes and articular cartilage chondrocytes, and its level is significantly increased during the entire cartilage development, whereas cartilage-specific ablation of PGRN showed a sharp reduction in skeletal length (29). Our study also showed that recombinant PGRN significantly enhanced the levels of COL2A1 and aggrecan, whereas down-regulation of endogenous PGRN inhibited COL2A1 and aggrecan expression and secretion. Therefore, it is reasonable to speculate that PGRN functions effectively in an autocrine manner in chondrocytes. Because PGRN is an autocrine growth factor, we first determined the level of endogenous PGRN in OA cartilage samples. We found the expression of PGRN was increased in the cartilage of lateral femoral condyles with mild OA, whereas its level was decreased in the cartilage of medial femoral condyles with severe OA. Although PGRN may increase as an adaptive response to protect chondrocytes against various stresses during the initial degenerative phase, it may be that elevated PGRN levels are not sufficient to completely neutralize the loss of articular cartilage, and failure to adapt may lead to decreased PGRN level and further progression of cartilage degeneration. Furthermore, the level of PGRN tended to be more increased in the superficial zone of the cartilage, where chondrocytes may undergo more stresses compared with the deep zone. We also observed that PGRN receptor EphA2 was decreased in severe OA cartilage, which may be the other reason why endogenous PGRN produced in OA could not ameliorate cartilage degradation.
Other endogenous autocrine factors in addition to PGRN have also exerted crucial roles within cartilage tissue. For instance, most bone morphogenetic proteins (BMPs), which are members of the transforming growth factor-β superfamily, play a protective role during OA development (30). Among the fibroblast growth factors family, FGF1, FGF2, and FGF8 may act as negative regulators of chondrocyte homeostasis, whereas FGF9 and FGF18 protect articular cartilage from degradation during OA progression (31). As with most anabolic growth factors, including BMPs and IGF-1, PGRN promotes chondrocytes proliferation and hypertrophy during the growth plate chondrogenesis, an essential process for both the long bone growth and bone fracture healing. Although BMP2 was the first trial in bone fracture healing given its promoting effect on new bone formation, its significant side effect of ectopic ossification hampers its further clinical application. Therefore, PGRN may be a novel therapeutic approach for application to bone fracture healing (32). Unlike other growth factors, however, PGRN still plays a protective role in the progression of OA, which is characterized by abnormal chondrocyte hypertrophy in articular cartilage followed by cartilage degradation. In the present study, we found that under the stimulation of TNFα, PGRN significantly inhibited MMP13 and ADAMTS5 expression, with such suppression being largely nullified in SIRT1−/− chondrocytes. Although PGRN exerts an anti-inflammatory function in inflammatory arthritis murine models, proteolytic cleavage of this precursor protein by serine proteases and metalloproteinases gives rise to individual 6-kDa granulin units, which are actually pro-inflammatory (33). Thus, the function of PGRN during inflammation is complex, with intact PGRN having anti-inflammatory properties, whereas granulins have been shown to promote inflammation. Furthermore, the function of PGRN might be tissue specific. Contrary to its positive effects on cartilage homeostasis, both our findings (34) and other studies (35, 36) indicate that PGRN is proinflammatory adipokine and involves in adipose insulin resistance, suggesting that PGRN may be a negative factor in regulation of energy metabolism. Different from other anabolic growth factors (BMP, insulin-like growth factors, fibroblast growth factors, etc.) that have already identified binding receptors, PGRN membrane receptor has not yet been clarified so far. Some studies reported PGRN action is not mediated through TNFR (37, 38), whereas more studies suggested that PGRN exerted its anti-inflammatory effect through antagonizing TNFα by binding to TNF receptors (5, 6). However, in our preliminary study, neither TNFR1 nor TNFR2 mediated PGRN function on cartilage homeostasis and STRT1 expression (data not shown). However, we found that expression pattern of EphA2 was similar to that of PGRN in OA cartilage and recombinant PGRN increased EphA2 level in chondrocytes, implicating a potential involvement of EphA2 in mediation of PGRN function. This finding may add value to the body of conflicting reports on PGRN functional receptor in chondrocyte. Consistent with what we have found, other groups indicated that PGRN could bind to EphA2 (39) and sortilin (40) in urothelial carcinoma cells and neuron, respectively. Thus, further investigation should be warranted to fully elucidate how PGRN acts through its membrane receptor to regulate cartilage homeostasis.
