

Abstract
Using the Nephrotic Syndrome Study Network Consortium data set and other publicly available transcriptomic data sets, we identified retinoic acid receptor responder protein 1 (RARRES1) as a gene whose expression positively correlated with renal function decline in human glomerular disease. The glomerular expression of RARRES1, which is largely restricted to podocytes, increased in focal segmental glomerulosclerosis (FSGS) and diabetic kidney disease (DKD). TNF-α was a potent inducer of RARRES1 expression in cultured podocytes, and transcriptomic analysis showed the enrichment of cell death pathway genes with RARRES1 overexpression. The overexpression of RARRES1 indeed induced podocyte apoptosis in vitro. Notably, this effect was dependent on its cleavage in the extracellular domain, as the mutation of its cleavage site abolished the apoptotic effect. Mechanistically, the soluble RARRES1 was endocytosed and interacted with and inhibited RIO kinase 1 (RIOK1), resulting in p53 activation and podocyte apoptosis. In mice, podocyte-specific overexpression of RARRES1 resulted in marked glomerular injury and albuminuria, while the overexpression of RARRES1 cleavage mutant had no effect. Conversely, podocyte-specific knockdown of Rarres1 in mice ameliorated glomerular injury in the setting of adriamycin-induced nephropathy. Our study demonstrates an important role and the mechanism of RARRES1 in podocyte injury in glomerular disease.
Keywords: Cell Biology, Nephrology
Keywords: Apoptosis, Chronic kidney disease, Signal transduction
Introduction
Podocytes are an integral component of the glomerular filtration unit, and its injury leads to proteinuria and glomerulosclerosis in multiple glomerular diseases, such as diabetic kidney disease (DKD) and focal segmental glomerulosclerosis (FSGS) (1). In DKD, the reduction in podocyte number or density correlates with disease progression (2–4), and their detachment from the glomerular basement membrane (GBM) and apoptosis upon injury are considered to be the 2 major mechanisms of podocyte loss in glomerular disease (5). As therapeutic options to prevent or reduce podocyte loss in glomerular disease are currently lacking, a better understanding of the mechanism of podocyte loss is warranted.
Retinoic acids (RA), the derivatives of vitamin A (retinol), are essential to the control of epithelial cell growth and differentiation as well as inhibition of inflammation (6). In addition to their established benefits in cancer chemoprevention (7), RA was shown to confer renoprotection in multiple experimental models of kidney disease (8, 9). However, these findings have not been translated into clinical use for kidney disease, largely because of the lack of insight into the effects of RA in human kidneys in the setting of glomerular disease and because RA can have significant toxicity in multiple tissues, particularly at high dosages (10, 11). Therefore, to better dissect the role of RA signaling in human glomerular disease, we used the transcriptomic data sets from patients with primary glomerular disease collected by the Nephrotic Syndrome Study Network Consortium (NEPTUNE) and examined the expression of RA-related signaling molecules and target genes (12). The analysis showed that several of the RA-related genes indeed correlated with the progression of glomerular disease. Among these, the expression of RA receptor responder protein 1 (RARRES1, also known as tazarotene-induced gene [TIG1]) (13) was positively associated with the progression of primary glomerular disease, suggesting that RARRES1 may be a risk gene for kidney disease. RARRES1, a type 1 transmembrane protein that was initially identified as an RA receptor–responsive gene (13), has since been reported as functioning as a tumor suppressor, as its expression is suppressed in various tumor cells (14, 15). However, the exact molecular mechanism of its antioncogenic effect is unclear, and the role of RARRES1 has not been explored in the context of kidney disease. Therefore, in this study, we examined the role and mechanism of RARRES1 signaling in the pathogenesis of glomerular diseases.
Results
Determination of RA-related signaling pathways and target gene expression in human glomerular transcriptomic data sets from NEPTUNE.
NEPTUNE is an NIH-funded consortium for studying rare primary glomerular diseases, including minimal change disease (MCD), membranous nephropathy (MN), and FSGS (12). Using the collected NEPTUNE data set of glomerular transcriptomic profiles from kidney biopsy samples of patients with primary glomerular disease, we examined the expression of a subset of RA-related signaling molecules and target genes that were identified in the previous studies with animal models (16, 17). Nearly 50% (26/52 genes) of RA-associated genes on the microarray chip were differentially regulated in the glomerular compartment in samples of at least one disease etiology in comparison with samples from 6 healthy living donors. We further evaluated the correlation between gene expression levels with clinical parameters of kidney function, estimated glomerular filtration rate (eGFR) and protein-to-creatinine ratio (PCR), and disease progression rate, as quantified using eGFR slope by a mixed-effects model (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI140155DS1). Among these parameters, RARRES1 expression correlated with eGFR slope (Supplemental Table 1) as well as with renal function, as assessed by eGFR, serum creatinine, and blood urea nitrogen (BUN) levels (Figure 1A and Supplemental Figure 1). Since the correlation of RARRES1 with eGFR slope suggested that RARRES1 might be associated with kidney longitudinal outcome, we evaluated the association between RARRES1 expression and composite endpoint (end-stage renal disease [ESRD] or a 40% reduction of baseline renal function using Cox’s regression model). During an average follow-up time of 25.6 months (range: 1 to 50 months), 28 out of 190 patients progressed to composite endpoint. Higher expression of RARRES1 was significantly associated with increased risk of progression to ESRD or a 40% reduction of baseline eGFR (Figure 1B), suggesting that RARRES1 is a risk gene associated with the progression of primary glomerular disease.
Figure 1. RARRES1 mRNA expression correlates with clinical outcomes.

(A) The association of glomerular RARRES1 expression levels with eGFR slope of the CKD cohort in the NEPTUNE data set. (B) Cumulative survival by tertiles of RARRES1 gene expression levels (Kaplan-Meier analysis). The lowest tertile correspond to RARRES1 gene expression lower than 3.103, the middle tertile to that between 3.103 and 3.857, and the highest tertile to that greater than 3.857. Twenty-seven out of 152 patients progressed to composite endpoint of ESRD or 40% baseline eGFR reduction.
Expression and localization of RARRES1 in the normal and diseased kidney.
To further validate the potential role of RARRES1 in kidney disease, we queried the published transcriptomic data sets in kidney disease compiled in the Nephroseq database (nephroseq.org). RARRES1 mRNA expression was significantly increased in the glomeruli of DKD patients compared with those of healthy controls (18), and its expression significantly correlated with eGFR in both CKD and DKD patients (Supplemental Figure 2). Notably, recent single-cell RNA-Seq data sets of mouse kidney or glomeruli (19, 20) indicated that Rarres1 is expressed predominantly in podocytes (Supplemental Figure 3, A and B, and refs. 19, 20). Our recent single-cell transcriptomic data of glomerular cells from diabetic mouse kidneys (21) confirmed that the expression of Rarres1 was largely restricted to podocytes in both the normal and diabetic kidneys (Supplemental Figure 3C and ref. 21). Immunohistochemical staining showed low abundance of RARRES1 in the kidney biopsy samples of patients with MCD, but significantly increased expression in the glomerular cells of the kidney biopsies from patients with DKD and FSGS (Figure 2, A and B; clinical patient characteristics are listed in Supplemental Table 2). Immunofluorescence staining showed that RARRES1 expression largely colocalized with the podocyte marker Podocin and further increased in DKD patients (Figure 2C).
