

Abstract
Focal adhesion kinase (FAK) is a key component of the membrane proximal signaling layer in focal adhesion complexes, regulating important cellular processes, including cell migration, proliferation, and survival. In the cytosol, FAK adopts an autoinhibited state but is activated upon recruitment into focal adhesions, yet how this occurs or what induces structural changes is unknown. Here, we employ cryo‐electron microscopy to reveal how FAK associates with lipid membranes and how membrane interactions unlock FAK autoinhibition to promote activation. Intriguingly, initial binding of FAK to the membrane causes steric clashes that release the kinase domain from autoinhibition, allowing it to undergo a large conformational change and interact itself with the membrane in an orientation that places the active site toward the membrane. In this conformation, the autophosphorylation site is exposed and multiple interfaces align to promote FAK oligomerization on the membrane. We show that interfaces responsible for initial dimerization and membrane attachment are essential for FAK autophosphorylation and resulting cellular activity including cancer cell invasion, while stable FAK oligomerization appears to be needed for optimal cancer cell proliferation in an anchorage‐independent manner. Together, our data provide structural details of a key membrane bound state of FAK that is primed for efficient autophosphorylation and activation, hence revealing the critical event in integrin mediated FAK activation and signaling at focal adhesions.
Keywords: cell adhesion; cryo‐electron microscopy; focal adhesion kinase; membrane complex; phosphatidylinositol‐4,5-bisphosphate
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Structural Biology
A cryo‐EM structure of membrane‐bound FAK reveals structural rearrangements underlying its oligomerization and activation at focal adhesions.

Introduction
Focal adhesions are large protein complexes that assemble at the cytoplasmic side of the plasma membrane and connect the actin cytoskeleton inside the cell via integrin trans‐membrane receptors to the extracellular matrix outside the cell. The complex plays an important mechanical role during mesenchymal cell migration, where contraction of actomyosin stress fibers attached to focal adhesions build up a force that is transmitted via the focal adhesion complex to the extracellular matrix to gain traction needed for cell motility (Sun et al, 2016). For this to occur in an orderly fashion, allowing for concerted focal adhesion assembly at the leading edge and disassembly at the rear (Wehrle‐Haller, 2012), an extensive signaling network is associated with focal adhesions, which at the systems level is integrated with other signaling networks that control proliferation and survival (Winograd‐Katz et al, 2014). To fulfill these multiple roles, focal adhesions adopt a layered architecture, where actin fibers attach to a membrane distal actin regulatory layer that is connected to integrin receptors and a membrane proximal signaling layer via a force transduction layer (Kanchanawong et al, 2010). Focal adhesion kinase (FAK) is a key component of the signaling layer and acts as a hub that integrates signaling at focal adhesions. FAK is both a scaffold protein and a non‐receptor tyrosine kinase that is essential during development and plays key functions in tissue repair and wound healing (Ilic et al, 1995; Ashton et al, 2010; Ransom et al, 2018; Toro‐Tapia et al, 2018). FAK is overexpressed in numerous human tumors and is in particular implicated in tumor invasion and metastasis (Provenzano & Keely, 2009; Sulzmaier et al, 2014). FAK also provides survival signals causing tumor resistance (Tavora et al, 2014; Hirata et al, 2015; Jiang et al, 2016; Diaz Osterman et al, 2019); hence, FAK has been considered an important target for anti‐cancer therapies (Sulzmaier et al, 2014; Lv et al, 2018).
Focal adhesion kinase shares its domain structure with the closely related proline‐rich tyrosine kinase 2 (Pyk2), both contain an N‐terminal FERM (band 4.1, ezrin, radixin, moesin homology) domain, a central kinase domain, and a C‐terminal focal adhesion targeting (FAT) domain (Fig 1A). Whereas the FAT domain is responsible for targeting FAK into focal adhesions (Arold et al, 2002; Hayashi et al, 2002; Gao et al, 2004), the FERM domain of FAK plays a central role in regulating FAK function (Cooper et al, 2003; Dunty et al, 2004) and is an interaction domain that allows FAK to integrate signals from various sources (Zaidel‐Bar et al, 2007; Frame et al, 2010; Horton et al, 2015). In the autoinhibited state, the FERM domain forms intramolecular interactions with the kinase domain that occludes the active site and sequesters important regulatory phosphorylation sites (Lietha et al, 2007). In this state, FAK can form FERM‐mediated dimers that are stabilized by FAT interactions with the FERM domain (Brami‐Cherrier et al, 2014). Signals emanating from growth factors and integrins lead to activation of FAK (Sulzmaier et al, 2014; Walkiewicz et al, 2015), but also mechanical inputs in form of stretching forces generated in focal adhesions appear important for FAK activation (Wang et al, 2001; Torsoni et al, 2003; Seong et al, 2013; Bauer et al, 2019). Located in the membrane proximal signaling layer in focal adhesions, FAK directly interacts with the membrane lipid phosphatidylinositol‐4,5‐bisphosphate (PI(4,5)P2) (Cai et al, 2008; Goni et al, 2014). PI(4,5)P2 levels are upregulated in focal adhesions by the PI(4)P‐5‐kinase type Iγ (Di Paolo et al, 2002; Ling et al, 2002), and we previously reported that interactions with PI(4,5)P2 guide FAK through a multistep activation sequence (Goni et al, 2014). This involves PI(4,5)P2 binding via a basic patch on the FERM domain, which induces FAK oligomerization and conformational changes that trigger autophosphorylation of Y397 in the linker between the FERM and the kinase domains. Subsequently, the Src kinase is recruited via its SH2 and SH3 domains that interact with the autophosphorylated linker in FAK and Src then phosphorylates residues Y576 and Y577 in the activation loop of the FAK kinase to induce full catalytic activity (Calalb et al, 1995; Lietha et al, 2007). Although previous work defined the main steps required for integrin‐mediated FAK activation (Goni et al, 2014), it did not provide a structural understanding of events occurring on the membrane.
Figure 1. Cryo‐EM structure of the FERM‐kinase region of FAK on a PI(4,5)P2 membrane.
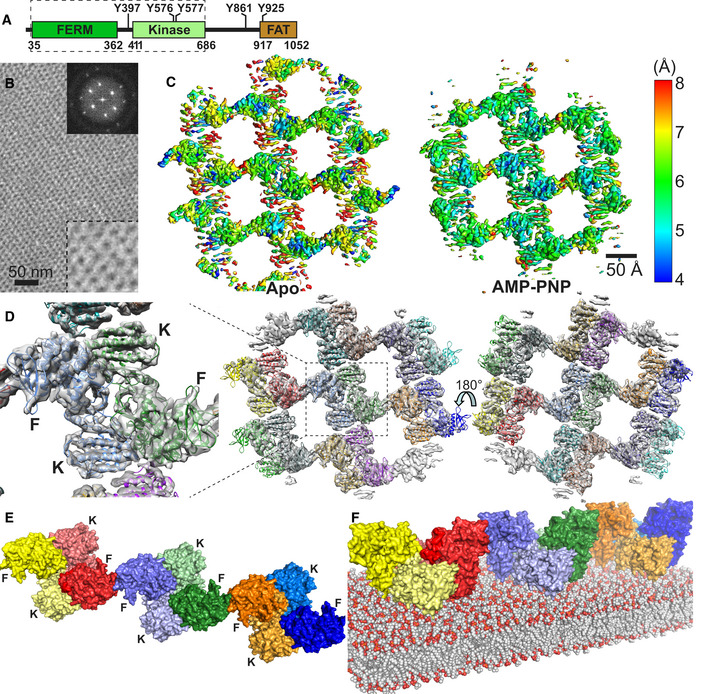
- Domain structure of FAK. Important phosphorylation sites are indicated, and the FAK region used in the cryo‐EM study is boxed with a dashed line.
- Negative stain image of 2D crystals containing the FERM‐kinase region of FAK bound to a PI(4,5)P2 monolayer. Top right insert: Fourier transform of the 2D crystal image. Bottom right insert: 2.5× zoom of the main image.
- Local resolution map of the refined Apo and AMP‐PNP particle, as calculated with Blocres.
- 14 FAK molecules, each containing a FERM and kinase domain, are fitted into the EM maps obtained from the AMP‐PNP dataset. Each molecule is in a different color. Middle: Membrane distal view; right: Membrane proximal view; left: zoomed view of central symmetric dimer from the membrane distal view with locations of FERM (F) and kinase (K) domains indicated. The equivalent figure for the Apo structure is shown in Fig EV1D.
- Linear oligomeric assembly of FAK formed by three symmetric FAK dimers in the AMP‐PNP particle shown from the membrane distal view. FERM (F) and Kinase (K) domains are labeled. FERM and kinase of the same molecule are in the same color with the kinases shaded in a lighter tone.
Here, we employ cryo‐electron microscopy (cryo‐EM) of FAK bound to a PI(4,5)P2 membrane to provide structural details of membrane‐induced conformational changes and FAK oligomerization. We observe that the oligomeric FAK assembly is formed via a tightly packed symmetric FAK dimer and these dimers are linked into an oligomer via FERM–FERM interactions that have previously been observed for FAK in solution (Brami‐Cherrier et al, 2014). Our structure suggests a membrane‐induced release of autoinhibition by binding of FERM‐mediated FAK dimers to PI(4,5)P2 via the basic patch in the FERM domain. Intriguingly, the released kinase domain reorients via a large conformational change placing the active site toward the membrane. As a result, the linker is exposed to promote efficient autophosphorylation. We show that interfaces observed in the cryo‐EM structure are important for cellular functions of FAK, including cell proliferation in 3D and cancer cell invasion.
Results
Cryo‐EM and overall architecture of FAK bound to a PI(4,5)P2 membrane
For structural studies of membrane‐bound FAK, we assembled purified FAK containing the FERM and kinase domains (FAK31‐686) on a lipid monolayer containing PI(4,5)P2 and analyzed samples by cryo‐EM. We showed previously that C‐terminal regions required for FAT do not affect PI(4,5)P2 binding, oligomerization, or PI(4,5)P2‐induced autophosphorylation (Goni et al, 2014). After sample optimization, we obtained regular 2D crystal‐like assemblies of FAK (Fig 1B). We prepared samples in the absence (Apo) or presence of the ATP analog AMP‐PNP, the latter in order to mimic an ATP‐bound state and rigidify relative movements between the N‐terminal (N) and C‐terminal (C) lobes of the kinase domain. We collected cryo‐EM datasets at different tilt angles for the Apo (0–60°) and AMP‐PNP bound state (0–50°). Crystallographic data processing using 2dx (Gipson et al, 2007) resulted in low‐resolution 3D maps with a resolution of ~ 20 Å (Fig EV1A). To improve the resolution, we next followed a single‐particle analysis procedure by extraction of particles from 2D crystal images (Righetto et al, 2019). A summary of cryo‐EM data collection and image processing parameters is shown in Table EV1. The particle exhibits a twofold symmetry and resulting 3D maps have an overall resolution of 6.32 Å (Apo) or 5.96 Å (AMP‐PNP; Fig EV1B), with local maximal resolution reaching ~ 4 Å (Fig 1C). We note that 2D crystals from the AMP‐PNP dataset could be classified into two different crystal packings as apparent from 2D classifications (Fig EV1C). Atomic models were generated by flexible fitting of the crystal structures of the FERM (PDB: 2AEH) and kinase domains of FAK (PDB:1MP8) into the cryo‐EM maps followed by manual adjustments (Figs 1D and EV1D, Table EV2). Secondary structure elements are well defined, but side chains rarely visible with a few exceptions (Fig EV1E). An important consideration during model building was the position and connectivity of the linker residues connecting the FERM and kinase domains (residues 362–411). For this, density of a twofold symmetric FAK dimer (hereafter referred to as symmetric dimer) at the center of the AMP‐PNP containing particle (zoomed in Fig 1D) was inspected, since maps are best defined in this region. Residues 362–394 are likely disordered and are not included in the model. Residues 395–401 (including the Y397 autophosphorylation site) are modeled in the apo and AMP‐PNP structures bound to the FERM F1 lobe, analogous to the position seen in autoinhibited FAK and FERM‐linker crystal structures (Ceccarelli et al, 2006; Lietha et al, 2007), based on density present at this position (Fig EV1F). Residues 402–411, leading to the kinase domain, are only modeled in the AMP‐PNP structure based on existing density, which is however not continuous (Fig EV1G). Although we consider less likely, we cannot exclude the possibility that residues 402–411 are disordered and connect to the other kinase domain in the dimer. Importantly, the main conclusions we draw apply with either connectivity. The final Apo and AMP‐PNP models are similar with an RMSD of 1.85 Å comparing FAK monomers (superimposing 593 Cα atoms) or 2.23 Å superimposing the symmetric FAK dimer (1,186 Cα atoms). Below, we describe the FAK‐AMP‐PNP structure unless stated otherwise.
Figure EV1. Expanded view on cryo‐EM structure.