Among the related publications, little is known about downstream events mediating PGRN in regulation of cartilage homeostasis, apart from the finding that PGRN promotes chondrogenesis through activation of Erk1/2 pathway in vitro (10, 29). Although this experimental evidence has shed light on the signaling pathways involved, it remains an exciting prospect to determine the one or more signaling pathways ultimately mediating the effects of PGRN on cartilage homeostasis. Here our study provides the novel mechanism of PGRN action in maintaining cartilage homeostasis. In our preliminary study, we first detected autophagy activity in chondrocytes cultured with PGRN, because our previous studies showed that PGRN disturbed autophagic balance in adipocytes and hepatocytes, respectively (34, 41). However, no statistical changes were observed in autophagy-related protein (LC3-II and p62) expression. The discrepancies may reflect cell-type specificities including differentiation state. As aging is a major risk factor of osteoarthritis, our latest study revealed a protective effect of SIRT1, a known anti-aging factor on chondrogenesis through PERK-eIF-2α-CHOP (20). More recently, Zhou et al. (21) have shown that PGRN increases SIRT1 expression, resulting in decreased level in acetylation of PGC-1α and FoxO1 in podocytes treated with high glucose. However, crosstalk between PGRN and SIRT1 in chondrocytes remain unknown. In the current study, the expression and activity of SIRT1 were dramatically increased in presence of PGRN, and PGRN-mediated chondroprotective function was largely impaired in SIRT1−/− cells, indicating that PGRN mediates its effects, at least in part, by regulating SIRT1 level. Although PGRN did not affect SOX9 expression (data not shown), we found PGRN could decrease the acetylation level of SOX9 and subsequently increase nuclear accumulation of SOX9 in chondrocytes. Because PGRN has no inherent deacetylation, it was thought that its effect on acetylation level and nuclear localization of SOX9 was mediated through SIRT1, which in turn, promoted COL2A1 and aggrecan expression. In a subsequent study, we have shown that the effects of PGRN on SOX9 acetylation and nuclear translocation were significantly diminished after silencing SIRT1 expression, further supporting that PGRN-mediated such effects depend on up-regulation of SIRT1.
TNFα is well accepted to act as a critical mediator of pathogenesis in cartilage degeneration and arthritis; therefore, TNFα inhibitors or antagonists have been extensively studied for therapy of RA (42) and OA (43). In our study, although PGRN alone did not affect levels of catabolic markers, a combination of PGRN with TNFα inhibited the expression of catabolic markers induced by TNFα. Moreover, TNFα inhibited SIRT1 activity, whereas co-treatment with PGRN reversed these inhibitory effects induced by TNFα. These findings are similar to a previous report that TNFα cleaved the C-terminal domain of SIRT1 and generated a smaller 75-kDa fragment, resulting in reduced SIRT1 activity (44). Given that TNFα is known to activate NF-κB signaling pathway, which in turn induces the expression of various MMPs and ADAMTS (24), it is plausible for us to hypothesize that PGRN inhibited chondrocyte catabolism induced by TNFα is mediated by regulating NF-κB–P65. Although PGRN did not much affect the total P65 protein level, it decreased the acetylation level of Lys-310 of P65 induced by TNFα, whereas transfection with SIRT1 siRNA almost abolished such inhibitory effects in PGRN-treated chondrocytes. Because the acetylation status of Lys-310 of P65 has been well established to be an important regulatory mechanism of the transcriptional activity of P65 (45, 46), we then determined whether PGRN affected P65 nuclear translocation by confocal microscopy. As expected, PGRN reduced translocation of P65 into the nucleus in chondrocytes treated with TNFα, whereas this effect was impaired in SIRT1−/− cells. Although Zhao and his colleagues thought that PGRN may mainly act as an antagonist of TNFα (10), we hereby propose that PGRN inhibited chondrocyte catabolism induced by TNFα through activation of SIRT1, and subsequently prevented P65 nuclear translocation by deacetylating P65, thereby decreasing the expression of MMP13 and ADAMTS5.
In conclusion, our study suggested that PGRN promoted cartilage-specific gene expression as well as inhibited the TNFα-induced expression of cartilage-degrading enzymes through activating SIRT1-SOX9/NF-κB–P65 signaling (Fig. 6i), indicating that PGRN may be a promising therapeutic target for managing OA as well as other cartilage degenerative diseases.