Figure 2. RARRES1 expression increased in FSGS and DKD.

(A) Immunohistochemical staining of RARRES1 in human renal biopsy specimens. Isotype IgG was used for negative control. Scale bar: 50 μm. (B) Quantification of mean RARRES1 intensity per glomerular cross section for human renal biopsy specimens as a fold change relative to MCD (n = 5 specimens per group, 10 glomeruli evaluated for each specimen). Data represent mean ± SD. **P < 0.01 and ****P < 0.0001 vs. MCD; ###P < 0.001 vs. FSGS, 1-way ANOVA with Tukey’s multiple comparison test. (C) Immunofluorescence costaining of RARRES1 and Podocin in renal biopsy specimen of DKD patients. Scale bar: 20 μm.
RARRES1 expression is induced by TNF-α in cultured human podocytes.
Since RARRES1 expression appears mostly in podocytes and is increased in the diseased kidney, we next determined how RARRES1 expression is upregulated using conditionally immortalized human podocytes. We first confirmed that RA induced RARRES1 expression in a dose-dependent manner (Supplemental Figure 4A). Interestingly, we found that TNF-α was more potent in the induction of RARRES1 than RA in human podocytes (Supplemental Figure 4, B and C), suggesting that in the kidney disease setting, such as with DKD and FSGS, RARRES1 expression is likely upregulated by TNF-α.
RARRES1 is cleaved and secreted extracellularly.
RARRES1, as a type 1 membrane protein, is composed of a short N-terminal intracellular region, a single membrane–spanning hydrophobic region, and a large C-terminal extracellular region containing a glycosylation signal (Supplemental Figure 5A). Cellular fractionation confirmed the membrane localization of RARRES1 in transfected podocytes expressing the V5-tagged RARRES1 (RARRES1-V5) (Figure 3A). Interestingly, a large extent of RARRES1 expression was found in the supernatant in RARRES1-V5 overexpressing podocytes (Figure 3B), suggesting that RARRES1 is cleaved and released extracellularly. The presence of cleaved RARRES1-V5 was confirmed by mass spectrometric analysis of supernatant proteins (data not shown). To further verify the cleavage on the extracellular domain of RARRES1, we overexpressed RARRES1 with either a C-terminal V5 tag or RARRES1 with a N-terminal FLAG tag, which would remain tethered to the plasma membrane (Supplemental Figure 5, A–C). As anticipated, while C-terminal V5-tagged RARRES1 was detected in the supernatant with both anti-V5 and anti-RARRES1 antibodies (Figure 3B), RARRES1 was detectable only by anti-RARRES1 antibody, but not by anti-FLAG antibody in the supernatant (Figure 3C), further confirming that the C-terminal portion of RARRES1 is cleaved and released extracellularly. The cleaved RARRES1 in the supernatant migrated as a larger protein than RARRES1 in the cell lysate (Figure 3B), suggesting that it may be highly glycosylated. Indeed, the deglycosylation treatment of the supernatant proteins resulted in the migration of RARRES1-V5 that was similar to the form found in the cell lysate (Figure 3D).
Figure 3. RARRES1 is cleaved and released into the supernatants as a highly glycosylated soluble form.

(A) Cultured human podocytes were transiently transfected to express control EGFP or C-terminal V5-tagged RARRES1 (RARRES1-V5). Expression of RARRES1 was probed with V5 antibody in the cytoplasmic (C) and membrane (M) fractions in comparison with the total cell lysate (T). Pan-cadherin was used to confirm the membrane protein enrichment in the M fraction. (B) Total cell lysate (CL) or supernatant (Supe) from cultured podocytes expressing control EGFP or C-terminal V5-tagged RARRES1 (RARRES1-V5) was probed for RARRES1 expression using V5 antibody. (C) Total cell lysate or supernatant from cultured podocytes expressing control EGFP or N-terminal FLAG-tagged RARRES1 (FLAG-RARRES1) was probed for RARRES1 expression using FLAG antibodies. (D) Total cell lysate or supernatant from cultured podocytes expressing RARRES1-V5 with or without deglycosylation treatment was probed with V5 antibody. Arrowheads indicate shift in the RARRES1-V5 migration with or without deglycosylation. (E) Total cell lysate or supernatant from cultured podocytes expressing EGFP control, C-terminal V5-tagged RARRES1 with the deletion of aa 43–76 (Δaa43-76-V5), deletion of aa 43–55 (Δaa43-55-V5), or WT RARRES1 (RARRES1-V5) was probed with V5 and RARRES1 antibodies. (F) Supernatant from cultured podocytes expressing C-terminal V5-tagged RARRES1 with the deletion of aa 68–71 (ΔFFNF-V5), alanine substitution of individual amino acids of aa 68–71 as indicated in underlined A residue (AAAA-V5, AFNF-V5, FANF-V5, FFAF-V5, and FFNA-V5), aspartic acid substitution of aa 70 (FFDF-V5), or WT RARRES1 (RARRES1-V5) was probed for expression with V5 antibody. (G) Total cell lysate or supernatant from cultured podocytes expressing EGFP control, C-terminal V5-tagged RARRES1 (RARRES1-V5), or RARRES1 FFAF mutant (FFAF-V5) with or without the treatment of broad-spectrum MMP inhibitor Marimastat (50 μM) were probed with V5 antibody. (H) Total cell lysate, supernatant, or isolated ectosomes (Ecto) from cultured podocytes expressing EGFP control, C-terminal V5-tagged RARRES1 (RARRES1-V5), or RARRES1 FFAF mutant (FFAF-V5) was probed with V5 antibody.
We next interrogated the potential cleavage site of RARRES1 by site-directed mutagenesis. We hypothesized that the cleavage may occur proximal to the transmembrane region near aa 21–42 (Supplemental Figure 5D) and constructed 2 deletion mutants of RARRES1 with a C-terminal V5-tag: RARRES1 with a larger deletion segment of aa 43–76 (Δaa43-76-V5) and RARRES1 with a smaller deletion of aa 43–55 (Δaa43-55-V5). The analysis of RARRES1 in the supernatant fraction in comparison with the cell lysate showed that while Δaa43-55-V5 was able to be cleaved, Δaa43-76-V5 was not, indicating that the cleavage site may be located between aa 56 and aa 76 (Figure 3E).
In silico analysis to predict cleavage site by enzymes from the metalloproteinase (MMP) family (ref. 22, 23; http://web.expasy.org/peptide_mass/) suggested that aa 68–71, encoding FFNF, may be the potential MMP cleavage site. Therefore, we constructed the mutant plasmid with the deletion of aa 68–71 (ΔFFNF-V5) or substitution of FFNF with AAAA (AAAA-V5). Both mutations abolished the cleavage of RARRES1 (Figure 3F), confirming that aa 68–71 are essential for the cleavage of RARRES1. We further made point mutations of aa 68–71 (encoding FFNF) as follows: AFNF, FANF, FFAF, FFNA, and FFDF (underlined aa changed to A or D in FFNF). The substitution of aa 70 (N) by either A or D (FFAF and FFDF) abolished the cleavage of RARRES1, while substitution by AFNF and FANA did not affect the cleavage, indicating that aa 70 is essential for the cleavage. (Figure 3F). Indeed, the cleavage of RARRES1-V5 was mitigated when cells were treated with the broad-spectrum MMP inhibitor marimastat (Figure 3G). Moreover, the cleaved RARRES1 in the supernatant fraction was not largely derived from ectosomes, since isolated ectosomes from transfected cells contained a minimal amount of WT or cleavage mutant RARRES1 (Figure 3H).