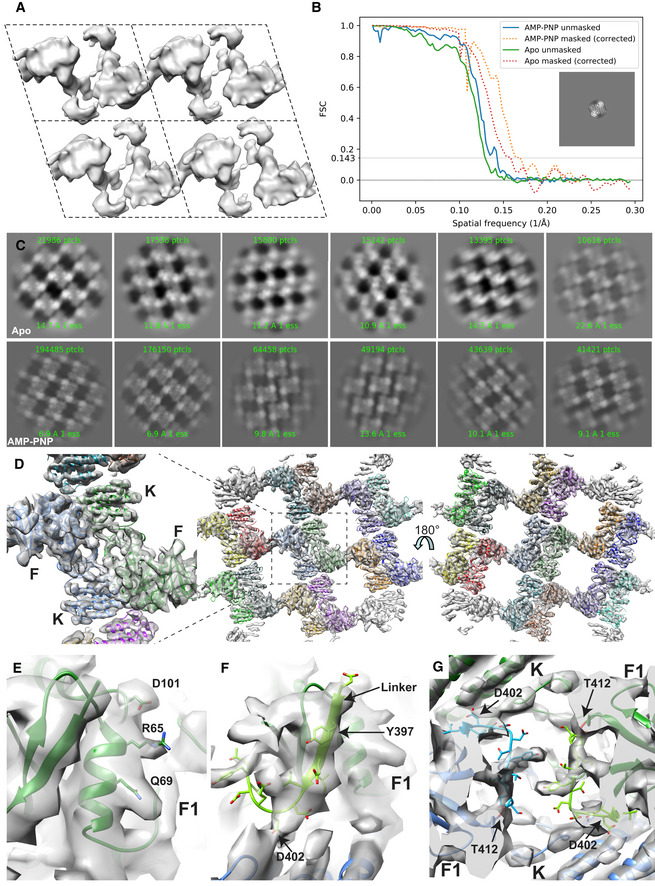
- EM maps obtained by crystallographic processing of images using 2dx (Gipson et al, 2007). Shown are four unit cells (dashed lines), each with parameters α = 110 Å, β = 88 Å and γ = 107° and p2 symmetry.
- Plot of the Fourier shell correlation over resolution. To avoid artificially inflating the FSC, the two half‐maps were reconstructed from particles randomly split on a per‐crystal basis. The inset shows a masked projection of the AMP‐PNP map.
- 2D averages of particles extracted from 2D crystal images. Note that the AMP‐PNP containing particles exhibit two different crystal packings.
- 14 FAK molecules, each containing a FERM and kinase domain are fitted into the EM maps obtained from Apo‐FAK data. Each molecule is in a different color. Middle: Membrane distal view; right: Membrane proximal view; left: zoomed view of central symmetric dimer from the membrane distal view with locations of FERM (F) and kinase (K) domains indicated. The equivalent figure for the AMP‐PNP structure is shown in Fig 1D.
- View of the FERM F1 lobe in the central region in the AMP‐PNP structure that exhibits density for side chains.
- Cryo‐EM maps show partial density for the linker region 402–412 (light green and light blue) connecting the F1 bound linker with the kinase (K) domain. The view is along a twofold symmetry axis; hence, two identical regions in two FAK molecules (green and blue) are seen. In both molecules, the ribbon of the N‐terminal residue in the kinase domain (T412) is colored in red.
The final structure contains 14 FAK molecules, each of them assembles into closely associated symmetric FAK dimers (Fig 1D). The symmetric dimers connect via FERM–FERM contacts to neighboring dimers to form a linear oligomeric assembly (Fig 1E). Lateral contacts to this linear oligomer are likely introduced crystallographically since they differ in the two crystal forms seen in the AMP‐PNP dataset (Fig EV1C), whereas the linear arrangement shown in Fig 1E is present in all crystals (AMP‐PNP and Apo). To model the FAK structure on the lipid membrane (which is for the most part not visible in 3D averaged maps), we considered the reported K216AKTLRK motif on the FERM domain as primary PI(4,5)P2‐binding site (Cai et al, 2008; Goni et al, 2014). As seen in Fig EV2A, this sequence aligns in all 14 FAK molecules in one plane allowing confident positioning of the lipid membrane relative to the FAK oligomer (Fig 1F). In further support of membrane placement, 3D maps show unmodeled density at the KAKTLRK motif, as well as at sites where the kinase domain contacts the modeled membrane (Fig EV2B), likely due to less mobile lipid molecules interacting with FAK at these sites.
Figure EV2. Known PI(4,5)P2‐binding sites in FAK support membrane placement.
- Unmodeled density at the KAKTLRK sites (red) in FERM domains (light arrowheads) and unmodeled density at K621/K627 residues (cyan) in the kinase domains (dark arrowheads) coincide with the position of the modeled membrane and likely represent lipid molecules with restricted mobility due to their interactions with FAK.
FAK oligomerizes on the membrane via multiple interfaces
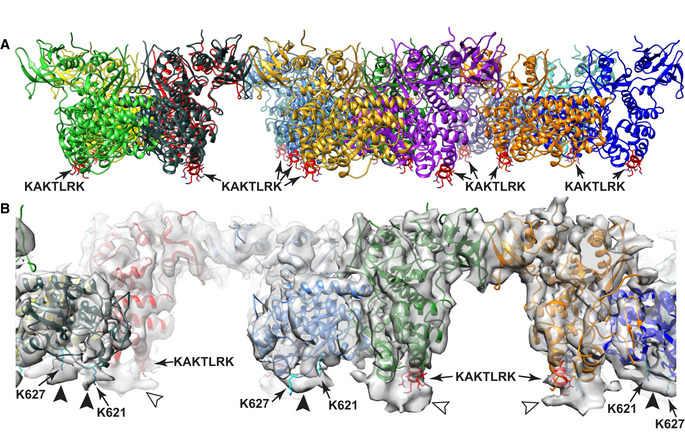
The resulting structural assembly reveals several new interfaces as well as interactions observed previously. Within the symmetric FAK dimer, new interactions are formed intramolecularly between the FERM and kinase domains (F–K and F’‐K’ in Fig 2A) and intermolecularly between FERM and kinase domains (F–K’ and F’‐K) as well as between two kinase domains (K–K’). Bridging dimers into an oligomeric assembly occurs via FERM–FERM interactions (F–F* and F′–F** in Fig 2A) in a manner described previously to occur for FAK in solution (Brami‐Cherrier et al, 2014). This interface involves W266 and is seen in 3D crystal structures that contain the FAK FERM domain. Superposition of this crystallographic FERM dimer with FERM domains in our cryo‐EM structure shows a highly similar FERM–FERM interaction (Fig 2B). For newly observed interactions within the symmetric dimer, EM maps provide limited details since sidechains are mostly not defined. We therefore performed Molecular Dynamics (MD) simulations on the symmetric FAK dimer to orient sidechains for nearby interactions, while the FAK backbone was restrained by EM maps. Analysis of the interactions throughout the simulations with PISA (Krissinel & Henrick, 2007) and SC (Lawrence & Colman, 1993) reveals rather extensive interactions of predominantly polar (intramolecular F–K), mixed (intermolecular F–K′), or hydrophobic (intermolecular K–K’) character and significant shape complementarity (Table 1; Fig 3A–C), with highest energy gain suggested to result from the K–K’ interaction. In addition to protein–protein interfaces, we also observe contacts with the modeled membrane, other than the KAKTLRK sequence in the FERM domain used for membrane positioning. The kinase domain is positioned lying flat on the membrane with main contacts made via the basic residues K621 and K627 in the kinase C‐lobe (Fig 3D). In this conformation, the active site of the kinase domain is facing toward the membrane. Inspection of the electrostatic potential of the membrane‐interacting surface of the symmetric FAK dimer reveals in addition to the KAKTLRK basic patch a large basic region on the kinase domain that includes K621 and K627 (Fig 3E). In agreement with our structural findings, these residues have been reported previously to contribute to PI(4,5)P2 binding in FAK (Hall & Schaller, 2017).
Figure 2. Interfaces in the FAK oligomeric assembly on the membrane.
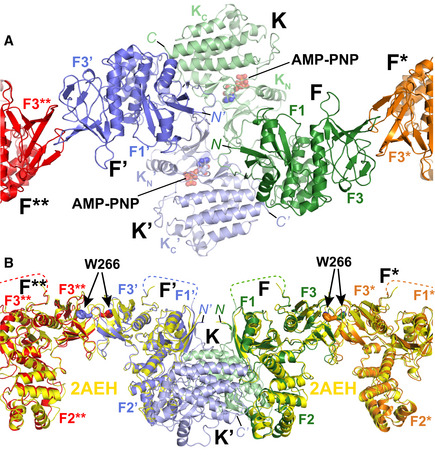
- Close‐up of the central symmetric dimer of the AMP‐PNP particle from the membrane distal view. Interfaces within the symmetric dimer are formed intramolecularly between the FERM and kinase domains (F and K; F’ and K’). Intermolecular interfaces are formed within the symmetric dimer between F and K’, F’ and K as well as K and K’. Intermolecular interactions that link symmetric dimers are formed between two FERM domains (F and F*; F’ and F**). Coloring is as in Fig 1E. The FERM subdomains F1 and F3 (F2 is not visible from this view), kinase N‐ and C‐lobes (KN, KC), AMP‐PNP and the N‐ and C‐termini (N, C) are labeled.
- Superposition of W266‐mediated FERM–FERM 3D‐crystallographic dimers (Ceccarelli et al, 2006) (yellow; PDB: 2AEH) and the FERM–FERM contacts seen in the cryo‐EM structure to link symmetric dimers (F–F*; F’–F**). The view is 90° rotated from panel (A). Subdomains and termini are labeled, and disordered regions in the linker are shown as dashed line.
Table 1.
FAK interfaces
Figure 3. Key interactions at interfaces.
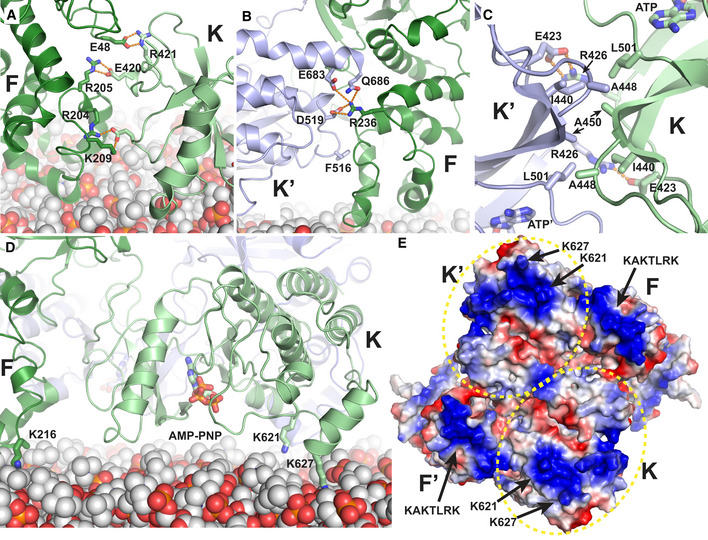
-
A–CDetails of intramolecular F–K (A) and intermolecular F–K’ (B) and K–K’ (C) interactions as seen in simulations restrained by EM maps. Side chains of main interacting residues are shown in stick representation, and hydrogen bonds are shown as orange dashed lines.
-
DThe kinase is oriented with the ATP‐binding site facing the membrane. Key interactions between the kinase and the membrane are made by K621 and K627. The side chains of K621, K627, and K216 (the latter is within the KAKTLRK motif of a neighboring FERM domain) as well as AMP‐PNP bound to the ATP‐binding site are shown in stick representation.
-
EElectrostatic surface potential of the symmetric FAK dimer as seen from the membrane proximal view. Both, the FERM (KAKTLRK) and the kinase (K621, K627) have extensive positive charges on this surface for interaction with negatively charged PI(4,5)P2 lipids. The kinase domains are encircled by a yellow dotted line.
We showed previously that PI(4,5)P2‐mediated membrane binding of FAK promotes autophosphorylation (Goni et al, 2014). To test whether autophosphorylation can occur in the observed membrane bound conformation, we modeled the linker residues in the FAK symmetric dimer to adopt a conformation as required for autophosphorylation, meaning that Y397 is located at the kinase active site according to known substrate‐kinase interactions (Hubbard, 1997). Although the active site points toward the membrane, we found that the linker can wrap around FAK such that Y397 is in an orientation susceptive for autophosphorylation, without the need to move the original position of the kinase domain from the cryo‐EM structure (Fig EV3A). The position of Y397 in the active site also remained stable during MD simulations of a FAK dimer placed on a PI(4,5)P2‐containing membrane, suggesting that autophosphorylation in this conformation is possible. Next, we compared the dynamics of this autophosphorylation‐ready FAK conformation to those observed in simulations of FAK models in which Y397 is not bound to the active site. We observed that the kinase domain moves closer to the membrane in the simulations of unbound compared to bound Y397 (Fig EV3B) and that residues from the activation loop, most notably K583, interact with lipid head groups thereby closing the active site entry (Fig EV3D). When the FERM‐kinase linker occupies this space, residues adjacent to Y397 form stable interactions with the same activation loop residues that otherwise interact with the membrane (Fig EV3C and D). The interaction between linker residue I400 and activation loop residue K583 remains stable in 11 out of 14 simulations or for 87% of the total simulation time (Fig EV3E). These results indicate that the linker could displace activation loop‐membrane interactions and in this way guide Y397 to the active site.