Experimental procedures
OA and normal cartilage sample obtaining and processing
OA human articular cartilage tissues were obtained from six patients (mean ± S.D. age 75.2 ± 5.7 years) undergoing total knee-joint replacement surgery. Normal human cartilage tissues were harvested from six age-matched patients (mean ± S.D. age 69.8 ± 7.1 years) undergoing surgery for femoral neck fracture without history of joint-related disease (inflammatory arthritis, OA, microcrystalline arthritis, or osteonecrosis).
Safranin-O and fast green staining
Cartilage tissues were fixed in 4% paraformaldehyde overnight and decalcified with 10% EDTA for 2 weeks, and 5-μm–thick sections from each cartilage tissue were obtained and stained with safranin-O and fast green. Briefly, sections were stained with hematoxylin for 2 min, followed by fast green staining for 3 min and safranin-O staining for 3 min.
Immunohistochemistry
To detect PGRN or EphA2 expression in articular cartilage, 5-μm–thick cartilage sections from each sample were obtained. Sections were immunostained with SPlink Detection Kits (Beijing Zhongshan Biotechnologies, SP-9001). Briefly, sections were treated with 0.25% trypsin for 15 min at 37°C for antigen retrieval and followed by 3% H2O2 for 10 min, and then were blocked using 5% goat serum for 15 min at room temperature (RT). Afterward, sections were incubated with rabbit polyclonal antibodies against PGRN at a dilution of 1:200 (Beijing Bioss Biotechnologies, bs-0823R) or rabbit monoclonal antibodies against EphA2 at a dilution of 1:200 (Cell Signaling Technology, 6997) overnight at 4°C. After three rinses with PBS, sections were incubated with rabbit secondary antibodies for 15 min at RT. Finally, sections were counterstained with hematoxylin. The percentage of designated gene-positive cells was calculated as the number of positive cells per grid divided by the total number of cells per grid quantified by Image-Pro Plus. The grid circumscribed a portion of the cartilage analyzed through a 10× objective and generally contained an average of 50 cells. For each sample, the fraction of positive cells in three distinct grid locations was calculated and averaged.
Primary chondrocyte culture
The articular cartilage tissues isolated from newborn Sprague-Dawley rat (3–5 days) were dissected, rinsed in PBS, then pretreated with 0.25% trypsin for 10 min at 37°C, followed by incubating in 0.2% collagenase (Sigma, C6885) for 1.5 h. The cell suspension was aspirated repeatedly and filtered through a 70-μm cell strainer, rinsed first in PBS and then in serum-free DMEM, and counted. Chondrocytes were seeded at a density of 2 × 106 cell/ml in DMEM with 100 units/ml penicillin, 100 μg/ml streptomycin, 50 µg/ml ascorbic acid, and 10% FBS (Gibco). The culture medium was changed at 48-h intervals. After reaching about 90% confluence, cells were treated with recombinant PGRN (R&D Systems, 2420-PG-050) (0–200 ng/ml) and/or TNFα (Peprotech, 315-01A).
siRNA transfection
Chondrocytes were transfected with three siRNA with distinctive sequences targeted for SIRT1 or PGRN, respectively. All siRNA were designed and synthesized by GenePharma Co. (Shanghai, China). The sequences of siRNA were as follows: For SIRT1 no. 1, sense strand 5′-CCA GUA GCA CUA AUU CCA ATT-3′, antisense active strand 5′-UUG GAA UUA GUG CUA CUG GTT-3′; for SIRT1 no. 2, sense strand 5′-CCC UGU AAA GCU UUC AGA ATT-3′, antisense active strand 5′-UUC UGA AAG CUU UAC AGG GTT-3′; for SIRT1 no. 3, sense strand 5′-GCG UCU UGA CGG UAA UCA ATT-3′, antisense active strand 5′-UUG AUU ACC GUC AAG ACG CTT-3′; for PGRN no. 1, sense strand 5′-GGG UGU AUC UUG UGA UGA UTT-3′, antisense active strand 5′-AUC AUC ACA AGA UAC ACC CTT-3′; for PGRN no. 2, sense strand 5′-GCU ACC CAC UGG GAA GUA UTT-3′, antisense active strand 5′-AUA CUU CCC AGU GGG UAG CTT-3′; for PGRN no.3, sense strand 5′-CCA GAC AAC UCU GCU CCA ATT, antisense active strand 5′-UUG GAG CAG AGU UGU CUG GTT-3′. The siRNA was introduced to cells using Lipofectamine 2000 (Invitrogen, 11668027), according to the procedure recommended by the manufacturer. The transfected cells were cultured in DMEM containing 10% FBS for 48 h after transfection.