Podocyte apoptosis caused by the overexpression of RARRES1 is mitigated by the mutation of its cleavage site.
To examine the role of RARRES1 in podocytes, we next performed RNA-Seq of immortalized human podocytes with overexpression of RARRES1-V5. The pathway analysis indicated that top pathways with differentially expressed genes (DEGs) were of cell migration and apoptosis (Supplemental Table 3). We also performed RNA-Seq of podocytes with RARRES1 knockdown and stimulated with or without TNF-α. The top DEGs stimulated by TNF-α but suppressed by RARRES1 knockdown were enriched in cell cycle–related pathways (Supplemental Table 4). Together, these analyses suggest that RARRES1 plays a key role in the regulation of cell cycle and apoptosis, consistent with its purported role as a tumor suppressor. Indeed, we found that the overexpression of WT RARRES1 (RARRES1-V5) in podocytes induced apoptosis as demonstrated by annexin V staining and Western blot analysis of cleaved caspase-3 levels (Figure 4, A and B). Importantly, the overexpression of cleavage mutant RARRES1 (FFAF-V5) did not induce apoptosis in transfected podocytes, indicating that the cleavage of RARRES1 is required for the proapoptotic effects of RARRES1 (Figure 4, A and B). The incubation of podocytes with 10 ng/mL of soluble RARRES1 (sRARRES1-V5) isolated from supernatant collected from RARRES1-V5–overexpressing HEK293T cells induced apoptosis (Figure 4, C and D). Interestingly, V5-tagged soluble RARRES1 in the supernatant of cultured HEK293T cells overexpressing RARRES1-V5 was also found in the cell lysates of the podocytes after treatment (Figure 4D), suggesting that the soluble RARRES1 was taken up by podocytes. To determine whether this uptake was occurring through the endocytic process, we pretreated cells with a dynamin inhibitor (Dynasore) to block endocytosis. Indeed, endocytosis inhibition blocked the uptake of exogenous soluble RARRES1 in cultured podocytes (Figure 4E) and blocked the podocyte apoptosis induced by soluble RARRES1 (Figure 4F). Together, these results indicate that podocyte apoptosis is a consequence of the endocytic uptake of cleaved RARRES1 that subsequently triggers an intracellular signaling cascade to activate the proapoptotic pathway.
Figure 4. Blockade of RARRES1 cleavage mitigates the apoptotic potential of RARRES1 overexpression in cultured human podocytes.

(A) Apoptosis assay on cultured podocytes expressing control EGFP, WT RARRES1 (RARRES1-V5), or RARRES1 cleavage mutant (FFAF-V5) at 24 hours after transfection by flow cytometry for annexin V and propidium iodide (PI). (B) Western blot analysis of cleaved caspase-3 (arrowhead) in transfected cells in A. (C) Annexin V flow cytometry assay on cultured podocytes incubated with soluble RARRES1 (sRARRES1-V5, 10 ng/mL, 24 hours of incubation) from supernatant of HEK293T cells overexpressing RARRES1-V5. (D) Western blot analysis of cleaved caspase-3 in podocytes in C. (E) Human podocytes were incubated with 10 ng/mL sRARRES1-V5 with or without endocytosis inhibitor Dynasore (80 μM) for 1 hour, and total cell lysates were immunoblotted with V5 antibody. (F) Human podocytes were incubated with 10 ng/mL sRARRES1-V5 with or without a lower dose of Dynasore (10 μM) for 12 hours. Lysates were probed with V5 and cleaved caspase-3 (Cl-Casp3) antibodies.
RARRES1 interacts with RIOK1 to induce apoptosis of podocytes.
To explore which membrane proteins or intracellular signaling molecules interact with RARRES1 to activate the apoptosis, we performed the immunoprecipitation/mass spectrometry (MS) analysis using cell lysates from podocytes overexpressing RARRES1-V5 or control vector. The immunoprecipitated proteins were largely intracellular proteins (Table 1; top 10 proteins are shown). Interestingly, RIO kinase 1 (RIOK1), an atypical protein kinase known to be involved in cell survival (24), was the fourth protein on the list (Table 1). We further confirmed the interaction between RARRES1-V5 and myc-DDK–tagged RIOK1 (RIOK1-myc-DDK) by coimmunoprecipitation and Western blotting. While the WT RARRES1-V5 interacted with RIOK1, the cleavage mutant FFAF-V5 did not (Figure 5A), suggesting that the cleavage of RARRES1 into soluble form is required for this interaction. As RIOK1 is an intracellular protein kinase, it is likely that cleaved RARRES1 that is endocytosed subsequently interacts with RIOK1. Consistent with the known function of RIOK1 to mediate the antiapoptotic effect by inhibition of p53 activation (24), the overexpression of RIOK1 in podocytes abolished the effect of RARRES1 on p53 phosphorylation and cleavage of caspase-3 (Figure 5B), suggesting that the interaction of RIOK1 with RARRES1 inhibits its activity, resulting in p53 activation and apoptosis induction. Consistent with this, knockdown of RARRES1 in cultured podocyte reduced TNF-α–induced p53 phosphorylation and cleavage of caspase-3 (Figure 5C).
Table 1. Top 10 RARRES1-interacting proteins identified by IP-MS.
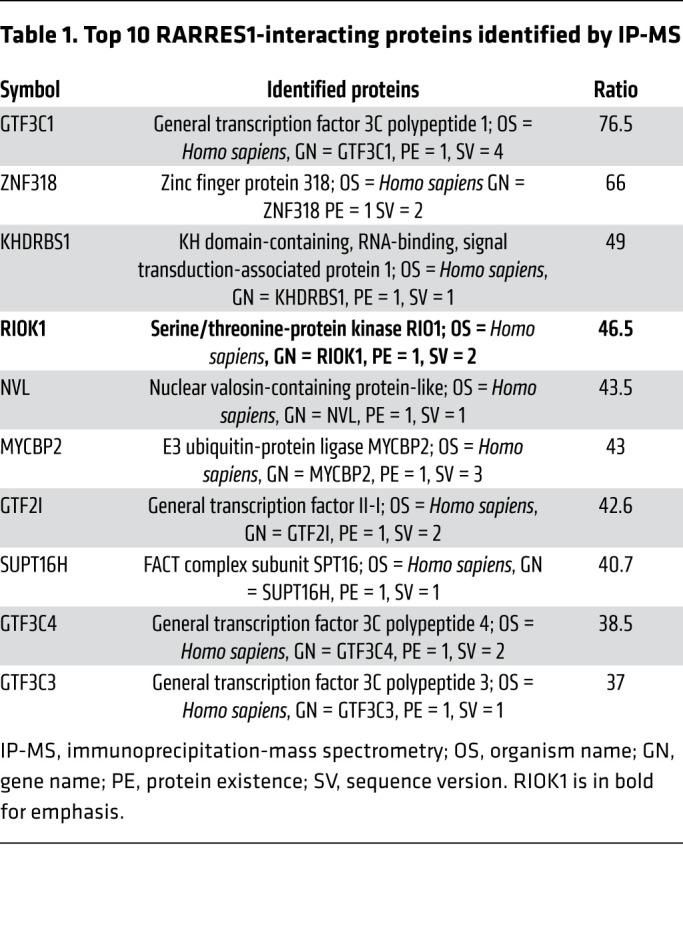
Figure 5. RIOK interacts with RARRES1 and TNF-α induces apoptosis through RARRES1 and RIOK1.