Figure EV3. Molecular dynamics simulations of the symmetric FAK dimer on a lipid membrane.
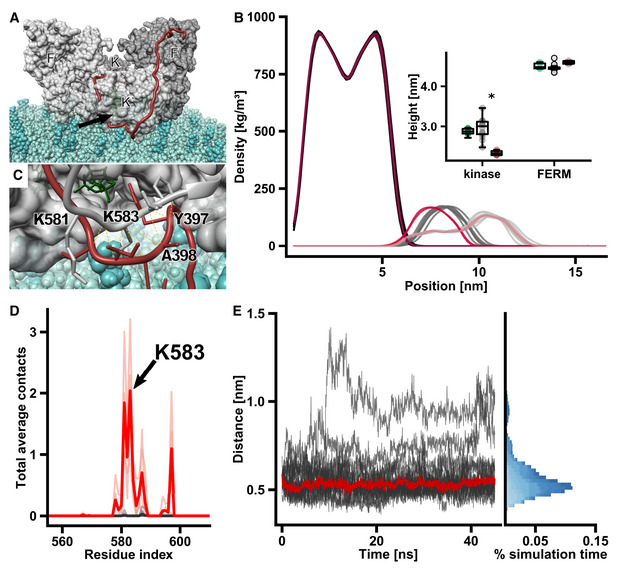
- FAK dimer with Y397 bound to the active site on a lipid bilayer. Monomers are shown in different shades of gray and FERM (F, F’) and kinase (K, K’) domains are labeled. The FERM‐kinase linker (red) makes a sharp turn at the active site cleft (arrow) due to the constraints from the lipid membrane. Note that we here show one of many putative positions of linker residues in between the active site and the domains, as the linker is likely very flexible.
- Mass density along the direction vertical to the membrane plane. From left to right, the densities of the lipid bilayer (darkest colors), kinase domains (medium shade colors), and FERM domains (lightest colors) are shown. Data from models with the Y397 substrate bound to the active site (n = 14) are depicted in gray, unbound active site models (n = 4) are in red. Insert: The heights are defined as the perpendicular distance between the center of mass (COM) of the domains and the phosphodiester bonds connecting lipid head groups and tails within a 1 nm radius from the respective COM. Shown are heights of starting models (bound + unbound, green), substrate bound simulations (gray), and unbound simulations (red). The central band of the boxplot represents the median and boxes the 25% (Q1) and 75% (Q3) quartiles. Lower and upper whiskers are defined as the lowest value above Q1–1.5*(Q3‐Q1), and the highest value below Q3 + 1.5*(Q3 − Q1), respectively. *P < 0.01, determined by an independent two‐sample t‐test.
- Residues from the activation loop (gray) interact with FERM‐kinase linker residues (red) adjacent to Y397, which is bound to the kinase active site. ATP is shown in green.
- Plot of membrane contacts. In FAK dimer simulations, in which Y397 is not bound to the active site, residues from the activation loop interact with lipid head groups (red lines). These contacts are not present in simulations, in which Y397 is bound to the active site (gray lines). Darker traces show mean number of contacts, and light traces show contacts from individual simulations.
- FERM‐kinase linker residues form stable interactions with activation loop residues during the course of the simulation. The distance between the backbone Cα of I400 in the linker and K583 in the activation loop is shown, with the red line representing the median distance. Different shades of blue in the histogram represent distinct simulations.
FERM dimerization and membrane interactions are key for FAK autophosphorylation
To test the relevance of the structural findings, we generated mutants at the observed interfaces (Table 2). The mutations do not appear to affect the protein integrity (Appendix Fig S1) and behave like FAK wild type (WT) as a monodisperse particle with a hydrodynamic radius compatible with a FAK monomer (Table EV3). We first tested the effect of the mutations on PI(4,5)P2‐induced autophosphorylation using an ELISA assay. As shown previously, autophosphorylation of FAK‐WT is strongly enhanced in the presence of PI(4,5)P2 containing vesicles (Goni et al, 2014) (Fig 4A), whereas catalytic turnover activity toward an exogenous substrate is not affected by PI(4,5)P2 (Fig 4B). The interface mutations do not affect catalytic turnover activity to a major extent, whereas the F–F* and both FAK–membrane interactions (KAKTLRK and Kinase‐Membrane) appear key to achieve high autophosphorylation rates (Fig 4A). Overall mutations within the symmetric dimer have little effect; however, mutation of hydrophobic residues at the K–K’ interface causes a reproducible reduction in autophosphorylation efficiency. We next asked whether autophosphorylation occurs in cis (enzyme and substrate in the same molecule) or trans (enzyme and substrate in different FAK molecules). For this, we incubated either FAK‐WT or a catalytically inactive kinase dead mutant (KD, contains the K454R mutation) in the presence of a catalytically active, but substrate deficient Y397F mutant. Combining KD and Y397F mutants exclusively allows autophosphorylation in trans, while FAK‐WT with Y397F can autophosphorylate in cis and trans (see schematic in Fig 4C). We observe that in the absence of PI(4,5)P2 the WT‐Y397F combination autophosphorylates significantly more efficiently, whereas the presence of PI(4,5)P2 renders the WT‐Y397 and KD‐Y397F combinations to equal and strongly increased autophosphorylation efficiency (Figs 4C and EV4A–B), indicating that in solution autophosphorylation occurs mainly in cis, but trans‐autophosphorylation appears to be dominant in the presence of PI(4,5)P2. We note that the cis‐autophosphorylation we observe in solution could be caused by the absence of the C‐terminal FAT domain which was shown to stabilize a F–F* FAK dimer for trans‐autophosphorylation also in solution (Brami‐Cherrier et al, 2014). However, C‐terminal regions are shown not to affect membrane‐associated autophosphorylation (Goni et al, 2014), likely because the FAT domain is released from the KAKTLRK region upon membrane binding of the FERM domain.
Table 2.
Interface mutants
| Name | Mutations | Mutated interface |
|---|---|---|
| Mut‐FK | Q418A, E420A, R421A, E492A, N493A | F–K |
| Mut‐FK′ | E683A, Q686A | F–K′ |
| Mut‐KK′ | A448D, M449A, A450D | K–K′ |
| Mut‐FF* | W266A | F–F* |
| Mut‐PPi | Q418A, E420A, R421A, E492A, N493A, E683A, Q686A, A448D, M449A, A450D, W266A | All protein–protein interfaces (F–K, F–K′, K–K′, F–F*) |
| KAKTLRK | K216A, K218A, R221A, K222A | F–M |
| Mut‐KM | K621A, K627A | K–M |
| Mut‐FF*/KM | W266A, K621A, K627A | F–F* and K–M |
Interfaces: F–K: intramolecular FERM–kinase; F–K′: intermolecular FERM–kinase; K–K′: intermolecular kinase–kinase; F–F*: intermolecular FERM–FERM; F–M: FERM–membrane; K–M: Kinase–Membrane.
Figure 4. In vitro phosphorylation and catalytic activity of interaction mutants.
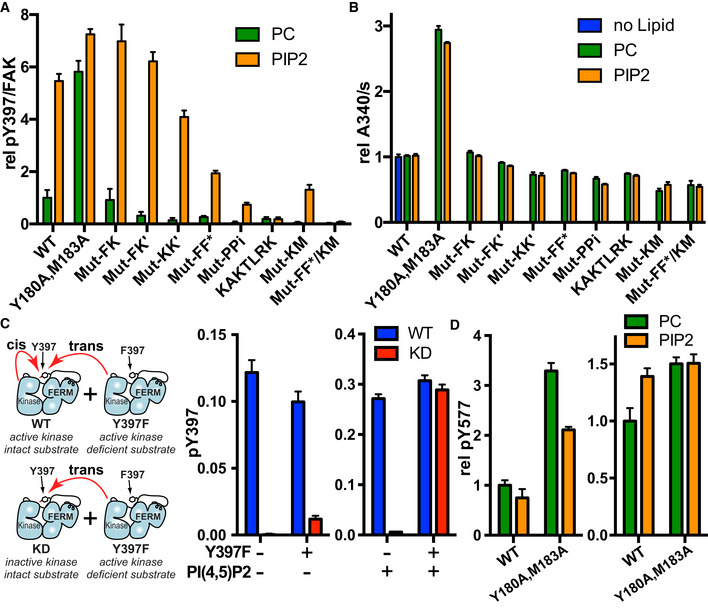
- Autophosphorylation as measured in an ELISA assay is shown for WT and interaction mutants in the presence of control vesicles (PC; phosphatidylcholine) or PI(4,5)P2 containing vesicles. Plotted are mean values from two independent autophosphorylation reactions, each determined in duplicates (n = 4). Mutant nomenclature is as shown in Table 2.
- Catalytic turnover activity as measured in a coupled kinase assay is shown. Plotted are mean values from at least three independent experiments.
- To determine whether autophosphorylation occurs in cis or trans, autophosphorylation was measured for WT or a kinase dead form of FAK (KD, contains K454R mutation) in the presence or absence of a substrate deficient FAK mutant (Y397F) and/or PI(4,5)P2 vesicles. PI(4,5)P2 negative experiments contain control PC vesicles instead. Note that the scales of the two plots are different, i.e., autophosphorylation is increased in the presence of PI(4,5)P2. Plotted are mean values from two independent phosphorylation reactions, each determined in duplicates (n = 4). Left: Schematic of possible cis‐ and trans‐autophosphorylation (red arrows) in mixtures of FAK‐WT/FAK‐Y397F and FAK‐KD/FAK‐Y397F. See also Fig EV4A and B for experiments at different protein concentrations and time points.
- Src phosphorylation of Y577 in FAK in the presence of PC or PI(4,5)P2 vesicles as determined by an ELISA method. The plot on the left is obtained in the presence of Src containing only the kinase domain (Src254‐536) and the plot on the right with Src containing additionally the SH3 and SH2 domains (Src84‐536). Plotted are mean values from two independent phosphorylation reactions, each determined in duplicates (n = 4).
Figure EV4. FAK autophosphorylation on PI(4,5)P2 membranes occurs in trans.
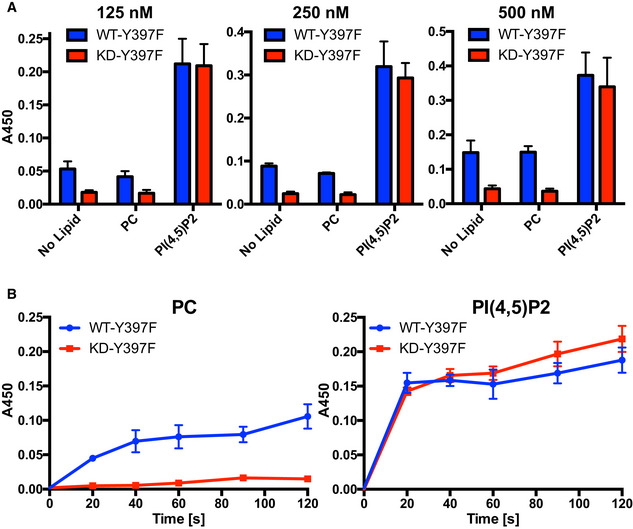
- To determine whether autophosphorylation occurs in cis or trans, autophosphorylation was measured for WT or a kinase dead form of FAK (KD, contains K454R mutation) in the presence of equimolar amounts of a substrate deficient FAK mutant (Y397F), either in buffer (no Lipid) or in the presence of PC or PI(4,5)P2 vesicles (for schematic of possible cis/trans‐autophosphorylation for the different combinations, see Fig 4C). The experiment was performed at different protein concentrations, the concentration of WT or KD is indicated. Plotted are mean values from two independent phosphorylation reactions, each determined in duplicates (n = 4). Error bars: s.d.
- Time‐course of autophosphorylation levels observed with the FAK‐WT/Y397F and KD/Y397F combinations in the presence of PC (left) or PI(4,5)P2 (right). Plotted are mean values from two independent phosphorylation reactions, each determined in duplicates (n = 4). Error bars: s.d.
Source data are available online for this figure.
We next examined the effect membrane binding of FAK has on activation loop phosphorylation by Src, using an ELISA assay to monitor Y577 phosphorylation of FAK in the presence of Src. Using only the Src kinase domain PI(4,5)P2 does not enhance Src phosphorylation of FAK‐WT while the open and activated Y180A, M183A mutant of FAK exhibits significantly reduced Src phosphorylation in the presence of PI(4,5)P2 (Fig 4D). On the other hand, when using a longer Src construct that can interact with autophosphorylated FAK (SH3‐SH2‐kinase) Src phosphorylation efficiency of WT‐FAK increases in the presence of PI(4,5)P2 (likely due to enhanced FAK autophosphorylation), however to a significantly lower extent than is seen for FAK autophosphorylation. Results using only the Src kinase domain agree with structural observations showing that the FAK active site and nearby activation loop are not exposed in the membrane bound state (Fig 3D).