Western blotting
Whole-cell or cartilage tissue extracts were prepared with the RIPA buffer. Proteins were separated by 8–15% SDS-PAGE gel; separated proteins were transferred onto PVDF membranes (Millipore) and were probed with the following primary antibodies: rabbit monoclonal antibodies against SIRT1 (Cell Signaling Technology, 9475), PGRN (Abcam, ab227816), and NF-κB–P65 (Cell Signaling Technology, 8242); rabbit polyclonal antibodies against COL2A1 (Abcam, ab34712), aggrecan (Abcam, ab3778), MMP13 (Abcam, ab39012), ADAMTS5 (Abcam, ab41037), and acetyl–NF-κB P65 (Lys-310) (Cell Signaling Technology, 3045); mouse monoclonal antibodies against GAPDH (Santa Cruz Biotechnology, sc-365062). At last, the blots were visualized by an ECL detection system (Millipore) with a horseradish peroxidase–conjugated secondary antibody. To recognize aggrecan optimal epitope, chondrocytes lysates per 10 µg are pretreated with 0.01 units Chondroitinase ABC (Sigma, C3667) for 2 h at 37°C. A representative blot from three or four independent experiments was presented for each protein. The relative levels of protein expression were calculated using densitometric scans by ImageJ software and were normalized to the GAPDH levels from three or four independent experiments. For each independent experiment, the relative levels of protein expression were calculated and averaged from three replicates.
RNA extraction and real time PCR
Total RNA from cultured chondrocytes or cartilage tissue were isolated by TRIzol reagent (Invitrogen, 15596–026) according to the manufacturer's instruction. The recovered RNA was further processed using RevertAid First Strand cDNA Synthesis kit (Thermo Fisher, K1621) to produce cDNA in accordance with the manufacturer's instructions. The cDNA products were directly used for PCR or stored at –80°C for later analysis. Real-time quantitative PCR was performed in MJ Mini Real-Time PCR Detection System using SYBR Premix Ex TaqTM II (Takara, RR047A). Primer sequence information is available upon request. Each experiment was performed in duplicate and experiments repeated three or four times independently. A dissociation curve analysis was conducted for each qPCR. Expression levels of the target gene were evaluated using a relative quantification approach (2−ΔΔCt method) against GAPDH levels.
Immunoprecipitation
Cytoplasmic lysate (200 μg) was incubated for 2 h at 4°C with the corresponding antibodies coupled to 20 μl of packed protein A + G Sepharose beads (Santa Cruz Biotechnology, sc-2002). Immune complexes were resolved by means of SDS-PAGE and immunoblotted with the indicated antibodies. To analyze the level of SOX9 acetylation, chondrocytes lysates were immunoprecipitated using anti-SOX9 antibody (Abcam, ab185230), and then immunoprecipitated proteins were run on SDS-PAGE and immunoblotted with anti-SOX9 and anti–acetyl lysine antibody (Santa Cruz Biotechnology, sc-32268), respectively. The relative level of SOX9 acetylation was calculated using densitometric scans by ImageJ software and was normalized to the SOX9 levels from three or four independent experiments. For each independent experiment, the relative level of SOX9 acetylation was calculated and averaged from three replicates.
Immunofluorescence
Primary cultured chondrocytes were seeded on 6-well chamber slides and fixed in 4% paraformaldehyde for 15 min at RT. Cells were then permeabilized with 0.2% Triton X-100 in PBS. After incubation in 5% goat serum, the permeabilized chondrocytes were incubated with anti-SOX9 (1:200) or anti–NF-κB–P65 (1:200) antibodies. For secondary reactions, species-matched Cy3-labeled secondary antibody at a dilution of 1:200 was used for 1 h at 37°C. Cell nuclei were stained with DAPI. Fluorescent images were collected on a Leica confocal microscope (SP5 II). The percentage of cells with nuclear SOX9 and P65 in slides was quantified by Image-Pro Plus. The data represent the percentage of cells from four independent experiments. For each independent experiment, the percentage of cells in five distinct grid locations was calculated and averaged.
SIRT1 deacetylase activity assay
To detect SIRT1 activity in cultured chondrocytes, SIRT1 was initially immunoprecipitated with a SIRT1 antibody (Cell Signaling Technology, 8469). Subsequently, SIRT1 deacetylase activity was measured by fluorometric SIRT1 Assay Kit (Sigma, CS1040), according to the procedure recommended by the manufacturer. Fluorescent intensity was measured at 460 nm (excitation 380 nm) and concentration was calculated using a standard curve.