(A) Podocytes were cotransfected with myc-DDK–tagged RIOK1 (RIOK1-myc-DDK) with either control EGFP vector, WT RARRES1-V5, or cleavage mutant FFAF-V5. Lysates were immunoprecipitated with V5 antibody to pull down the RARRES1-interacting proteins and probed with FLAG and RIOK-1 antibodies. Input lysates were probed with FLAG, RIOK1, V5, and GAPDH antibodies. (B) Human podocytes were transfected with overexpression plasmids of RARRES1-V5 and/or RIOK1-myc-DDK. Western blot analysis was performed 24 hours after transfection for phospho-p53 (ser46), cleaved caspase-3, and RARRES1. (C) Podocytes stably transduced with lentiviral vector expressing scrambled control shRNA or RARRES1 shRNA were treated with TNF-α (10 ng/mL for 24 hours). Lysates were probed for phospho-p53 (Ser46), RARRES1, and cleaved caspase-3. Representative blots of 3 independent experiments are shown.
Overexpression of RARRES1WT, but not RARRES1MT, induces proteinuria and podocyte loss in vivo.
We next examined the role of RARRES1 in vivo by inducing the podocyte-specific overexpression of WT RARRES1 or cleavage FFAF mutant in mice (RARRES1WT or RARRES1MT, respectively, as described in Supplemental Figure 6). The podocyte-specific expression of RARRES1WT and RARRES1MT was achieved by feeding the transgenic mice with doxycycline-supplemented (Dox-supplemented) chow from 8 weeks to 20 weeks of age. WT littermates fed with Dox-supplemented chow were used as controls (RARRESCL). All mice were sacrificed at 20 weeks of age (12 weeks after Dox induction). RARRES1WT mice developed significant albuminuria, while RARRES1CL and RARRES1MT mice did not (Figure 6A). Consistent with this, RARRES1WT kidneys showed the development of glomerulosclerosis that was absent in the kidneys of RARRES1CL and RARRES1MT mice (Figure 6, B and C). We did not detect significant change in BUN among all groups at 20 weeks of age. However, given the extent of glomerular injury in RARRES1WT mice, the renal function decline may become more significant in RARRES1WT mice at a later time point.
Figure 6. Podocyte overexpression of RARRESWT induces glomerular injury.

(A) Albumin excretion over 24 hours. (B) Representative images of PAS-stained kidneys. Scale bars: 50 μm (upper panels); 20 μm (lower panels). (C) Average glomerulosclerosis (GS) score per glomerular cross section per mouse (n = 6 mice per group, 25 glomeruli evaluated for each mouse). Data are represented as mean ± SD.****P < 0.0001 vs. RARRES1CL; ####P < 0.0001 vs. RARRES1WT, 1-way ANOVA with Tukey’s multiple comparisons test.
The ultrastructural analysis of podocyte foot processes showed significant effacement only in RARRES1WT mice at 20 weeks of age (Figure 7, A and C). Moreover, quantification of WT-1+ cells also indicated a significant loss of podocytes in RARRES1WT mice, but not in RARRES1CL and RARRES1MT mice (Figure 7, B and C). The TUNEL+ podocytes were more readily detected in the glomeruli of RARRES1WT mice, but not in those of RARRES1CL and RARRES1MU mice (Figure 7, B and C). These results are consistent with the above in vitro results and indicate that the overexpression of RARRES1WT, but not RARRES1MT, induces podocyte loss through increased apoptosis.
Figure 7. Podocyte overexpression of RARRESWT induces podocyte injury and loss.

(A) Representative transmission electron microscopy images for low and high magnifications. Scale bars: 1 μm. (B) Representative images of WT1 immunostaining (red) and TUNEL staining (green). Scale bars: 20 μm. Arrowhead shows the WT1+ and TUNEL+ cell in RARRESWT kidney, and magnified view is shown in the dotted inset. (C) Quantification of average foot process (n = 4 mice per group evaluated) from transmission electron microscopy images (n = 4 mice per group) and average TUNEL+ and WT1+ cells per glomerular cross section (gcs) per mouse (n = 6 mice per group, 10 glomeruli evaluated for each mouse). All data are represented as mean ± SD. ****P < 0.0001 vs. RARRES1CL; ###P < 0.001 and ####P < 0.0001 vs. RARRES1WT, 1-way ANOVA with Tukey’s multiple comparisons test.
The knockdown of Rarres1 attenuates albuminuria and glomerular injury in mice with adriamycin-induced nephropathy.
Given that the podocyte overexpression of RARRES1 induced glomerulosclerosis and proteinuria in vivo, we next determined whether the reduced RARRES1 expression might confer renoprotection in the setting of podocyte injury in vivo. We generated inducible podocyte-specific Rarres1 knockdown mice as described in our previous study (25) and selected 2 independent lines with approximately 80%–90% knockdown of RARRES1 expression for our studies, referred to as RARRES1KD (Supplemental Figure 7). Knockdown of Rarres1 was induced by feeding of Dox-supplemented chow, and WT littermates fed with Dox-supplemented chow were used as controls (RARRESCL). RARRES1KD mice did not develop albuminuria or any histological changes at the baseline up to the age of 6 months (data not shown). To establish adriamycin-induced (ADR-induced) nephropathy, RARRES1KD and RARRES1CL mice were first fed with Dox-supplemented chow starting at 6 weeks of age and ADR was administered 2 weeks after Dox supplementation. All mice were sacrificed at 12 weeks of age. We found that RARRES1KD mice with ADR had attenuated albuminuria and glomerular injury when they were compared with RARRES1CL mice with ADR (Figure 8, A–D). Quantification of WT1+ and TUNEL+ podocytes also indicated that RARRES1 knockdown led to the mitigation of podocyte loss and apoptosis in ADR mice (Figure 8, C and D), confirming a role of RARRES1 in inducing podocyte injury in mice with experimental FSGS.
Figure 8. Podocyte Rarres1 knockdown attenuates albuminuria and glomerular injury in ADR-induced nephropathy in mice.