We further tested how the mutations affect binding to PI(4,5)P2 membranes. As expected, mutations at membrane interaction sites are deficient in PI(4,5)P2 binding, with KAKTLRK mutations in the FERM domain resulting in somewhat lower residual binding than mutation in the Kinase C‐lobe (Fig 5). Surprisingly, mutation of the F–F* interface also significantly reduces binding to PI(4,5)P2. This is likely due to loss of avidity since our structural data indicate that the mutated W266 affects intermolecular F–F* interactions, but is distant to the membrane. Further, mutation of hydrophobic residues at the K–K’ interface modestly reduce PI(4,5)P2 binding. Taken together, mutational effects on PI(4,5)P2 binding closely mirror effects on PI(4,5)P2‐induced autophosphorylation (Fig 4A).
Figure 5. PI(4,5)P2 binding.
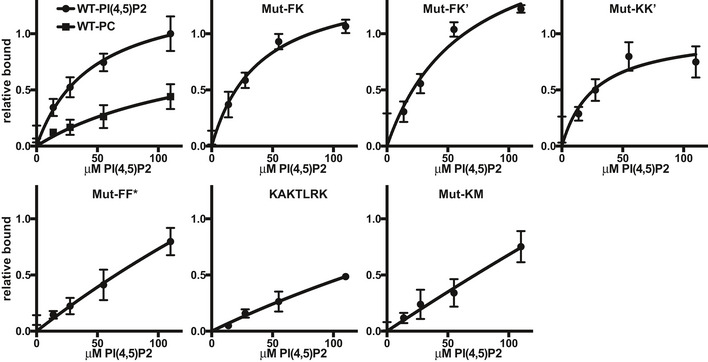
Interactions between WT or interface mutant FAK with a PI(4,5)P2 containing membrane was probed using a pull‐down assay with membrane coated silica beads. Plotted are bound protein relative to the binding for WT at the highest PI(4,5)P2 concentration. Mutant naming is as in Table 2. For WT, the non‐specific binding to a PC membrane is shown for comparison. Plotted are mean values from at least three independent experiments. Error bars: s.d. Source data are available online for this figure.
FERM dimerization and PI(4,5)P2 binding are together essential for cellular FAK activation and cancer cell invasion
To test the importance of the observed interfaces in cells, we introduced FAK mutants that affected autophosphorylation and binding to PI(4,5)P2 vesicles into FAK‐deficient squamous cell carcinoma (SCC) cells, in which the gene encoding fak was deleted by Cre‐mediated recombination (McLean et al, 2004). As observed with purified proteins, the most severe effects are caused by mutations at FAK‐membrane (KAKTLRK and Mut‐KM) and F–F* interfaces, resulting in strongly reduced FAK autophosphorylation on Y397 and other tyrosine phosphorylation sites, including Y576/Y577 and Y925 (Fig 6A). We note that the KM mutations cause reduced steady state levels of FAK, complicating quantifications for some of the phosphorylation sites. In these SCC cells, FAK appears upstream of Src, since the mutations can also cause reduced Src phosphorylation on Y416, a surrogate of the Src activation state (Fig 6B). On the other hand, mutation at the symmetric K–K’ dimer interface does not lead to detectable changes in FAK or Src phosphorylation, but effects appear amplified downstream at the level of Akt and ERK phosphorylation (Fig 6B). Remarkably, combining mutations at the F–F* and K–M interfaces has an additive effect and leads to a complete loss of FAK‐Src association (Fig 6C).
Figure 6. FAK signaling in SCC cells.
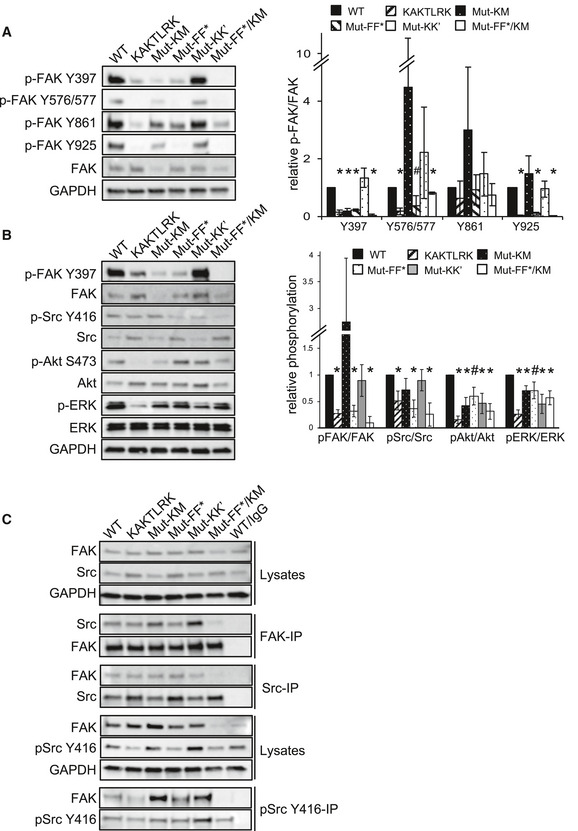
- FAK phosphorylation of WT or interface mutant FAK in SCC cells. Whole cell lysates were subjected to Western blot analysis with anti‐pFAK Y397, anti‐pFAK Y576/577, anti‐pFAK Y861, anti‐pFAK Y925, and anti‐FAK. Anti‐GAPDH served as a loading control. The graph shows densitometric analysis of relative pFAK/FAK ratios (mean) from three independent experiments. Student's t‐test was carried out to calculate the statistical significance. Error bars: s.d. *P < 0.01, # P < 0.05. For full blots see source data.
- Downstream signaling in SCC cells expressing WT or interface mutant FAK. Whole cell lysates are analyzed by Western blot analysis with the indicated antibodies. The graph shows densitometric analysis of relative phospho/total protein ratios (mean) from three independent experiments. Student's t‐test was carried out to calculate the statistical significance. Error bars: s.d. *P < 0.01, # P < 0.05. For full blots see source data.
- Src phosphorylation and interactions between FAK and Src or pSrc Y416 in SCC cells. FAK, Src or pSrc Y416 were immunoprecipitated from whole cell SCC lysates using anti‐pSrc Y416, anti‐FAK, anti‐Src, or non‐specific IgG‐agarose, followed by Western blot analysis with anti‐FAK, anti‐pSrc Y416, and anti‐Src. Anti‐GAPDH served as a loading control. For full blots, see source data.
Source data are available online for this figure.
We next analyzed the effect of the mutations on FAK and Src localization and intracellular trafficking. The FAK mutants still localize to focal adhesions as visualized by confocal microscopy (Fig 7A); however, phosphorylated Src is internalized in cells expressing the FERM‐membrane and F–F* interface mutants and targeted into puncta that we have identified previously as autophagosomes (Sandilands et al, 2011) (Fig 7B). This is indicative of defective FAK signaling at focal adhesions as a result of partners of FAK, in this case p‐Src, not being properly tethered into protein complexes at focal adhesions, likely due to impaired scaffolding functions or positioning. The removal of p‐Src to the autophagy pathway under such circumstances signifies that FAK is dysfunctional (Sandilands et al, 2011). Furthermore, of the tested interface mutants only the KAKTLRK FERM‐membrane mutant reduces nuclear localization of FAK (Fig 7C), likely due to a nuclear localization sequence within the KAKTLRK sequence (Lim et al, 2008), implying nuclear translocation is not perturbed by altering the FAK assembly on the membrane.
Figure 7. FAK and Src localization in SCC cells expressing interface mutant FAK .
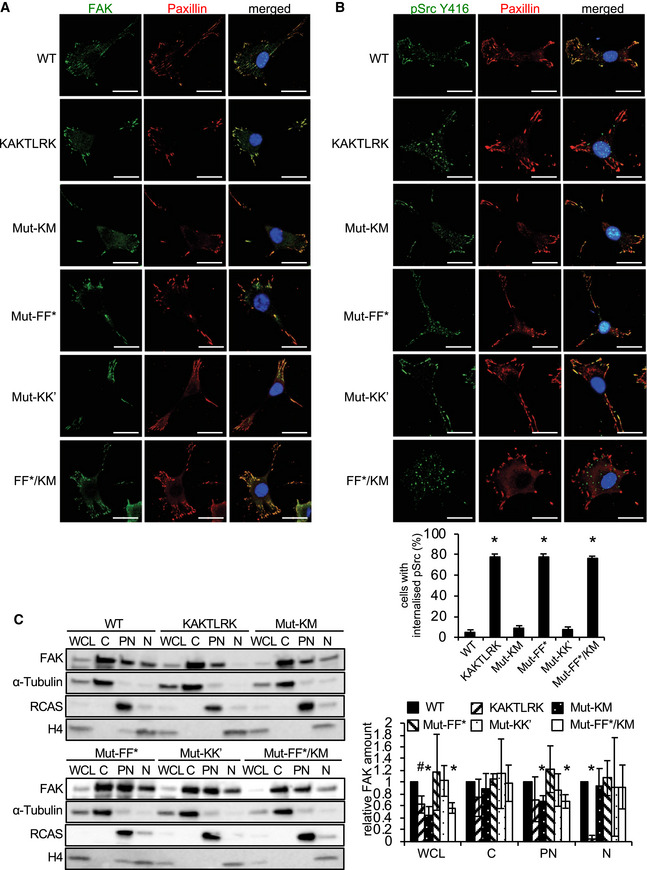
-
A, BFAK‐expressing SCC cells were grown on glass coverslips, fixed, and stained with anti‐FAK (A), or anti‐pSrc Y416 (B), anti‐Paxillin and DAPI (A, B). Representative immunofluorescence images are shown. Scale bars, 20 μm. The graph (B) shows the quantification of internalized active Src from three independent experiments. Student's t‐test was carried out to calculate the statistical significance. Error bars, s.d. *P < 0.01.
-
CSubcellular fractionation of SCC cells expressing FAK‐WT or interface mutant FAK. Whole cell lysates (WCL) and subcellular fractions were subjected to Western blot analysis with anti‐FAK, anti‐Tubulin (marker for cytoplasmic fraction; C), anti‐RCAS (marker for perinuclear fraction; PN), and anti‐H4 (marker for nuclear fraction; N). The graph shows densitometric analysis of relative FAK amount of the FAK mutants in the different fractions from three independent experiments. Student's t‐test was carried out to calculate the statistical significance. Error bars: s.d. *P < 0.01, # P < 0.05. For full blots see source data.
Source data are available online for this figure.
We further analyzed the effect of FAK mutants on SCC cell proliferation in 3D cultures and invasion through Matrigels. All tested interface mutants exhibit a significant reduction in cell proliferation in 3D cultures (Fig 8A). This is in contrast to SCC cell invasion, where mutation of the K–K’ interface has no effect, however, combining mutations at membrane and F–F* interaction sites severely hampers invasion, while mutation of the kinase–membrane interface alone enhances invasion by a mechanism that is unknown (Fig 8B). Therefore, our mutational data in SCC cells demonstrate that different interfaces observed in the EM structure differentially affect specific cellular FAK functions. On the one hand, FERM and kinase interactions with the membrane as well as FERM dimerization via the F–F* interface are crucial for FAK signaling, FAK/Src dynamics, cell proliferation, and invasive migration. On the other hand, FAK oligomerization via the K–K’ interface has more subtle effects with mutations causing impaired downstream signals at the level of MAP and Akt kinases, likely contributing to suboptimal FAK‐mediated cancer cell proliferation under anchorage‐independent condition. The strong effects in SCC cells seen with membrane and F–F* interaction mutants, but subtle changes caused by K–K’ mutations agree well with data obtained with purified mutants (Figs 4A and 5).
Figure 8. FAK mutant SCC cells in 3D proliferation and invasion.
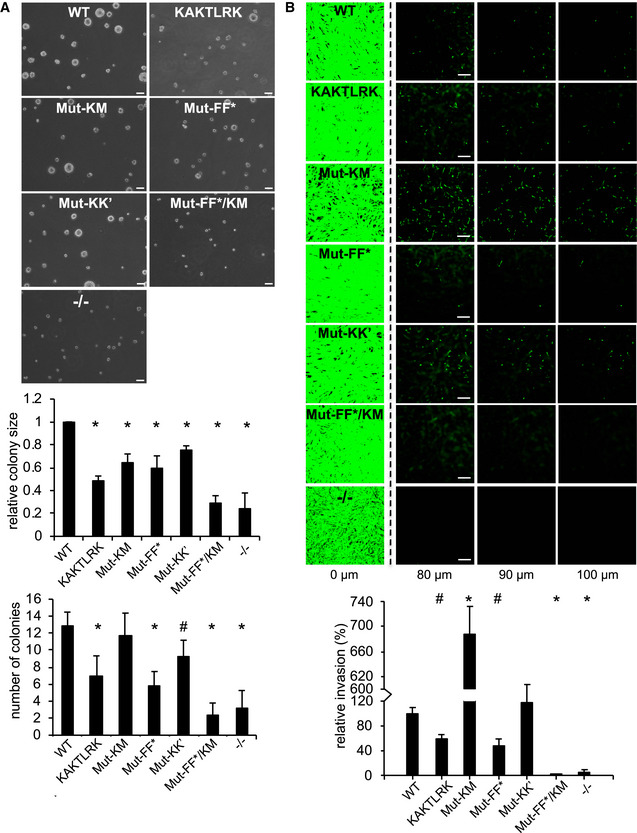
- FAK‐WT, interface mutant FAK or FAK−/− SCC cells were resuspended in methylcellulose solution in growth medium on a layer of agarose and observed for 3D sphere formation. Images were taken from 10 random fields after 9 days (representative images are shown) and the relative colony size (top graph) and the number of colonies (bottom graph) were assessed from three independent experiments. Scale bars, 200 μm. Student's t‐test was carried out to calculate the statistical significance. Error bars s.d. *P < 0.01, # P < 0.05.