Collagen quantification and analysis
Collagen quantification was performed using fluorometric Soluble Collagen Quantification Assay Kit (Sigma, CS0006) following the manufacturer's protocol. Fluorescent intensity was measured at 465 nm (excitation 375 nm) and concentration was calculated using a standard curve.
ELISA
Collected conditional media of chondrocytes underwent ELISA using rat ELISA kits according to the manufacturers' instructions. PGRN- and aggrecan-specific ELISA kits were obtained from Jiangsu MEIMIAN Industrial Co., Ltd. (MM-70087R2 and MM-0772R2).
ChIP
ChIP analysis was performed using a SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, 9002) following the manufacturer's protocol. The chromatin sample was subjected to immunoprecipitation with P65 or the control normal rabbit IgG. Real-time PCR was performed, and primer sets that amplify the area included the NF-κB motif (−478/−328 for −424/−415) within the 5′ region of genomic human ADAMTS5 (NC_000021.9) based on previous reports (47). The primers used were as follows: Forward: 5′-CAA AGG GGA AAA ACT TGC GG-3′; reverse: 5′-AAT TGG GGT TGG CTA ACC TTG-3′. Values obtained from each immunoprecipitation are expressed as a percent of the total input chromatin from three independent experiments.
Ethics approval
Animal care was approved by the Animal Experiment Administration Committee of the Medicine of Xi'an Jiaotong University. Human samples were collected after obtaining written informed consent as approved by the Ethics Committee of the Xi'an Jiaotong University and the study was conducted in compliance with the ethical principle of the Declaration of Helsinki.
Statistics
Statistical analysis was performed with the SPSS 17.0 software (SPSS Inc., Chicago, IL). All the experiments were repeated three or four times independently, and data were presented as mean ± S.D. A Shapiro-Wilk normality test was performed to evaluate the Gaussian distribution of the data. All groups of data were assessed for the homogeneity of variance using the Fisher test. Differences between two groups were analyzed by unpaired two-tailed Student's t tests. Differences among more than two groups were analyzed by one-way analysis of variance (ANOVA) and Tukey's multiple comparison test. p values less than 0.05 were considered statistically significant.
Data availability
All data are contained within the manuscript and supporting information.
Supplementary Material
This article contains supporting information.
Author contributions—D. F., R. W., W. F., H. L., and S. W. data curation; D. F., X. K., and S. W. formal analysis; D. F., X. K., Y. Z., and S. W. investigation; D. F., X. K., H. C., and K. Z. methodology; D. F. and X. K. writing-original draft; X. K. and S. W. funding acquisition; S. W. conceptualization.
Funding and additional information—This work was supported by the First Affiliated Hospital of Xi'an Jiaotong University College of Medicine Foundation Grant 2018QN-01, the National Natural Science Foundation of China Grant 81672221, 30971392 and 81071440/H0601 and Natural Science Basic Research Program of Shaanxi Programs 2020JM-689 and 2020JQ-507.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- PGRN
- progranulin
- OA
- osteoarthritis
- RA
- rheumatoid arthritis
- BMP
- bone morphogenetic protein
- RT
- room temperature
- ANOVA
- analysis of variance.