Nphs1-rtTA;TRE-Rarres1KD mice were given either control chow (RARRES1CL) or Dox-supplemented chow (RARRES1KD) for 2 weeks before ADR (+ADR) or vehicle (–ADR) injection. All mice were sacrificed 4 weeks after injection. (A) Urinary albumin-to-creatinine ratio (UACR) after ADR or vehicle injection, where week 0 indicates the baseline before injection. Data are represented as mean ± SEM. n = 5 mice per group. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. respective –ADR control; ####P < 0.0001 vs. RARRES1CL+ADR by 2-way ANOVA with Tukey’s multiple comparisons test. (B) Representative images of PAS-stained kidneys. Original magnification, ×200 (upper panels); ×400 (lower panels). Scale bars: 20 μm. (C) Representative images of WT1 (red) and TUNEL (green) coimmunostaining. Scale bars: 20 μm. A magnified view of WT1+ and TUNEL+ cells is shown in the inset. Arrowhead shows the colocalization of WT1 and TUNEL cells. (D) Average glomerulosclerosis score per glomerular cross section per mouse (n = 5 mice per group, 25 glomeruli evaluated for each mouse) and TUNEL+ and WT1+ cells per glomerular cross section per mouse (n = 5 mice per group, 15 glomeruli evaluated for each mouse). Data are represented as mean ± SD. ***P < 0.001 and ****P < 0.0001 vs. –ADR control; ##P < 0.01 and ####P < 0.0001 vs. RARRES1CL+ADR, 1-way ANOVA with Tukey’s multiple comparisons test.
Discussion
In this study, using the NEPTUNE data sets, we sought to determine the RA-related genes and signaling pathways in human glomerular disease. We found that many RA-related genes were highly regulated in human glomerular disease and correlated with the progression of kidney disease, further underscoring the importance of the RA signaling pathway in human glomerular disease. Interestingly, we found that several genes in the RA pathway, such as RARRES1, correlated positively with the progression of kidney disease, suggesting that, although RA signaling largely confers renoprotection, as demonstrated in several experimental models (16, 17), few of the specific RA-induced downstream genes may concurrently elicit harmful effects in the kidney. Among these, we focused on RARRES1 for further analysis because its expression correlated with the severity of human glomerular disease and predicted glomerular disease progression based on the NEPTUNE data sets. The correlation of increased glomerular expression of RARRES1 with kidney disease was further corroborated by previously collected transcriptome data sets in patients with CKD and DKD, and, together these data suggested that RARRES1 is a risk gene for the progression of kidney disease. Interestingly, its mRNA expression in the recently published single-cell data sets (19–21) was shown to be largely limited to podocytes, a terminally differentiated and quiescent cell type in the glomeruli (1). RARRES1, whose role is implicated as a tumor suppressor (14, 15), may therefore contribute to podocyte quiescence in normal conditions.
We found TNF-α to be a potent inducer of RARRES1 expression in cultured podocytes, and in turn, RARRES1 mediated the proapoptotic effect of TNF-α in podocytes. Since the TNF-α/NF-κB pathway is activated in diseased podocytes (26, 27), this is likely the main mechanism of increased RARRES1 expression in the diseased kidney. The role of TNF-α in FSGS and DKD has been well demonstrated. Both circulating and local TNF-α levels increase in patients with FSGS and DKD (27). A clinical trial using TNF-α inhibitor for FSGS patients is ongoing (28). These findings together with our data suggest that either circulating or local TNF-α, which is increased in FSGS or DKD, could induce RARRES1 expression and apoptosis in podocytes. Our in vitro data indicated that the overexpression of RARRES1 induces podocyte apoptosis by the activation of the p53 pathway, which is consistent with its previously reported tumor suppressor effect. We further validated in vivo that the overexpression of RARRES1 in podocytes alone is sufficient to induce albuminuria and podocyte loss in mice. Together, these in vitro and in vivo data suggest that high expression levels of RARRES1 induce podocyte apoptosis and loss. Since podocyte loss is considered a major cause of progression of kidney disease, including DKD and FSGS (2–4), the high expression level of RARRES1 likely contributes to the progression of DKD and FSGS.
A notable finding from our study is that the cleavage of RARRES1 is required for its proapoptotic function in cultured podocytes and that sequence flanking aa 70 in its extracellular domain is a key cleavage site. In silico analysis suggests that this cleavage is mediated by a protease in the MMP family, and broad inhibition of MMPs reduced the cleavage of RARRES1. However, as there are 23 human MMPs with varying substrate specificities (29), future studies are required to dissect which MMPs are involved in the cleavage of RARRES1. Consistent with the in vitro data, mice with overexpression of RARRES1WT in podocytes had soluble RARRES1 in the urine and developed albuminuria and podocyte loss, while the mice with overexpression of RARRES1MT in podocytes did not, further demonstrating that the cleavage of RARRES1 is required for its effect on podocyte loss in vivo. The utilization of RARRES1MT overexpression in mice also served as a control for RARRES1WT overexpression for potential protein overexpression–induced cell toxicity in vivo. Conversely, the knockdown of RARRES1 attenuates albuminuria and podocyte loss in mice with ADR-induced nephropathy, further supporting the role of RARRES1 in podocyte injury. Our data showed that knockdown of RARRES1 in podocytes did not cause any renal phenotype at the baseline, indicating that RARRES1 might not be essential or that the low amount of RARRES1 in these knockdown mice is sufficient to maintain normal podocyte function. Future studies are required to determine whether knockout of RARRES1 in podocytes causes any renal phenotype in mice and whether soluble RARRES1 in the urine could be developed as a potential biomarker for progression of glomerular disease.
Interestingly, the cleavage of RARRES1 and subsequent endocytic uptake of the soluble RARRES1 were required for the proapoptotic effect in cultured podocytes, indicating that RARRES1 likely needs to interact with intracellular signaling molecules to induce apoptosis. One such signaling molecule identified as an RARRES1-interacting protein was RIOK1. RIOK1 has been show to regulate cell survival and promote tumor growth, and the inhibition of RIOK1 is shown to induce apoptosis through the activation of the p53 pathway (24, 30, 31). We confirmed that the apoptotic effect of RARRES1 was abolished when we overexpressed RIOK1, suggesting that RARRES1 interacts and suppresses RIOK1 function. Future studies are required to determine whether soluble RARRES1 promotes apoptosis in the neighboring cells, such as glomerular endothelial cells or tubular cells, via a crosstalk mechanism. In addition, it would be informative to validate these mechanisms in cancer cells.
Previous studies suggest that a low dose of RA induces podocyte differentiation (16, 32). However, our data showed that RA induced RARRES1 expression in a dose-dependent manner. These data suggest that, while a low dose of RA might be beneficial, a high dose of RA could be toxic for podocytes. It has been shown that the systemic adverse effects of RA are mediated by RA-induced apoptosis (33), and it would be interesting to determine whether RARRES1 also mediates the systemic adverse effects of RA. Therefore, we have to be cautious in considering RA as a drug to treat patients with glomerular disease. Our study also suggests that additional screening of drug-related pathways in human data sets could help the translation of the findings from animal studies to human disease.
In summary, we demonstrate a potentially novel mechanism by which RARRES1, a transmembrane protein, acts on podocytes through releasing its cleaved form that subsequently interacts with an intracellular protein kinase to induce a proapoptotic effect, as summarized in Figure 9. Future studies are required to understand whether any genetic variants of RARRES1 are associated with glomerular disease and how RARRES1 cleavage is regulated in disease conditions in order to develop a potential therapeutic approach in kidney disease.
Figure 9. Summary of the mechanism by which RARRES1 regulates podocyte apoptosis.