- FAK‐WT, interface mutant FAK or FAK ‐/- SCC cells were seeded on growth factor‐reduced Matrigel in serum‐free media. After 72 h, invasion toward a serum gradient was analyzed by staining the cells with calcein and taking images at 10 μm increments through the Matrigel. Representative images of the invasion assay are shown. Scale bars, 200 μm. The graph shows relative invasion from five independent experiments. Student's t‐test was carried out to calculate the statistical significance. Error bars, s.e.m. *P < 0.01, # P < 0.05.
Discussion
Focal adhesion kinase constitutes an important element in the complex multilayered architecture of focal adhesions and localizes into the most membrane proximal layer in cells (Kanchanawong et al, 2010). The membrane at focal adhesion sites has increased levels of PI(4,5)P2 (Di Paolo et al, 2002; Ling et al, 2002) and our new structural insights provide a detailed view of how FAK spontaneously assembles on a membrane containing PI(4,5)P2. Importantly, the cellular data presented here (Figs 6, 7, 8) as well as reported previously (Cai et al, 2008; Brami‐Cherrier et al, 2014; Hall & Schaller, 2017) support that our cryo‐EM structure represents a state that is important for FAK to convert into an active signaling kinase in focal adhesions. In cells, FAK is known to undergo large conformational changes when integrated into focal adhesions (Cai et al, 2008) and the cryo‐EM structure presented here provides the structural basis for this change. Superposition of the autoinhibited FERM‐kinase structure of FAK (Lietha et al, 2007) with the FERM domain in our membrane‐bound structure shows a large movement of the kinase domain (~ 80 Å at the C‐terminus) and a rotation of ~ 90°, approximately around the kinase N‐terminus (Fig 9A). Our structure also immediately suggests a mechanism that is responsible for triggering the conformational change. When visualizing the above superposition in the presence of the membrane, it is apparent that the kinase in the autoinhibited conformation would cause a significant steric clash with the membrane (Fig 9B). This clash occurs as F–F*‐mediated FAK dimers approach and bind the membrane via their KAKTLRK lipid‐binding sites. Hence, formation of a F–F*‐mediated dimer is essential to trigger the conformational change in agreement with mutational data showing a critical role for this site in PI(4,5)P2‐induced FAK autophosphorylation and cellular function (Brami‐Cherrier et al, 2014) (Figs 4A and 6, 7, 8). We present a model where membrane‐induced release of FAK autoinhibition in a F–F*‐mediated FAK dimer represents a first and essential step in a multistep activation sequence of FAK (Fig 9C). It is interesting to note that PI(4,5)P2 binds and induces functionally important conformational changes in other FERM containing proteins, including in ERM proteins, talin, and merlin. However, surprisingly the PI(4,5)P2‐binding site is not conserved in these FERM domains (Hamada et al, 2000; Saltel et al, 2009; Moore et al, 2012; Chinthalapudi et al, 2018).
Figure 9. Membrane‐induced FAK activation.
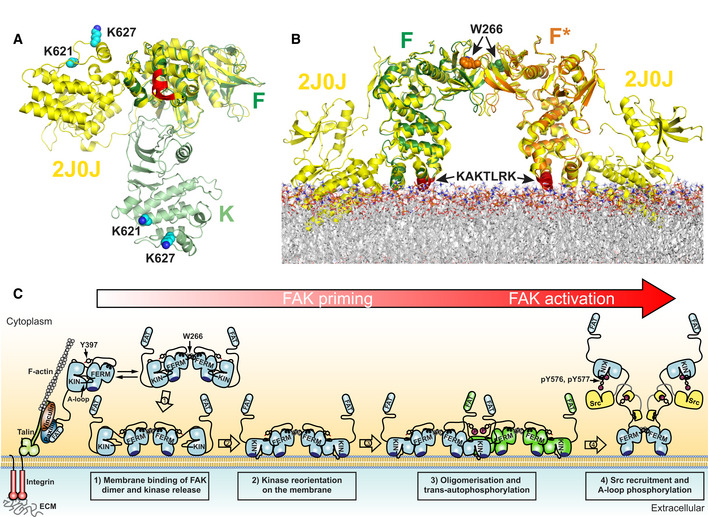
- FERM superposition of autoinhibited FAK (Lietha et al, 2007) (yellow, PDB: 2J0J) with a membrane bound FAK monomer as observed in the cryo‐EM structure (green) shown from the membrane proximal view. The KAKTLRK membrane‐binding site on the FERM domain is colored red and membrane interacting residues K621 and K627 in the kinase domain are in cyan with side chains in space fill representation.
- FERM superpositions of autoinhibited FAK (yellow) with a F–F*‐interacting FERM dimer bound via the KAKTLRK motifs (red) to a lipid membrane. Membrane‐bound kinase domains connected to the FERM domains F and F* are removed for clarity.
- Schematic model for PI(4,5)P2 induced FAK activation in focal adhesions. Locally increased concentrations of FAK upon focal adhesion targeting promotes formation of FERM mediated FAK dimers. Binding of FAK dimers via their KAKTLRK basic region (dark blue) to PI(4,5)P2‐rich membranes in focal adhesions causes dissociation of the autoinhibitory interaction between the FERM and Kinase (KIN) domains (step 1). The released kinase domains reorient to interact via their own basic residues with the membrane (step 2). This aligns a large surface (F–K’, K–K’, and F’–K in Fig 2A) for formation of the symmetric dimer and exposes the autophosphorylation site for efficient trans‐autophosphorylation across the symmetric dimer (step 3). Src is recruited to the autophosphorylation site and phosphorylates Y576 and Y577 in the activation loop (A‐loop) of FAK to induce catalytic activation of FAK (step 4). Possibly an additional mechanism is involved in removing the kinase domain from the membrane to expose the Src phosphorylation sites, thereby potentiating the activating step 4 (see text).
Once the kinase domain of FAK is released from the FERM domain, it is free to rearrange. As one would expect, the kinase then rotates with its own basic patch containing K621 and K627 toward the membrane. This requires an approximately 90° rotation of the kinase, which if rotated around the kinase N‐terminus brings the kinase into a position very close to the one observed in the cryo‐EM structure (Fig EV5). Kinase rearrangement and membrane interaction via K621 and K627 are an essential second step in the FAK activation sequence (Fig 9C), as demonstrated by mutations at the kinase–membrane interface (Hall & Schaller, 2017) (Figs 4, 5, 6, 7, 8). Indeed, it appears that kinase–membrane interactions are required to keep FAK stably on the membrane and hence prevent reformation of the autoinhibited FERM‐kinase conformation, as mutation of K621 and K627 results in significantly reduced binding to PI(4,5)P2 membranes (Hall & Schaller, 2017) (Fig 5). A similar kinase–membrane interaction was observed in one of two possible membrane‐binding modes seen in coarse grain MD simulations of FAK on a PI(4,5)P2 membrane (Herzog et al, 2017). However, in these simulations the autoinhibitory FERM–kinase interaction remained intact and as a consequence the two basic patches on FERM and kinase domains are not able to form ideal membrane interactions simultaneously. Together, kinase release from the FERM domain and subsequent attachment to the membrane keep the kinase in a position that expose the Y397 site for efficient autophosphorylation, as occurs in the constitutively open Y180A, M183A mutant (Lietha et al, 2007; Goni et al, 2014) (Fig 4A). Combining mutations that prevent both of the two‐first steps (Mut‐FF*/KM) is detrimental to any cellular activity we tested (Figs 6, 7, 8). Once FAK interacts with the membrane via FERM and kinase domains a large surface of ~ 1,200 A2 over both domains aligns for association of the symmetric dimer via F–K’, K–K’, and K‐F’ interfaces, resulting in formation of FAK oligomers (step 3 in Fig 9C).
Figure EV5. Kinase rearrangement on the membrane.
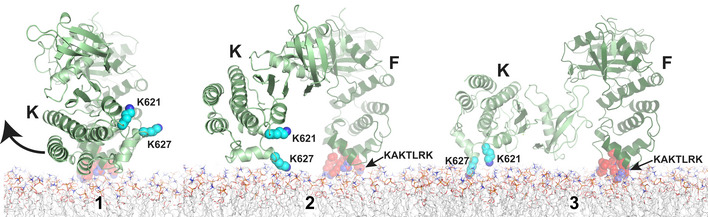
Once steric hindrance has caused kinase detachment from the membrane bound FERM domain (state 1; see schematic in Fig 9C of the main manuscript), the kinase rotates by ~ 90° (via state 2 to state 3) to allow the basic residues K621/K627 (cyan, side chains shown in space fill) in the kinase domain to interact with the PI(4,5)P2 membrane, as seen in the cryo‐EM structure (state 3). Membrane interacting KAKTLRK residues in the FERM domain are colored in red and side chains shown in space fill.
Our data suggest that FAK autophosphorylation on the membrane occurs in trans (Figs 4C, and EV4A and B), likely within the symmetric dimer, which has the active site of one molecule within 42 Å of Y397 bound to the FERM domain in the other molecule. This distance would be 57 Å for cis‐autophosphorylation and 105 Å for trans‐autophosphorylation across a F–F*‐mediated dimer, which even the released linker cannot reach. Notably, K–K’ mutations have relatively subtle effects, possibly because a partially intact interface is sufficient for formation of a transient enzyme–substrate complex. Nevertheless, K–K’ mutations consistently affect FAK function in vitro and in cells (Figs 4A, 5, 6B and 8A). Interestingly, K‐K mutations affect 3D cell proliferation but not invasion. As shown in other signaling systems, different signaling levels can control different biological outputs (Benschop et al, 1999; Zhou et al, 2014; Gottschalk et al, 2016). It is possible that reduced FAK oligomerization by disruption of the K–K’ interface lowers signaling levels below a threshold required for efficient cell proliferation, but is still sufficient for cell invasion (Fig 8). Surprisingly, the kinase–membrane interface mutant exhibits substantially increased cell invasion capability in our assay. The reason for this is unclear, but an intriguing speculation would be that in the setting of this experiment this mutant can still enter the activation sequence shown in Fig 9C, but due to failure of the kinase domain to interact with the membrane the active site is exposed, causing increased kinase activation and SCC invasion.
It is indeed a key observation in our cryo‐EM structure that the active site of the FAK kinase domain is not exposed in the membrane bound state. We believe this explains why the turnover activity toward an exogenous substrate is not increased by binding to PI(4,5)2 (Goni et al, 2014) (Fig 4B), even though autoinhibitory FERM‐kinase interactions are released. This raises the question of the relevance of keeping FAK activity low upon PI(4,5)P2 binding. Our hypothesis is that the membrane bound state we observe represents an additional layer of regulation, where PI(4,5)P2‐binding primes FAK for activation, but an additional event is required to trigger catalytic activity. Full activity requires phosphorylation of the activation loop residues Y576 and Y577 by Src (step 4 in Fig 9C) (Calalb et al, 1995; Lietha et al, 2007), yet these sites, being close to the active site, are still occluded by kinase–membrane interactions and membrane binding inefficiently enhances their phosphorylation (Fig 4D). In our in vitro system, increased recruitment of Src containing SH2 and SH3 domains to autophosphorylated FAK is sufficient to boost Src phosphorylation of Y576/Y577 in the presence of PI(4,5)P2 to some extent. However, it is tempting to speculate that in the crowded environment of the cell an additional stimulus could be required to expose the FAK active site and activation loop to trigger full FAK activity. Several triggers might exist and could be important physiologically depending on the cellular context. One stimulus that structurally seems plausible is the generation of tension force that builds up upon contraction of actomyosin fibers that are attached to focal adhesions. Stretching of the FAK molecule via FERM attachment to the membrane and C‐terminal cytoskeletal anchoring could expose the FAK active site for activation, as shown by single molecule force spectroscopy (Bauer et al, 2019). Further, FAK is activated in cells with increasing tension at focal adhesions (Torsoni et al, 2003; Wong et al, 2011; Seong et al, 2013) and FAK can act as a cellular force sensor that is required for cellular outputs in response to force (Wang et al, 2001). The structural assembly we observe of FAK on the membrane is fully compatible with a scenario where force applied to FAK via C‐terminal attachment to cytoskeletal components could be responsible for detachment of the kinase domain from membrane and FERM interactions to expose the active site and nearby activation loop to induce activation.
In conclusion, we present new structural information of an oligomeric membrane bound state of FAK, providing atomic details on a key component of the signaling layer in focal adhesions. The structure demonstrates that FAK undergoes large domain rearrangements upon membrane binding and suggests membrane binding as a trigger that releases autoinhibition. A surprising feature in the structure is that the kinase active site is facing toward the membrane and we propose that this represents a primed state of FAK that requires an additional trigger for full catalytic activation. Whereas dimerization and kinase attachment to the membrane are essential for any biological activity, stable oligomerization further increases the signaling level above a threshold required for cell proliferation. These insights could help in the design of novel agents targeting either all or specific functions of FAK.