References
- 1. Loeser R. F., Goldring S. R., Scanzello C. R., and Goldring M. B. (2012) Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 64, 1697–1707 10.1002/art.34453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goldring M. B., and Marcu K. B. (2009) Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 11, 224 10.1186/ar2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim H. A., Lee Y. J., Seong S. C., Choe K. W., and Song Y. W. (2000) Apoptotic chondrocyte death in human osteoarthritis. J. Rheumatol. 27, 455–462 [PubMed] [Google Scholar]
- 4. Blagojevic M., Jinks C., Jeffery A., and Jordan K. P. (2010) Risk factors for onset of osteoarthritis of the knee in older adults: A systematic review and meta-analysis. Osteoarthritis Cartilage 18, 24–33 10.1016/j.joca.2009.08.010 [DOI] [PubMed] [Google Scholar]
- 5. Liu C. J. (2011) Progranulin: A promising therapeutic target for rheumatoid arthritis. FEBS Lett. 585, 3675–3680 10.1016/j.febslet.2011.04.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tang W., Lu Y., Tian Q. Y., Zhang Y., Guo F. J., Liu G. Y., Syed N. M., Lai Y., Lin E. A., Kong L., Su J., Yin F., Ding A. H., Zanin-Zhorov A., Dustin M. L., et al. (2011) The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 332, 478–484 10.1126/science.1199214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. He Z., Ong C. H., Halper J., and Bateman A. (2003) Progranulin is a mediator of the wound response. Nat. Med. 9, 225–229 10.1038/nm816 [DOI] [PubMed] [Google Scholar]
- 8. Bateman A., and Bennett H. P. (2009) The granulin gene family: From cancer to dementia. Bioessays 31, 1245–1254 10.1002/bies.200900086 [DOI] [PubMed] [Google Scholar]
- 9. Zhao Y. P., Tian Q. Y., Frenkel S., and Liu C. J. (2013) The promotion of bone healing by progranulin, a downstream molecule of BMP-2, through interacting with TNF/TNFR signaling. Biomaterials 34, 6412–6421 10.1016/j.biomaterials.2013.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao Y. P., Liu B., Tian Q. Y., Wei J. L., Richbourgh B., and Liu C. J. (2015) Progranulin protects against osteoarthritis through interacting with TNF-α and β-Catenin signalling. Ann. Rheum. Dis. 74, 2244–2253 10.1136/annrheumdis-2014-205779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo F., Lai Y., Tian Q., Lin E. A., Kong L., and Liu C. (2010) Granulin-epithelin precursor binds directly to ADAMTS-7 and ADAMTS-12 and inhibits their degradation of cartilage oligomeric matrix protein. Arthritis Rheum. 62, 2023–2036 10.1002/art.27491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamamoto Y., Takemura M., Serrero G., Hayashi J., Yue B., Tsuboi A., Kubo H., Mitsuhashi T., Mannami K., Sato M., Matsunami H., Matuo Y., and Saito K. (2014) Increased serum GP88 (progranulin) concentrations in rheumatoid arthritis. Inflammation 37, 1806–1813 10.1007/s10753-014-9911-4 [DOI] [PubMed] [Google Scholar]
- 13. Andrés Cerezo L., Kuklová M., Hulejová H., Vernerová Z., Kaspříková N., Veigl D., Pavelka K., Vencovský J., and Šenolt L. (2015) Progranulin is associated with disease activity in patients with rheumatoid arthritis. Mediators Inflamm. 2015, 740357 10.1155/2015/740357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen L., Li Q., Wang J., Jin S., Zheng H., Lin J., He F., Zhang H., Ma S., Mei J., and Yu J. (2017) MiR-29b-3p promotes chondrocyte apoptosis and facilitates the occurrence and development of osteoarthritis by targeting PGRN. J. Cell. Mol. Med. 21, 3347–3359 10.1111/jcmm.13237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dvir-Ginzberg M., Gagarina V., Lee E. J., and Hall D. J. (2008) Regulation of cartilage-specific gene expression in human chondrocytes by SirT1 and nicotinamide phosphoribosyltransferase. J. Biol. Chem. 283, 36300–36310 10.1074/jbc.M803196200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bar Oz M., Kumar A., Elayyan J., Reich E., Binyamin M., Kandel L., Liebergall M., Steinmeyer J., Lefebvre V., and Dvir-Ginzberg M. (2016) Acetylation reduces SOX9 nuclear entry and ACAN gene transactivation in human chondrocytes. Aging Cell 15, 499–508 10.1111/acel.12456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takayama K., Ishida K., Matsushita T., Fujita N., Hayashi S., Sasaki K., Tei K., Kubo S., Matsumoto T., Fujioka H., Kurosaka M., and Kuroda R. (2009) SIRT1 regulation of apoptosis of human chondrocytes. Arthritis Rheum. 60, 2731–2740 10.1002/art.24864 [DOI] [PubMed] [Google Scholar]