TNF-α induces expression of RARRES1, which is cleaved into soluble RARRES1, potentially mediated by inflammation-stimulated MMP. The soluble RARRES1 is then endocytosed and interacts with intracellular RIOK1. The interaction between RIOK1 and RARRES1 leads to the inactivation of RIOK1, thereby activating p53 and the subsequent apoptosis pathway. Podocyte loss is a key event for progression of DKD and FSGS.
Methods
Cell culture.
HEK293T cells were obtained from ATCC (CRL-3216) and cultured according to their specifications. Conditionally immortalized human podocytes were obtained from Moin Saleem (University of Bristol, Bristol, United Kingdom) and grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 1× insulin-transferrin-selenite (ITS) media supplement (MilliporeSigma). Podocytes were cultured on collagen I–coated plates and maintained at 33°C (5% CO2, 90% humidity). Before experiments, cells were moved to a 37°C incubator and cultured for at least 5 days to fully induce differentiation. All experiments were repeated at least 3 times for each indicated condition. Podocytes between passages 9 and 20 were used in all experiments.
hRARRES1 WT and mutant protein expression.
Human RARRES1 expression plasmids were constructed using the RARRES1 ORF (NM_206963). pTRE-Tight-hRARRES1 was constructed by ligating the hRARRES1 fragment into pTRE-Tight vector (Clontech, catalog 631059). FLAG-tagged RARRES1 expression plasmid (pTRE-Tight-FLAG-hRARRES1) was constructed by using the linker primers (forward: 5′-GAATTCGACTACAAGGACGACGATGACAAAGCCGGC-3′; reverse: 5′-GCCGGCTTTGTCATCGTCGTCCTTGTAGTCGAATTC-3′). N-terminal–tagged human RARRES1 overexpression plasmid pCDNA4-FLAG-hRARRES1 was derived from pTRE-Tight-FLAG-hRARRES1 by subcloning the FLAG-hRARRES1 into the pCDNA4 expression plasmid. C-terminal V5–tagged human RARRES1 overexpression plasmid (pN1-hRARRES1-V5) was constructed in a similar manner. Primer linkers used to generate various V5-tagged RARRES1 plasmids are shown in Supplemental Table 5. All constructs were verified with DNA sequencing.
Pull-down and MS analysis.
hRARRES1-V5 plasmid or control V5 plasmids were transfected to human podocyte cell lines with ViaFect transfection reagent (Promega, E4981). At 36 hours after transfection, the cells were harvested and total protein was extracted from the cell lysate with IP lysis buffer (Thermo Scientific, 87787) with protease inhibitor cocktail (MilliporeSigma, P8340) and phosphatase inhibitor (MilliporeSigma P0044). The protein lysates were incubated with anti–V5-tag mAb Magnetic Beads (MBL International Corp., M167-11) at room temperature for 2 hours. Bound proteins after washing with IP lysis buffer were resuspended with 2× loading buffer and boiled for 2 minutes at 95°C. Protein gels were stained with Gel Code Blue Stain Reagent (Thermo Scientific, 24590) for MS analysis. The protein identification using liquid chromatography–MS/MS (LC-MS/MS) was performed at the Center for Advanced Proteomics Research Core at Rutgers New Jersey Medical School (Newark, New Jersey, USA).
Flow cytometry analysis.
Human podocytes were transfected with human RARRES1 overexpression plasmid with ViaFect transfection reagent (Promega, E4981). At 36 hours after transfection, cells were harvested and stained further with the Annexin V Apoptosis Detection Kit (Thermo Fisher Scientific, 88-8007). The number of cells labeled with annexin V–FITC and propidium iodide were quantified using the FACSCalibur Flow cytometer, and the data were analyzed with CellQuest software (BD Biosciences).
TUNEL assay.
The DeadEnd Colorimetric TUNEL System (Promega) was used to detect apoptotic cells on frozen kidney sections following the manufacturer’s instructions. Cy5-conjugated streptavidin (Promega) was used to detect the apoptotic cells, and sections were then mounted with ProLong Gold Antifade Reagent with DAPI (Life Technologies).
Western blot analysis.
Lysate preparation and Western blotting were performed according to the standard protocol. Each lane contained 30 to 60 μg of total protein. Complete, unedited Western blot images are available in the supplemental material. Band density for the protein of interest was normalized to either GAPDH or β-actin. The following antibodies were used in this study: human RARRES1 antibody (R&D Systems, catalog AF4255), V5 (GenScript, catalog A01724), FLAG (MilliporeSigma, catalog F3165), human RIOK1 (Abcam, catalog ab176005), cleaved caspase-3 (Cell Signal Technology, catalog 9664), phospho-p53 (Ser46) (Cell Signaling Technology, catalog 2521), pan-Cadherin (Invitrogen, catalog 71-7100), GAPDH (Cell Signaling Technology, catalog A2188), and β-actin (MilliporeSigma, catalog A4700).
mRNA sequencing of cultured human podocytes.
Total mRNA from cultured podocytes were harvested, sequenced, and analyzed as previously described (34). All original data were deposited in the NCBI’s Gene Expression Omnibus database (GEO GSE151484).
Quantitative real-time PCR.
Real-time PCR was performed according to the standard protocol. Ct values of the gene targets were normalized to GAPDH. Fold change in expression of target genes compared with the reference group was calculated using the 2-ΔΔCT method, with GAPDH as the calibrator. Sequences of primers used were as follows: hRARRES1, forward, 5′-AGGTGTCACACTACTACTTGG-3′; reverse, 5′-AGCTGTTGACAGTGGTACTTC-3′; mRARRES1, forward, 5′-TCGGCAGCTCATACGTGATGT-3′, reverse, 5′-GTACCAGACCAAGTGAATACG-3′.
Preparation of the soluble human RARRES1.
Human RARRES1 overexpression plasmid was transfected to 293T cells. At 12 hours after transfection, the medium was changed to serum-free DMEM medium with 1% penicillin-streptomycin antibiotics. The supernatant was collected after 24 hours. Soluble RARRES1 was concentrated with Amicon Ultra-15 Centrifugal filters (Millipore Sigma, C7715). The RARRES1 concentration was determined by the Enzyme-Linked Immunosorbent Assay Kit for Retinoic Acid Receptor Responder 1 (RARRES1) of human RARRES1 (MyBioSource, MBS2019555), and 10 ng/mL soluble RARRES1 was added into culture medium to treat podocytes.
Establishment of the inducible human WT and mutant RARRES1 overexpression mice and inducible RARRES1 knockdown mice.
The human RARRES1 cDNA was subcloned with the C-terminal V5 tag into the inducible pTRE-Tight vector (Clontech, 631059). For the construction of the inducible mutant overexpression vectors, the purified fragment of the human mutant RARRES1 from the previously constructed pN1-RARRES1(FFAF)-V5 vector was subcloned into the pTRE-Tight vector. The plasmids were digested with restriction enzymes to release the RARRES1-V5 encoding fragments for transgenic mouse derivation in surrogate FVB mice, generating the RARRES1 inducible mice (TRE-RARRES1WT or TRE-RARRES1MT mice). TRE-RARRES1 mice were crossed with Nphs1-rtTA mice (a gift from Jeffrey Miner, Washington University School of Medicine, St. Louis, Missouri, USA). The podocyte-specific expression of RARRES1 proteins was induced by the administration of Dox-supplemented chow (625 g/kg, Envigo). For Rarres1 knockdown mice, we used a protocol similar to that described in our previous study (25). Briefly, TRE-Rarres1 shRNA knockdown mice were obtained from Mirimus Inc. and crossed with Nphs1-rtTA mice to generate Nphs1-rtTA;TRE-Rarres1KD mice. Knockdown was induced with Dox-supplemented chow as described above. Overexpression or knockdown of RARRES1 was verified by either quantitative PCR (qPCR) or Western blot analysis of isolated glomeruli from transgenic mice.