Materials and Methods
Protein expression and purification
Focal adhesion kinase‐WT and mutants were expressed and purified as described earlier (Goni et al, 2014). In brief, FAK from Gallus gallus that contains the FERM and the kinase domains (chFAK31‐686) was expressed in HEK293GnT1 cells by transfecting the pOPIN‐F vector (Berrow et al, 2007) containing the chFAK31‐686 sequence with a N‐terminal 6xHis tag using polyethyleneimine (PEI) as a carrier. Cells were harvested, lysed, and purified by Ni‐affinity chromatography (HisTrap column, GE Healthcare), ionic exchange chromatography (Source15Q, GE Healthcare), and size exclusion chromatography (Superdex200, GE Healthcare). The poly‐His tag was removed by cleavage with 3C PreScission protease. Recombinant human Src proteins containing either only the kinase domain (hSrc254‐536) or the SH3, SH2, and kinase domains (hSrc84‐536) with an N‐terminal 6xHis tag were expressed in Escherichia coli BL21(DE3) by coexpression with YopH as reported by Seeliger et al (2005). Pure protein was obtained by Ni‐chelate affinity chromatography (HisTrap), ion exchange chromatography (Source 15Q), and size exclusion chromatography (Superdex200). After the Ni‐chelating column, the 6xHis tag was removed by incubation with TEV protease.
Generation and optimization of FAK 2D crystals
For 2D crystallization, lipid monolayers were obtained by adding a 1:1 mixture of unsaturated porcine brain PI(4,5)P2 (Avanti Polar Lipids) with saturated PI(4,5)P2diC16 (Echelon Biosciences) to a well containing FAK31‐686 at 0.05 mg/ml in 300 mM lithium sulfate, 50 mM sodium acetate pH 5.5, and 1 mM TCEP. Two‐dimensional crystals were formed over 5–7 days at 4°C. For initial crystal evaluation FAK 2D crystals, samples were negatively stained on carbon coated grids with 2% uranyl acetate and imaged using a 120 kV Tecnai G2 Spirit electron microscope, equipped with the TVIPS CMOS detector. AMP‐PNP containing crystals were obtained as for Apo crystals, but adding 1 mM AMP‐PNP and 2 mM MgCl2.
Cryo‐EM data collection
Apo dataset
Grids containing vitrified 2D crystals of apo‐FAK were imaged using both a Titan Krios (Thermo Fischer Scientific) transmission electron microscope (TEM) operating at 300 kV equipped with a Gatan Quantum‐LS energy filter (slit width 20 eV; Gatan Inc.) and a K2 Summit direct electron detector (Gatan Inc.), and a Tecnai G2 Polara (Thermo Fischer Scientific) TEM operating at 300 kV also equipped with a K2 Summit direct electron detector, without energy filter. In both microscopes, SerialEM (Mastronarde, 2005) was used for automated data collection at each tilt angle, which was manually varied from 0 to 60 degrees in steps of 15 degrees. Movies were recorded at the Titan in super‐resolution mode with a physical pixel size at the sample level of 1.058 Å/px (0.529 Å/px in super‐resolution), and at the Polara in counting mode with a pixel size at the sample of level 1.7 Å/px. Each of the 515 movies from the Titan dataset comprised an exposure of 40 e‐/Ų fractionated into 80 frames over 10 s. For the Polara dataset, each of the 97 movies acquired comprised an exposure of 40 e‐/Ų fractionated into 50 frames over 10 s. Movie frames were aligned for drift correction and averaged with dose‐weighting using MotionCor2 (Zheng et al, 2017), with down‐sampling to the physical pixel size in the case of the Titan dataset. Defocus measurement was carried out using CTFFIND4 (Rohou & Grigorieff, 2015) and CTFTILT (Mindell & Grigorieff, 2003) to estimate the tilt geometry via 2dx (Gipson et al, 2007).
AMP‐PNP dataset
Grids containing vitrified 2D crystals of FAK in the presence of AMP‐PNP were imaged using a Titan Krios TEM operating at 300 kV equipped with a Gatan Quantum‐LS energy filter (slit width 20 eV) and a K2 Summit direct electron detector. SerialEM was used for automated data collection at each tilt angle, which was manually varied from 0 to 50 degrees in steps of 10 degrees. A total of 2,608 movies were recorded in super‐resolution mode at the Titan with a physical pixel size at the sample level of 1.058 Å/px (0.529 Å/px in super‐resolution). Each movie from the Titan dataset comprised an exposure of 80 e‐/Ų fractionated into 40 frames over 8 s. The acquired data were pre‐processed in real time using FOCUS (Biyani et al, 2017). Super‐resolution movie frames were aligned for drift correction, downsampled to the physical pixel size, and averaged with dose‐weighting using MotionCor2. Defocus measurement was carried out using CTFFIND4 and CTFTILT to estimate the tilt geometry.
3D reconstruction
Apo dataset
Initial 2D crystal structure determination was carried out in 2dx following conventional electron crystallography protocols, revealing unit cell parameters of α = 110 Å, β = 88 Å and γ = 107°. The FAK 2D crystals were found to be highly disordered, with no visible diffraction spots beyond ~ 20 Å in the non‐tilted images. Consequently, a low‐resolution electron potential map with p2 symmetry imposed resulted from this processing (Fig EV1A), after pruning and merging data from 595 selected 2D crystal images, being 508 movies from the Titan and 87 from the Polara datasets. This map allows placing of the FERM domain but shows weak density for the kinase domain. Attempting a higher resolution reconstruction, we then applied the single‐particle approach to 2D crystals implemented in the FOCUS package. Briefly, the approach consists of extracting patches of the 2D crystal images and treating them as single particles, therefore allowing for local corrections of lattice distortions (Righetto et al, 2019). We extracted 361,796 particles from the 595 images previously selected, using a box size of 238 × 238 pixels and a pixel size of 1.7 Å/px. Particles extracted from Titan images were downsampled accordingly at the time of extraction to match this pixel size. For each particle, FOCUS calculates the expected defocus at the center of the box based on the known tilt geometry for the 2D crystal and exports the metadata in STAR format. The particle stack was then imported into cryoSPARC (Punjani et al, 2017) and subject to 2D classification into 50 classes. Particles from class averages not clearly displaying views of the FAK 2D crystals (i.e., “bad” particles) were excluded from further analysis, resulting in a new stack with 146,430 particles. Then, ab initio 3D map generation was performed with two classes without symmetry, one of which displayed densities for the FERM and kinase domains and comprised 106,785 particles. This subset and respective 3D reference were further used for a homogeneous refinement imposing C2 symmetry, with a FAK dimer at the center of the box. Finally, the particle stack with the refined orientations and shifts was imported into cisTEM (Grant et al, 2018) for per‐particle CTF refinement without alignments, using a resolution limit of 7 Å.
AMP‐PNP dataset
After initial processing in FOCUS for tilt geometry and lattice estimation, the same unit cell parameters as the apo dataset were found for the AMP‐PNP dataset. This time, 2,666,154 particles were extracted from 2,513 selected movies after dataset pruning in FOCUS. Particle boxes were downsampled by 2× at extraction time to a pixel size of 2.116 Å/px in a box of 200 × 200 pixels. The particle stack was then imported into cryoSPARC and subject to 2D classification into 50 classes. Again, particles from class averages not clearly displaying views of the FAK 2D crystals were excluded from further analysis, resulting in a new stack with 871,180 particles. Interestingly, 2D classification revealed the co‐existence of two different types of crystal packing in this dataset (Fig EV1C). Both crystal packings were kept at this stage, as they cannot be interpreted from the 2D class averages of tilted images. As in the Apo case, ab initio 3D map generation was performed with two classes without symmetry, at which stage we could disentangle the two crystal packings. One class contained 548,394 particles (63% of the data) and displayed clear densities for the FERM and kinase domains, in the same crystal packing as the apo state, while the other comprised 322,786 particles (37% of the data) in a different crystal packing that was not as well resolved. The subset of the best resolved 3D class was then refined imposing C2 symmetry. Finally, the particle stack of the refined map was imported into cisTEM for CTF refinement without alignments like in the apo case.
Resolution estimation
Because cryoSPARC always shuffles the order of the particles for its internal half‐set assignments (this order was not changed by cisTEM), resolution estimates were inflated due to the overlap between particles extracted from the same 2D crystal. In order to obtain a more reliable resolution estimate, a Python script was written to re‐assign the particles into half‐sets in a per‐crystal rather than per‐particle manner, and each half‐set was reconstructed using the program relion_reconstruct (He & Scheres, 2017). No refinement of the alignment parameters was performed at this stage. Resolution was estimated based on the Fourier shell correlation curve (Harauz & van Heel, 1986) using the 0.143 threshold criterion (Rosenthal & Henderson, 2003) after applying a soft‐edged spherical mask on the central FAK dimer of each map, which could not be isolated from the map without cutting through the density of the neighboring dimer linked via the FERM domain (Fig EV1B). Mask creation and postprocessing operations were carried out using the program focus.postprocess (Righetto et al, 2019), including FSC calculation, correction for artificial correlations introduced by the mask (Chen et al, 2013), spectral signal‐to‐noise ratio weighting and B‐factor sharpening based on the Guinier plot (Rosenthal & Henderson, 2003). Finally, the maps were low‐pass filtered at the global cutoffs of 6.32 Å (apo) and 5.96 Å (AMP‐PNP). Local resolution was estimated using the program blocres from Bsoft (Cardone et al, 2013; Heymann & Belnap, 2007) (Fig 1C). A summary of EM data collection and processing is given in Table EV1.
Flexible fitting of atomic models
Flexible fitting of existing atomic models for the FERM domain (PDB: 2AEH, residues 33–363) and the kinase domain (PDB: 1MP8, residues 414–686) was performed into the obtained EM maps of FAK in the apo and AMP‐PNP states. After manual placing and rigid‐body fitting of the PDB coordinates using UCSF Chimera (Pettersen et al, 2004), they were combined into a single PDB file and flexibly fit using normal mode analysis in iMODFIT (Lopez‐Blanco & Chacon, 2013), followed by real space refinement in PHENIX (Afonine et al, 2018) and manual adjustments in Coot (Emsley & Cowtan, 2004) to improve the model geometry. Initially, a single FAK molecule was fit following this protocol, with more molecules later included in order to complete the dimer and the 2D crystal supercell, always followed by a real space refinement in PHENIX to optimize the chain interfaces. Please refer to Table EV2 for geometry and map‐to‐model validation scores. For the central dimer, exhibiting the best density, linker residues 395 to 400 were fitted bound to the FERM domains, as in 2AL6 and 2J0J (Ceccarelli et al, 2006; Lietha et al, 2007) (Fig EV1F). The connection from 401 to 413 (beginning of the kinase was built manually based on existing density that is not continuous (Fig EV1G).
Molecular dynamics restrained by EM maps
Targeted conformational search was performed with AMBER 16 using sander.MPI, considering the cryo‐EM density map as an external potential added to the force field using EMAP implementation and self‐guided Langevin dynamics (MapSLGD) (Wu et al, 2013) to accelerate the conformational search. A constant force of 0.005 kcal/g was applied only in the backbone atoms (CA, C, N, O) to allow the side chains to interact freely. The total simulation time was 10 ns and 1 fs time step in the NVT ensemble considering implicit solvent and a 0.9 nm cut‐off using the Particle Mesh Ewald method.