- 18. Fujita N., Matsushita T., Ishida K., Kubo S., Matsumoto T., Takayama K., Kurosaka M., and Kuroda R. (2011) Potential involvement of SIRT1 in the pathogenesis of osteoarthritis through the modulation of chondrocyte gene expressions. J. Orthop. Res. 29, 511–515 10.1002/jor.21284 [DOI] [PubMed] [Google Scholar]
- 19. Matsuzaki T., Matsushita T., Takayama K., Matsumoto T., Nishida K., Kuroda R., and Kurosaka M. (2014) Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann. Rheum. Dis. 73, 1397–1404 10.1136/annrheumdis-2012-202620 [DOI] [PubMed] [Google Scholar]
- 20. Kang X., Yang W., Wang R., Xie T., Li H., Feng D., Jin X., Sun H., and Wu S. (2018) Sirtuin-1 (SIRT1) stimulates growth-plate chondrogenesis by attenuating the PERK-eIF-2α-CHOP pathway in the unfolded protein response. J. Biol. Chem. 293, 8614–8625 10.1074/jbc.M117.809822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou D., Zhou M., Wang Z., Fu Y., Jia M., Wang X., Liu M., Zhang Y., Sun Y., Lu Y., Tang W., and Yi F. (2019) PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 10, 524 10.1038/s41419-019-1754-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. He Z., and Bateman A. (2003) Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J. Mol. Med. 81, 600–612 10.1007/s00109-003-0474-3 [DOI] [PubMed] [Google Scholar]
- 23. Matsushita T., Sasaki H., Takayama K., Ishida K., Matsumoto T., Kubo S., Matsuzaki T., Nishida K., Kurosaka M., and Kuroda R. (2013) The overexpression of SIRT1 inhibited osteoarthritic gene expression changes induced by interleukin-1β in human chondrocytes. J. Orthop. Res. 31, 531–537 10.1002/jor.22268 [DOI] [PubMed] [Google Scholar]
- 24. Kapoor M., Martel-Pelletier J., Lajeunesse D., Pelletier J. P., and Fahmi H. (2011) Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 7, 33–42 10.1038/nrrheum.2010.196 [DOI] [PubMed] [Google Scholar]
- 25. Elayyan J., Lee E. J., Gabay O., Smith C. A., Qiq O., Reich E., Mobasheri A., Henrotin Y., Kimber S. J., and Dvir-Ginzberg M. (2017) LEF1-mediated MMP13 gene expression is repressed by SIRT1 in human chondrocytes. FASEB J. 31, 3116–3125 10.1096/fj.201601253R [DOI] [PubMed] [Google Scholar]
- 26. Zhou J., Gao G., Crabb J. W., and Serrero G. (1993) Purification of an autocrine growth factor homologous with mouse epithelin precursor from a highly tumorigenic cell line. J. Biol. Chem. 268, 10863–10869 [PubMed] [Google Scholar]
- 27. Anakwe O. O., and Gerton G. L. (1990) Acrosome biogenesis begins during meiosis: Evidence from the synthesis and distribution of an acrosomal glycoprotein, acrogranin, during guinea pig spermatogenesis. Biol. Reprod. 42, 317–328 10.1095/biolreprod42.2.317 [DOI] [PubMed] [Google Scholar]
- 28. Baba T., Hoff H. B. 3rd, Nemoto H., Lee H., Orth J., Arai Y., and Gerton G. L. (1993) Acrogranin, an acrosomal cysteine-rich glycoprotein, is the precursor of the growth-modulating peptides, granulins, and epithelins, and is expressed in somatic as well as male germ cells. Mol. Reprod. Dev. 34, 233–243 10.1002/mrd.1080340302 [DOI] [PubMed] [Google Scholar]
- 29. Feng J. Q., Guo F. J., Jiang B. C., Zhang Y., Frenkel S., Wang D. W., Tang W., Xie Y., and Liu C. J. (2010) Granulin epithelin precursor: A bone morphogenic protein 2-inducible growth factor that activates Erk1/2 signaling and JunB transcription factor in chondrogenesis. FASEB J. 24, 1879–1892 10.1096/fj.09-144659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deng Z. H., Li Y. S., Gao X., Lei G. H., and Huard J. (2018) Bone morphogenetic proteins for articular cartilage regeneration. Osteoarthritis Cartilage 26, 1153–1161 10.1016/j.joca.2018.03.007 [DOI] [PubMed] [Google Scholar]
- 31. Chen T. M., Chen Y. H., Sun H. S., and Tsai S. J. (2019) Fibroblast growth factors: Potential novel targets for regenerative therapy of osteoarthritis. Chin. J. Physiol. 62, 2–10 10.4103/CJP.CJP_11_19 [DOI] [PubMed] [Google Scholar]