Mouse kidney histology.
Kidney samples were fixed in 10% formalin, embedded in paraffin, and sectioned to 4 μm thickness. Periodic acid–Schiff–stained (PAS-stained) kidney sections were used for kidney histology. Histological scoring was performed in a blinded manner by the renal pathologist. Assessment of the mesangial and glomerular cross-sectional areas was performed by the operator blinded to the identity of the experimental groups by measuring the pixel counts on an average of a minimum of 10 glomeruli per section under ×400 magnification (Zeiss AX10 microscope). Glomerulosclerosis was graded on a semiquantitative scale (0 to 3+), as follows: 0 (absent), 1+ (involving 1%–25% of all glomeruli sampled), 2+ (involving 26%–50% of glomeruli), and 3+ (involving >50% of glomeruli), as described (17). An average of 25 glomeruli sampled per animal were evaluated.
Transmission electron microscopy.
Tissues were fixed in 2.5% glutaraldehyde with 0.1M sodium cacodylate (pH 7.4) for 72 hours at 4°C. Samples were further incubated with 2% osmium tetroxide and 0.1M sodium cacodylate (pH 7.4) for 1 hour at room temperature. Ultrathin sections were stained with lead citrate and uranyl acetate and viewed on a Hitachi H7650 microscope. Briefly, negatives were digitized, and images with a final magnitude of up to ×10,000 were obtained. ImageJ 1.26t software (NIH) was used to measure the length of the peripheral GBM, and the number of slit pores overlying this GBM length were counted. The arithmetic mean of the foot process width (WFP) was calculated as follows: WFP = π/4×(ΣGBM length)/(Σsilts), where Σslits indicates the total number of slits counted, ΣGBM length indicates the total GBM length measured in 1 glomerulus, and π/4 is the correction factor for the random orientation by which the foot processes were sectioned (35).
Immunofluorescence.
Mouse kidney tissues were harvested following perfusion with 4% paraformaldehyde (PFA) in PBS, post-fixed for 2 hours on ice, equilibrated in 30% sucrose overnight, and frozen in OCT (Thermo Fisher Scientific) before sectioning. Kidney sections from human kidney biopsies were prepared accordingly. Then, sections of 4 μm were blocked with 2% horse serum and 2% BSA in PBS and incubated overnight at 4°C with the following primary antibodies: anti-RARRES1 (Santa Cruz Biotechnology Inc., catalog sc-390461) and anti-NPSH2 antibody (Abcam, catalog ab50339). Secondary antibodies conjugated with fluorochromes were obtained from Jackson ImmunoResearch Labs. DAPI was used as a nuclear counterstain. Immunostained kidney sections were mounted with ProLong Gold Antifade Reagent with DAPI (Life Technologies).
Immunohistochemistry.
Formalin-fixed and paraffin-embedded sections were deparaffinized, and endogenous peroxidase was inactivated with H2O2. Sections were then blocked in 2% goat serum in PBS for 1 hour at room temperature and then incubated with anti-RARRES1 (Santa Cruz Biotechnology Inc., catalog sc-390461) at 4°C overnight. The next day, sections were washed 3 times with PBS and then incubated with secondary antibody for 30 minutes. Positive staining was revealed by peroxidase-labeled streptavidin and diaminobenzidine substrate,with a fixed exposure time of 3 minutes for all experiments among the groups. The control included a section stained with only secondary antibody.
Quantification of immunostaining.
Immunostained images with a final magnitude of approximately ×400 were obtained. ImageJ 1.26t software was used to measure the level of immunostaining in the glomeruli. First, the images were converted to 8-bit grayscale. Glomerular regions were selected for measurement of area and integrated density, and background intensity was measured by selecting 3 distinct areas in the background with no staining. The corrected optical density (COD) was determined as follows: COD = ID – (A × MGV), where ID is the integrated density of the selected glomerular region, A is the area of the selected glomerular region, and MGV is the mean gray value of the background readings (36).
Statistics.
Data are expressed as mean ± SD or SEM as indicated. Unpaired, 2-tailed t test was used to analyze data between 2 groups. One-way or 2-way ANOVA with Tukey’s multiple comparisons test was used when more than 2 groups were present, as indicated. All experiments were repeated at least 3 times, and representative experiments are shown. Statistical significance was defined as P < 0.05.
Study approval.
The human kidney biopsy sample collection was approved by the Medical Ethics Committee of Zhongshan Hospital, affiliated to Xiamen University (2020-054). All animal procedures were performed according to protocols approved by the IACUC at the Icahn School of Medicine at Mount Sinai (no. 06-1098).
Author contributions
AC, KL, and JCH designed and conducted experiments, acquired and analyzed data, and wrote the manuscript. YF, HL, WJ, ZL, YL, AW, QH, FZ, CW, JF, TG, BL, and MK acquired and analyzed data.
Supplementary Material
Acknowledgments
AC is supported by the National Natural Science Foundation of China (grant 81800637), the Natural Science Foundation of Fujian Province (2019J01560), and the Xiamen Science and Technology Project (grant 3502Z20194014). JCH is supported by VA Merit Award IBX000345C, NIH 1R01DK078897, NIH 1R01DK088541, and NIH P01DK56492. KL is supported by NIH R01DK117913. For further information, see Supplemental Acknowledgments.
Version 1. 07/07/2020
In-Press Preview
Version 2. 09/14/2020
Electronic publication
Version 3. 10/01/2020
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(10):5523–5535.https://doi.org/10.1172/JCI140155.
See the related Commentary at The yin and yang of retinoic acid signaling in kidney diseases.
Contributor Information
Anqun Chen, Email: anqunchen@163.com.
Ye Feng, Email: ye.feng@mssm.edu.
Han Lai, Email: hanlai91917@gmail.com.
Wenjun Ju, Email: wenjunj@umich.edu.
Zhengzhe Li, Email: zhengzhe.li@mssm.edu.
Yu Li, Email: isleeyu@stu.xmu.edu.cn.
Andrew Wang, Email: 7andrewwang@gmail.com.
Quan Hong, Email: hongquan@301hospital.com.cn.
Fang Zhong, Email: fang.zhong@mssm.edu.
Chengguo Wei, Email: chengguo.wei@mssm.edu.
Jia Fu, Email: jia.fu@mssm.edu.
Matthias Kretzler, Email: kretzler@umich.edu.
Kyung Lee, Email: kim.lee@mssm.edu.
John Cijiang He, Email: cijiang.he@mssm.edu.