Molecular dynamics of FAK dimer on the membrane
To create models, in which Y397 is not bound to the kinase active site, we started from the central FAK dimer from the EM structure and filled in linker residues that were not defined using the SWISS‐MODEL server (Waterhouse et al, 2018). This is likely to represent only one of multiple conformations of the disordered FERM‐kinase linker. To model the linker in a way that Y397 is positioned for autophosphorylation, we added residues 391–400 into the kinase active site based on homology to peptides bound to other tyrosine kinases (PDB accession codes: 1IR3, 1K3A, 2PVF, 3FYG, 5CZH) using Modeller v9.20 (Sali & Blundell, 1993) and UCSF Chimera (Pettersen et al, 2004). We inserted the remaining residues as above to yield models that represent either cis‐ or trans‐binding modes. We could not distinguish between the cis and trans models regarding their propensity for autophosphorylation during subsequent simulations. Therefore, we only show results of the trans models, based on experimental findings (Figs 4C, and EV4A and B). We attached PI(4,5)P2 lipids to each FERM basic patch (K216AKTLRK) of the FAK dimers by superimposing FERM‐F2 lobes with docked PI(4,5)P2 head groups (as described here: (Goni et al, 2014)) and adding fatty acid tails to the head groups. We incorporated the resulting FAK‐bound PI(4,5)P2 lipids into a POPC lipid bilayer comprising 10% PI(4,5)P2 in the upper leaflet using the Charmm‐GUI Membrane Builder (Jo et al, 2007, 2008; Wu et al, 2014; Lee et al, 2016). Using Gromacs 2016.4 (Van Der Spoel et al, 2005), we placed the FAK dimer models including ATP and Mg2 + on a membrane in a simulation box with dimensions 16.1 × 16.1 × 15.14 nm for bound and 16.1 × 16.1 × 16.3 nm for unbound models, solvated using the TIP3P water model and neutralized the charge with NaCl. Subsequently, we performed steepest descent minimization until energy convergence and Fmax below 103 kJ/mol/nm. Using Gromacs version 2018.5 and the Charmm36 force field (Klauda et al, 2010; Best et al, 2012; Huang & MacKerell, 2013), we equilibrated the minimized systems for 50 ps in the NVT ensemble with a 1 fs time steps followed by 325 ps equilibration in the NPT ensemble with a 2 fs time step using the velocity rescaling thermostat with a 1 ps coupling constant to set the temperature to 303.15 K, and the Berendsen barostat with a 5 ps coupling constant and a compressibility of 4.5 × 10−5 to adjust the pressure to 1 bar. For production simulations, we kept the temperature at 303.15 K using the Nosé‐Hoover thermostat with a coupling constant of 1 ps, and the pressure at 1 bar using the Parrinello‐Rahman barostat with a coupling constant of 5 ps. Equations of motion were solved using the leapfrog integrator. We froze hydrogen bonds using the LINCS algorithm and treated the long‐range electrostatics with the Particle Mesh Ewald method with a 1.2 nm cut‐off. For the calculation of van der Waals attraction, we used a 1.2 nm cutoff transitioning the potential to zero at the cutoff distance using the force‐switch modifier. The Verlet neighbor list was updated every 20 integration steps. In simulations of bound FAK dimer models, we restrained the distance between Pγ of ATP and the hydroxyl oxygen of Y397 to > 4.2 Å for the first 15 ns using a flat bottom harmonic potential with a force constant of 103 kJ/mol/nm.
To determine distances and densities, we used Gromacs tools and post‐processed using the Python 3 scipy package. Contacts between protein residues and lipids were analyzed using ConAn (Mercadante et al, 2018).
Generation of vesicles
Vesicles were prepared at 2 mM total lipid concentration containing either 20% (mol/mol) DPPC (Echelon Biosciences) (PC vesicles) or 20% (mol/mol) PI(4,5)P2 diC16 (Echelon Biosciences) (PI(4,5)P2 vesicles), both with 80% (mol/mol) chicken egg‐PC (Avanti Polar Lipids). Solvent was removed on a rotary evaporator and lipid films were hydrated with 20 mM Hepes (pH 7.5), 150 mM NaCl, and 1 mM Tris (2‐carboxyethyl) phosphine (TCEP) and sonicated until resuspended.
In vitro FAK auto and Src phosphorylation by ELISA
Focal adhesion kinase (WT or mutants) were first treated with 100 nM of phosphatase YopH in order to remove residual phosphorylation and YopH is subsequently inactivated with 1 mM sodium orthovanadate. FAK autophosphorylation reactions were performed using 500 nM dephosphorylated FAK proteins, 1 mM ATP, 5 mM MgCl2 in the presence of 125 μM PC or PI(4,5)P2 vesicles (12.5 μM PI(4,5)P2 on outer leaflet). Autophosphorylation reactions were stopped after 1 min with 50 mM EDTA and phosphorylation levels quantified by a standard ELISA method using anti‐pFAK Y397 (Cell Signaling) and anti‐FAK (Cusabio) as primary antibodies, a HRP‐conjugated secondary antibody (Dako) and TMB (Calbiochem) for detection. After addition of 0.18 M sulfuric acid absorption was measured at 450 nm. For Src phosphorylation reactions, either only the Src kinase domain (hSrc254‐536) or Src with SH3, SH2, and kinase domains (hSrc84‐536) was added equimolar at 500 nM during the phosphorylation reactions and ELISA detection was carried out with an anti‐pFAK Y577 antibody (Thermo Fisher).
In vitro kinase assays
An enzyme‐coupled spectrophotometric assay was used to determine ATP turnover of FAK proteins as described previously (Lietha et al, 2007). In brief, reactions were performed with 1 μM kinase, 1 mM phosphoenolpyruvate, 0.25 mM NADH, 0.08 units/l pyruvate kinase, 0.1 units/l lactate dehydrogenase, and 0.5 mM E4Y (as polyGlu‐Tyr, 4:1 Glu/Tyr; Sigma) in the presence of 125 μM PC or PI(4,5)P2 vesicles. Reactions were initiated by adding 1 mM ATP/5 mM MgCl2, and NADH depletion was monitored by continuous absorption reading at 340 nm.
In vitro PI(4,5)P2‐binding assay
Lipid‐binding assays were performed by pull‐down assays using lipid‐coated silica beads. PC and PI(4,5)P2 vesicles were generated as described above, with the inclusion of 10% (mol/mol) dansyl‐phosphatidylethanolamine (Avanti Polar Lipids) for quantification of lipid coating. Silica beads with 0.61 um diameter (Spherotech) were coated at 0.05 μmol lipids/μg beads by vortexing 2 min and incubation at 45°C on a thermomixer at 600 rpm for 30 min and removal of unbound lipids by extensive washes. Attached lipids were quantified on a small aliquot by removal of lipids with 1% SDS and measuring fluorescence of the dansyl dye (excitation: 336 nm, emission: 513 nm). For pull‐down assays, coated beads containing 1.1 mM total lipid (for PI(4,5)P2‐coated beads: 110 μM PI(4,5)P2 on outer leaflet), or twofold dilutions thereof, were incubated with 4 μM FAK proteins (WT or mutants) for 2 h at 4°C, centrifuged at 1,500 g for 60s and beads washed three times with binding buffer: 20 mM HEPES pH 7.5, 150 mM NaCl, and 1 mM TCEP. Bound proteins were removed from beads with buffer containing 1% SDS and protein amounts determined using a BCA assay (Thermo Scientific).
Dynamic light scattering
Dynamic light scattering (DLS) experiments were carried out at the Molecular Interactions Facility at the CIB Margarita Salas. The experiments were performed using a Protein Solutions DynaPro MS/X instrument (Protein Solutions) at 20°C and a 90° light scattering cuvette. The samples were centrifuged during 10 min at 12,000 g and 20°C immediately before measurements. Data were collected and analyzed with the Dynamics V6 Software.
CD spectroscopy
CD spectroscopy (CD) spectra were collected on a Jasco J715 instrument between 250 and 200 nm at a protein concentration of 2.5 μM in 20 mM Tris pH 8.0, 100 mM NaCl, 5% glycerol at 20°C. Ellipticities corrected for the buffer contribution were converted to mean residue ellipticities using a mean molecular mass per residue of (M w/N aa).
Antibodies for cell biology
Antibodies used were as follows: anti‐Paxillin (BD Transduction Laboratories, NJ, USA), anti‐FAK, anti‐pFAK Y397, anti‐pFAK Y576/577, anti‐pFAK Y925, anti‐Src, anti‐pSrc Y416, anti‐α‐Tubulin, anti‐RCAS, anti‐H4, and anti‐GAPDH (Cell Signaling Technologies, Danvers, MA, USA), as well as anti‐pFAK Y861 (Invitrogen, Paisley, UK). Anti‐rabbit or mouse peroxidase‐conjugated secondary antibodies were purchased from Cell Signaling Technologies.
Cell culture
Focal adhesion kinase‐depleted SCC cell lines were generated as described previously (Serrels et al, 2010). Briefly, SCCs were cultured in Glasgow MEM containing 10% FCS, 2 mM l‐glutamine, non‐essential amino acids, sodium pyruvate and MEM vitamins at 37°C, 5% CO2. SCC FAK‐WT and FAK mutant cells were maintained in 1 mg/ml hygromycin B.
Whole cell lysates
Cells were washed twice in ice‐cold PBS and then lysed in RIPA buffer (50 mM Tris‐HCl pH 8.0, 150 mM NaCl, 1% Triton X‐100, 0.1% SDS, and 0.5% sodium deoxycholate) supplemented with PhosStop and Complete Ultra Protease Inhibitor tablets (Roche, Welwyn Garden City, UK). Lysates were cleared by centrifugation.
Perinuclear fractionation
The protocol was adapted and optimized from Shaiken and Opekun (2014). All buffers were supplemented with PhosStop and Complete Ultra Protease Inhibitor tablets (Roche, Welwyn Garden City, UK). Cells were washed twice in ice‐cold PBS and then lysed in buffer A (20 mM Tris‐HCl pH 7.5, 1 mM MgCl2, 1 mM EGTA, 0.03% NP‐40), incubated for 5 min at 4°C with rotation and then cleared by centrifugation at 800 g for 4 min. NP‐40 was added to the supernatants (cytoplasmic fraction) to give a final concentration of 1% and lysates were cleared by centrifugation. Pellets were washed once with NP‐40 containing buffer A and once with buffer A lacking NP‐40. The washed pellets were resuspended in Buffer B (10 mM Tris‐HCl pH 7.4, 1 mM KCl, 2.5 mM MgCl2, 0.2 M LiCl, 0.1% Triton X‐100, 0.1% sodium deoxycholate), incubated for 15 min at 4°C with rotation and then cleared by centrifugation at 2,000 g for 5 min. The supernatants (perinuclear fraction) were cleared by centrifugation. Pellets were washed once in buffer B, resuspended in RIPA buffer, sonicated, and cleared by centrifugation (nuclear fraction).
Immunoblotting and immunoprecipitation
Protein concentrations were calculated using a BCA protein assay kit (Thermo Scientific, Loughborough, UK). For immunoprecipitation, 1 mg cell lysate was incubated with 2 μg of unconjugated anti‐pSrc Y416 at 4°C overnight with agitation or with 10 μl of agarose coupled anti‐FAK or anti‐Src (Millipore, Billerica, MA, USA). Unconjugated antibody samples were incubated with 10 μl of Protein A agarose for 1 h at 4°C. Beads were washed three times in lysis buffer and once in 0.6 M LiCl (for pSrc Y416‐IP) or twice in lysis buffer and twice in PBS (for FAK‐ and Src‐IPs), resuspended in 20 μl 2× sample buffer (130 mM Tris pH 6.8, 20% glycerol, 5% SDS, 8% β‐mercaptoethanol, bromphenol blue) and heated for 5 min at 95°C. Samples were then subjected to SDS‐PAGE analysis using the Bio‐Rad TGX pre‐cast gel system. Immunoblotting of the proteins occurred using the Bio‐Rad Trans‐blot Turbo transfer system, blocked in 5% BSA/TBS supplemented with 1% Tween‐20, and incubated with primary antibodies overnight at 4°C. Blots were washed three times in TBST, incubated with peroxidase‐conjugated secondary antibody for 45 min at room temperature and washed as before. The blots were developed using Clarity Western ECL Substrate (Bio‐Rad, Hemel Hempstead, UK) and imaged using a Bio‐Rad ChemiDoc MP Imaging System (Bio‐Rad, Hemel Hempstead, UK).
Immunofluorescence microscopy and image analysis
Cells were grown on glass coverslips for 24 h and washed once in TBS prior to fixation (3.7% formaldehyde, 100 mM PIPES pH 6.8, 10 mM EGTA, 1 mM MgCl2, and 0.2% Triton X‐100) for 10 min. The fixed cells were washed twice in TBStx (TBS supplemented with 0.1% Triton X‐100) and blocked in TBStx block (TBStx supplemented with 3% BSA). Primary antibodies were incubated in TBStx block over night at 4°C, followed by three washes in TBStx, incubated with Alexa‐fluor labeled secondary antibodies diluted 1:200 in TBStx block (Life Technologies, Paisley, UK), and washed as before. Samples were mounted in vectashield mounting medium containing DAPI (Vector Labs, Peterborough, UK) and imaged using a Nikon A1+ confocal microscope (Nikon, Amsterdam, the Netherlands).
Invasion assays
Invasion was analyzed as described previously in (Serrels et al, 2010). Briefly, growth factor‐reduced Matrigel (BD Biosciences, Oxford, UK) was diluted 1:1 in ice‐cold PBS and allowed to set at 37°C in transwells. 2 × 104 cells were seeded onto the underside of the transwell. After 4 h, the transwells were washed in PBS and placed into serum‐free SCC growth medium with full growth medium containing 10% FCS on top of the Matrigel. After 72 h, cell invasion was evaluated by staining with 5 μM calcein (Life Technologies, Paisley, UK) for 1 h. Horizontal z sections through the Matrigel were acquired at 10 μm intervals with an Nikon A1+ confocal microscope and analyzed using ImageJ software.
3D proliferation assay
5 × 105 cells were resuspended in a 1.4% methylcellulose solution in growth medium, plated on a layer of 0.9% agarose and incubated at 37°C, 5% CO2. After 9 days, images were taken from 10 random fields (10× magnification). Colonies were counted and the colony size measured using ImageJ software.
Statistical tests
For all experiments shown, n = 3. Error bars for the graphs show s.d. Student's t‐test was carried out to calculate the statistical significance. *P < 0.01, # P < 0.05.