- 32. Wei J., Zhang L., Ding Y., Liu R., Guo Y., Hettinghouse A., Buza J., De La Croix J., Li X., Einhorn T. A., and Liu C. J. (2020) Progranulin promotes diabetic fracture healing in mice with type 1 diabetes. Ann. N. Y. Acad. Sci. 1460, 43–56 10.1111/nyas.14208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kessenbrock K., Frohlich L., Sixt M., Lammermann T., Pfister H., Bateman A., Belaaouaj A., Ring J., Ollert M., Fassler R., and Jenne D. E. (2008) Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J. Clin. Invest. 118, 2438–2447 10.1172/JCI34694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li H., Zhou B., Xu L., Liu J., Zang W., Wu S., and Sun H. (2014) Circulating PGRN is significantly associated with systemic insulin sensitivity and autophagic activity in metabolic syndrome. Endocrinology 155, 3493–3507 10.1210/en.2014-1058 [DOI] [PubMed] [Google Scholar]
- 35. Matsubara T., Mita A., Minami K., Hosooka T., Kitazawa S., Takahashi K., Tamori Y., Yokoi N., Watanabe M., Matsuo E., Nishimura O., and Seino S. (2012) PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab. 15, 38–50 10.1016/j.cmet.2011.12.002 [DOI] [PubMed] [Google Scholar]
- 36. Youn B. S., Bang S. I., Klöting N., Park J. W., Lee N., Oh J. E., Pi K. B., Lee T. H., Ruschke K., Fasshauer M., Stumvoll M., and Blüher M. (2009) Serum progranulin concentrations may be associated with macrophage infiltration into omental adipose tissue. Diabetes 58, 627–636 10.2337/db08-1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen X., Chang J., Deng Q., Xu J., Nguyen T. A., Martens L. H., Cenik B., Taylor G., Hudson K. F., Chung J., Yu K., Yu P., Herz J., Farese R. V. Jr., Kukar T., et al. (2013) Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J. Neurosci. 33, 9202–9213 10.1523/JNEUROSCI.5336-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Etemadi N., Webb A., Bankovacki A., Silke J., and Nachbur U. (2013) Progranulin does not inhibit TNF and lymphotoxin-α signalling through TNF receptor 1. Immunol. Cell Biol. 91, 661–664 10.1038/icb.2013.53 [DOI] [PubMed] [Google Scholar]
- 39. Neill T., Buraschi S., Goyal A., Sharpe C., Natkanski E., Schaefer L., Morrione A., and Iozzo R. V. (2016) EphA2 is a functional receptor for the growth factor progranulin. J. Cell Biol. 215, 687–703 10.1083/jcb.201603079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu F., Padukkavidana T., Vægter C. B., Brady O. A., Zheng Y., Mackenzie I. R., Feldman H. H., Nykjaer A., and Strittmatter S. M. (2010) Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68, 654–667 10.1016/j.neuron.2010.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu J., Li H., Zhou B., Xu L., Kang X., Yang W., Wu S., and Sun H. (2015) PGRN induces impaired insulin sensitivity and defective autophagy in hepatic insulin resistance. Mol. Endocrinol. 29, 528–541 10.1210/me.2014-1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Furst D. E. (2010) Development of TNF inhibitor therapies for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 28, S5–S12 [PubMed] [Google Scholar]
- 43. Buckland J. (2013) Osteoarthritis: Positive feedback between ADAMTS-7 and TNF in OA. Nat. Rev. Rheumatol. 9, 566 10.1038/nrrheum.2013.135 [DOI] [PubMed] [Google Scholar]
- 44. Dvir-Ginzberg M., Gagarina V., Lee E. J., Booth R., Gabay O., and Hall D. J. (2011) Tumor necrosis factor α-mediated cleavage and inactivation of SirT1 in human osteoarthritic chondrocytes. Arthritis Rheum. 63, 2363–2373 10.1002/art.30279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen L., Fischle W., Verdin E., and Greene W. C. (2001) Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293, 1653–1657 10.1126/science.1062374 [DOI] [PubMed] [Google Scholar]
- 46. Kiernan R., Brès V., Ng R. W., Coudart M. P., El Messaoudi S., Sardet C., Jin D. Y., Emiliani S., and Benkirane M. (2003) Post-activation turn-off of NF-κB-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 278, 2758–2766 10.1074/jbc.M209572200 [DOI] [PubMed] [Google Scholar]
- 47. Kobayashi H., Hirata M., Saito T., Itoh S., Chung U. I., and Kawaguchi H. (2013) Transcriptional induction of ADAMTS5 protein by nuclear factor-κB (NF-κB) family member RelA/p65 in chondrocytes during osteoarthritis development. J. Biol. Chem. 288, 28620–28629 10.1074/jbc.M113.452169 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript and supporting information.