References
- 1.Durvasula RV, Shankland SJ. Podocyte injury and targeting therapy: an update. Curr Opin Nephrol Hypertens. 2006;15(1):1–7. doi: 10.1097/01.mnh.0000199012.79670.0b. [DOI] [PubMed] [Google Scholar]
- 2.Pagtalunan ME, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99(2):342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steffes MW, Schmidt D, McCrery R, Basgen JM, International Diabetic Nephropathy Study Group Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001;59(6):2104–2113. doi: 10.1046/j.1523-1755.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- 4.Nishizono R, et al. FSGS as an adaptive response to growth-induced podocyte stress. J Am Soc Nephrol. 2017;28(10):2931–2945. doi: 10.1681/ASN.2017020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tharaux PL, Huber TB. How many ways can a podocyte die? Semin Nephrol. 2012;32(4):394–404. doi: 10.1016/j.semnephrol.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Evans TR, Kaye SB. Retinoids: present role and future potential. Br J Cancer. 1999;80(1-2):1–8. doi: 10.1038/sj.bjc.6690312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okuno M, et al. Retinoids in cancer chemoprevention. Curr Cancer Drug Targets. 2004;4(3):285–298. doi: 10.2174/1568009043333023. [DOI] [PubMed] [Google Scholar]
- 8.Xu Q, Lucio-Cazana J, Kitamura M, Ruan X, Fine LG, Norman JT. Retinoids in nephrology: promises and pitfalls. Kidney Int. 2004;66(6):2119–2131. doi: 10.1111/j.1523-1755.2004.66002.x. [DOI] [PubMed] [Google Scholar]
- 9.Mallipattu SK, He JC. The beneficial role of retinoids in glomerular disease. Front Med (Lausanne) 2015;2:16. doi: 10.3389/fmed.2015.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montesinos P, et al. Differentiation syndrome in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline chemotherapy: characteristics, outcome, and prognostic factors. Blood. 2009;113(4):775–783. doi: 10.1182/blood-2008-07-168617. [DOI] [PubMed] [Google Scholar]
- 11.Piersma AH, Hessel EV, Staal YC. Retinoic acid in developmental toxicology: Teratogen, morphogen and biomarker. Reprod Toxicol. 2017;72:53–61. doi: 10.1016/j.reprotox.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 12.Gadegbeku CA, et al. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int. 2013;83(4):749–756. doi: 10.1038/ki.2012.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagpal S, Patel S, Asano AT, Johnson AT, Duvic M, Chandraratna RA. Tazarotene-induced gene 1 (TIG1), a novel retinoic acid receptor-responsive gene in skin. J Invest Dermatol. 1996;106(2):269–274. doi: 10.1111/1523-1747.ep12340668. [DOI] [PubMed] [Google Scholar]
- 14.Sahab ZJ, Hall MD, Zhang L, Cheema AK, Byers SW. Tumor Suppressor RARRES1 Regulates DLG2, PP2A, VCP, EB1, and Ankrd26. J Cancer. 2010;1:14–22. doi: 10.7150/jca.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oldridge EE, et al. Retinoic acid represses invasion and stem cell phenotype by induction of the metastasis suppressors RARRES1 and LXN. Oncogenesis. 2013;2:e45. doi: 10.1038/oncsis.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He JC, et al. Retinoic acid inhibits HIV-1-induced podocyte proliferation through the cAMP pathway. J Am Soc Nephrol. 2007;18(1):93–102. doi: 10.1681/ASN.2006070727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ratnam KK, et al. Role of the retinoic acid receptor-α in HIV-associated nephropathy. Kidney Int. 2011;79(6):624–634. doi: 10.1038/ki.2010.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60(9):2354–2369. doi: 10.2337/db10-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park J, et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. 2018;360(6390):758–763. doi: 10.1126/science.aar2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karaiskos N, et al. A single-cell transcriptome atlas of the mouse glomerulus. J Am Soc Nephrol. 2018;29(8):2060–2068. doi: 10.1681/ASN.2018030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu J, et al. Single-cell RNA profiling of glomerular cells shows dynamic changes in experimental diabetic kidney disease. J Am Soc Nephrol. 2019;30(4):533–545. doi: 10.1681/ASN.2018090896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clemente C, et al. MT4-MMP deficiency increases patrolling monocyte recruitment to early lesions and accelerates atherosclerosis. Nat Commun. 2018;9(1):910. doi: 10.1038/s41467-018-03351-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, et al. Correction: CleavPredict: a platform for reasoning about matrix metalloπroteinases proteolytic events. PLoS ONE. 10(6):e0131952. doi: 10.1371/journal.pone.0131952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Read RD, et al. A kinome-wide RNAi screen in Drosophila Glia reveals that the RIO kinases mediate cell proliferation and survival through TORC2-Akt signaling in glioblastoma. PLoS Genet. 2013;9(2):e1003253. doi: 10.1371/journal.pgen.1003253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chuang PY, et al. In vivo RNA interference models of inducible and reversible Sirt1 knockdown in kidney cells. Am J Pathol. 2014;184(7):1940–1956. doi: 10.1016/j.ajpath.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiggins JE, et al. NFkappaB promotes inflammation, coagulation, and fibrosis in the aging glomerulus. J Am Soc Nephrol. 2010;21(4):587–597. doi: 10.1681/ASN.2009060663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedigo CE, et al. Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J Clin Invest. 2016;126(9):3336–3350. doi: 10.1172/JCI85939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joy MS, et al. Phase 1 trial of adalimumab in focal segmental glomerulosclerosis (FSGS): II. Report of the FONT (Novel Therapies for Resistant FSGS) study group. Am J Kidney Dis. 2010;55(1):50–60. doi: 10.1053/j.ajkd.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69(3):562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Zhang T, et al. The atypical protein kinase RIOK3 contributes to glioma cell proliferation/survival, migration/invasion and the AKT/mTOR signaling pathway. Cancer Lett. 2018;415:151–163. doi: 10.1016/j.canlet.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 31.Weinberg F, et al. The atypical kinase RIOK1 promotes tumor growth and invasive behavior. EBioMedicine. 2017;20:79–97. doi: 10.1016/j.ebiom.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vaughan MR, et al. ATRA induces podocyte differentiation and alters nephrin and podocin expression in vitro and in vivo. Kidney Int. 2005;68(1):133–144. doi: 10.1111/j.1523-1755.2005.00387.x. [DOI] [PubMed] [Google Scholar]
- 33.Melnik BC. Overexpression of p53 explains isotretinoin’s teratogenicity. Exp Dermatol. 2018;27(1):91–93. doi: 10.1111/exd.13420. [DOI] [PubMed] [Google Scholar]
- 34.Fu J, et al. Gene expression profiles of glomerular endothelial cells support their role in the glomerulopathy of diabetic mice. Kidney Int. 2018;94(2):326–345. doi: 10.1016/j.kint.2018.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koop K, et al. Expression of podocyte-associated molecules in acquired human kidney diseases. J Am Soc Nephrol. 2003;14(8):2063–2071. doi: 10.1097/01.ASN.0000078803.53165.C9. [DOI] [PubMed] [Google Scholar]
- 36.Potapova TA, Sivakumar S, Flynn JN, Li R, Gorbsky GJ. Mitotic progression becomes irreversible in prometaphase and collapses when Wee1 and Cdc25 are inhibited. Mol Biol Cell. 2011;22(8):1191–1206. doi: 10.1091/mbc.e10-07-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.