Author contributions
IA and PR cloned, expressed, and purified FAK variants and Src proteins; IA, PR, and DL performed their biochemical characterization. IA and JB formed, optimized, and imaged FAK 2D crystals by negative stain EM; IA, MC, and HS optimized and performed cryo‐EM data collection. IA, RDR, NB, and HS processed cryo‐EM data and RDR performed single‐particle analysis from 2D crystal images and flexible model fitting. CFR ran EM map restrained MD simulations; SdB, CD, and FG performed and analyzed MD simulations of FAK on the membrane. CS and JC performed cell biology experiments and together with AB and MCF analyzed the results. IA and DL wrote the first draft of the paper; all authors read and edited the paper. OL, FG, JB, MCF, HS, and DL coordinated the joint project; OL, FG, MCF, HS, and DL provided funding.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
Cryo‐EM data processing calculations were performed at sciCORE (http://scicore.unibas.ch/) scientific computing center at the University of Basel. We thank Ana Herrero from the CRUK Edinburgh Center/IGMM for helping to optimize the perinuclear fractionation protocol in SCC cells as well as Ruth Matesanz Rodríguez and Juan Roman Luque Ortega from the CIB Margarita Salas (CIB‐CSIC) for help with collecting CD spectra and DLS data, respectively. I.A. acknowledges support from the José Castillejo Mobility Grant issued by the Spanish Ministry of Education, Culture and Sports. R.D.R. acknowledges funding from the Fellowships for Excellence program sponsored by the Werner‐Siemens Foundation and the University of Basel. H.S. acknowledges support by the Swiss National Science Foundation (grants 205320_166164 and 185544 (NCCR TransCure)). M.C.F., A.B., J.C., and C.S. were supported by a Cancer Research UK Programme Grant (C157/A24837) to M.C.F. F.G. acknowledges funding by the Klaus Tschira Foundation, by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – 2082/1 – 390761711, and the state of Baden‐Württemberg through bwHPC and the DFG through grant INST 35/11341 FUGG. S.d.B. thanks the Carl Zeiss Foundation for financial support. D.L. acknowledges support from the Spanish Ministry of Economy, Industry and Competitiveness for the Retos Grant BFU2016‐77665‐R and the Ministry of Science, Innovation and Universities for the Spanish State Research Agency Retos Grant RTI2018‐099318‐B‐I00, both co‐funded by the European Regional Development Fund (FEDER) and from an intramural grant from the Spanish National Research Council (CSIC, Ref: 201820I124). O.L. is funded by SAF2017‐82632‐P grant from the Spanish Ministry of Science, Innovation and Universities (MCIU/AEI), co‐funded by the European Regional Development Fund (ERDF), by the National Institute of Health Carlos III and by projects Y2018/BIO4747 and P2018/NMT4443 from the Autonomous Region of Madrid and co‐funded by the European Social Fund and the European Regional Development Fund. C.F.R. is funded by a BES‐2015‐071348 PhD fellowship by the Spanish Ministry of Science, Innovation and Universities (MCIU/AEI).
The EMBO Journal (2020) 39: e104743
See also: KM McAndrews & R Kalluri (October 2020)
Contributor Information
Margaret C Frame, Email: m.frame@ed.ac.uk.
Henning Stahlberg, Email: henning.stahlberg@unibas.ch.
Daniel Lietha, Email: daniel.lietha@cib.csic.es.
Data availability
The final structures and cryo‐EM maps as well as raw data images are available in the following databases:
Protein Data Bank accession codes 6TY3 (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TY3; Apo) and 6TY4 (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TY4; AMP‐PNP)
Electron Microscopy Data Bank accession codes EMD‐10615 (http://www.ebi.ac.uk/pdbe/entry/EMD-10615; apo) and EMD‐10616 (http://www.ebi.ac.uk/pdbe/entry/EMD-10616; AMP‐PNP)
Electron Microscopy Public Image Archive accession code EMPIAR‐10347 https://www.ebi.ac.uk/pdbe/emdb/empiar/entry/10347/)
References
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, Adams PD (2018) Realspace refinement in PHENIX for cryo‐EM and crystallography. Acta Crystallogr D Struct Biol 74: 531–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arold ST, Hoellerer MK, Noble ME (2002) The structural basis of localization and signaling by the focal adhesion targeting domain. Structure 10: 319–327 [DOI] [PubMed] [Google Scholar]
- Ashton GH, Morton JP, Myant K, Phesse TJ, Ridgway RA, Marsh V, Wilkins JA, Athineos D, Muncan V, Kemp R et al (2010) Focal adhesion kinase is required for intestinal regeneration and tumorigenesis downstream of Wnt/c‐Myc signaling. Dev Cell 19: 259–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer MS, Baumann F, Daday C, Redondo P, Durner E, Jobst MA, Milles LF, Mercadante D, Pippig DA, Gaub HE et al (2019) Structural and mechanistic insights into mechanoactivation of focal adhesion kinase. Proc Natl Acad Sci USA 116: 6766–6774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benschop RJ, Melamed D, Nemazee D, Cambier JC (1999) Distinct signal thresholds for the unique antigen receptor‐linked gene expression programs in mature and immature B cells. J Exp Med 190: 749–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrow NS, Alderton D, Sainsbury S, Nettleship J, Assenberg R, Rahman N, Stuart DI, Owens RJ (2007) A versatile ligation‐independent cloning method suitable for high‐throughput expression screening applications. Nucleic Acids Res 35: e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, Mackerell AD Jr (2012) Optimization of the additive CHARMM all‐atom protein force field targeting improved sampling of the backbone phi, psi and side‐chain chi(1) and chi(2) dihedral angles. J Chem Theory Comput 8: 3257–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biyani N, Righetto RD, McLeod R, Caujolle‐Bert D, Castano‐Diez D, Goldie KN, Stahlberg H (2017) Focus: the interface between data collection and data processing in cryo‐EM. J Struct Biol 198: 124–133 [DOI] [PubMed] [Google Scholar]
- Brami‐Cherrier K, Gervasi N, Arsenieva D, Walkiewicz K, Boutterin MC, Ortega A, Leonard PG, Seantier B, Gasmi L, Bouceba T et al (2014) FAK dimerization controls its kinase‐dependent functions at focal adhesions. EMBO J 33: 356–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Lietha D, Ceccarelli DF, Karginov AV, Rajfur Z, Jacobson K, Hahn KM, Eck MJ, Schaller MD (2008) Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol 28: 201–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calalb MB, Polte TR, Hanks SK (1995) Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol 15: 954–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone G, Heymann JB, Steven AC (2013) One number does not fit all: mapping local variations in resolution in cryo‐EM reconstructions. J Struct Biol 184: 226–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli DF, Song HK, Poy F, Schaller MD, Eck MJ (2006) Crystal structure of the FERM domain of focal adhesion kinase. J Biol Chem 281: 252–259 [DOI] [PubMed] [Google Scholar]
- Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SH, Henderson R (2013) High‐resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135: 24–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinthalapudi K, Mandati V, Zheng J, Sharff AJ, Bricogne G, Griffin PR, Kissil J, Izard T (2018) Lipid binding promotes the open conformation and tumor‐suppressive activity of neurofibromin 2. Nat Commun 9: 1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper LA, Shen TL, Guan JL (2003) Regulation of focal adhesion kinase by its amino‐terminal domain through an autoinhibitory interaction. Mol Cell Biol 23: 8030–8041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, Pellegrini L, Letinic K, Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR, De Camilli P (2002) Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature 420: 85–89 [DOI] [PubMed] [Google Scholar]
- Diaz Osterman CJ, Ozmadenci D, Kleinschmidt EG, Taylor KN, Barrie AM, Jiang S, Bean LM, Sulzmaier FJ, Jean C, Tancioni I et al (2019) FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. Elife 8: e47327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunty JM, Gabarra‐Niecko V, King ML, Ceccarelli DF, Eck MJ, Schaller MD (2004) FERM domain interaction promotes FAK signaling. Mol Cell Biol 24: 5353–5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Frame MC, Patel H, Serrels B, Lietha D, Eck MJ (2010) The FERM domain: organizing the structure and function of FAK. Nat Rev Mol Cell Biol 11: 802–814 [DOI] [PubMed] [Google Scholar]
- Gao G, Prutzman KC, King ML, Scheswohl DM, DeRose EF, London RE, Schaller MD, Campbell SL (2004) NMR solution structure of the focal adhesion targeting domain of focal adhesion kinase in complex with a paxillin LD peptide: evidence for a two‐site binding model. J Biol Chem 279: 8441–8451 [DOI] [PubMed] [Google Scholar]
- Gipson B, Zeng X, Zhang ZY, Stahlberg H (2007) 2dx–user‐friendly image processing for 2D crystals. J Struct Biol 157: 64–72 [DOI] [PubMed] [Google Scholar]
- Goni GM, Epifano C, Boskovic J, Camacho‐Artacho M, Zhou J, Bronowska A, Martin MT, Eck MJ, Kremer L, Grater F et al (2014) Phosphatidylinositol 4,5‐bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc Natl Acad Sci USA 111: E3177–E3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk RA, Martins AJ, Angermann BR, Dutta B, Ng CE, Uderhardt S, Tsang JS, Fraser ID, Meier‐Schellersheim M, Germain RN (2016) Distinct NF‐kappaB and MAPK activation thresholds uncouple steady‐state microbe sensing from anti‐pathogen inflammatory responses. Cell Syst 2: 378–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A, Grigorieff N (2018) cisTEM, user‐friendly software for single‐particle image processing. Elife 7: e35383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JE, Schaller MD (2017) Phospholipid binding to the FAK catalytic domain impacts function. PLoS ONE 12: e0172136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada K, Shimizu T, Matsui T, Tsukita S, Hakoshima T (2000) Structural basis of the membrane‐targeting and unmasking mechanisms of the radixin FERM domain. EMBO J 19: 4449–4462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauz G, van Heel M (1986) Exact filters for general geometry three dimensional reconstruction. Optik 73: 146–156 [Google Scholar]
- Hayashi I, Vuori K, Liddington RC (2002) The focal adhesion targeting (FAT) region of focal adhesion kinase is a four‐helix bundle that binds paxillin. Nat Struct Biol 9: 101–106 [DOI] [PubMed] [Google Scholar]
- He S, Scheres SHW (2017) Helical reconstruction in RELION. J Struct Biol 198: 163–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog FA, Braun L, Schoen I, Vogel V (2017) Structural insights How PIP2 imposes preferred binding orientations of FAK at lipid membranes. J Phys Chem B 121: 3523–3535 [DOI] [PubMed] [Google Scholar]
- Heymann JB, Belnap DM (2007) Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol 157: 3–18 [DOI] [PubMed] [Google Scholar]
- Hirata E, Girotti MR, Viros A, Hooper S, Spencer‐Dene B, Matsuda M, Larkin J, Marais R, Sahai E (2015) Intravital imaging reveals how BRAF inhibition generates drug‐tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 27: 574–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton ER, Byron A, Askari JA, Ng DHJ, Millon‐Fremillon A, Robertson J, Koper EJ, Paul NR, Warwood S, Knight D et al (2015) Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol 17: 1577–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, MacKerell AD Jr (2013) CHARMM36 all‐atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 34: 2135–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard SR (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J 16: 5572–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T (1995) Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK‐deficient mice. Nature 377: 539–544 [DOI] [PubMed] [Google Scholar]
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT et al (2016) Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22: 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Im W (2007) Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2: e880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W (2008) CHARMM‐GUI: a web‐based graphical user interface for CHARMM. J Comput Chem 29: 1859–1865 [DOI] [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM (2010) Nanoscale architecture of integrin‐based cell adhesions. Nature 468: 580–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, Mondragon‐Ramirez C, Vorobyov I, MacKerell AD Jr, Pastor RW (2010) Update of the CHARMM all‐atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114: 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Colman PM (1993) Shape complementarity at protein/protein interfaces. J Mol Biol 234: 946–950 [DOI] [PubMed] [Google Scholar]
- Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y et al (2016) CHARMM‐GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput 12: 405–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ (2007) Structural basis for the autoinhibition of focal adhesion kinase. Cell 129: 1177–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, Larocque N, Fisher SJ, Schlaepfer DD, Ilic D (2008) Nuclear FAK promotes cell proliferation and survival through FERM‐enhanced p53 degradation. Mol Cell 29: 9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling K, Doughman RL, Firestone AJ, Bunce MW, Anderson RA (2002) Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature 420: 89–93 [DOI] [PubMed] [Google Scholar]
- Lopez‐Blanco JR, Chacon P (2013) iMODFIT: efficient and robust flexible fitting based on vibrational analysis in internal coordinates. J Struct Biol 184: 261–270 [DOI] [PubMed] [Google Scholar]
- Lv PC, Jiang AQ, Zhang WM, Zhu HL (2018) FAK inhibitors in cancer, a patent review. Expert Opin Ther Pat 28: 139–145 [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (2005) Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152: 36–51 [DOI] [PubMed] [Google Scholar]
- McLean GW, Komiyama NH, Serrels B, Asano H, Reynolds L, Conti F, Hodivala‐Dilke K, Metzger D, Chambon P, Grant SG et al (2004) Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev 18: 2998–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercadante D, Grater F, Daday C (2018) CONAN: a tool to decode dynamical information from molecular interaction maps. Biophys J 114: 1267–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N (2003) Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142: 334–347 [DOI] [PubMed] [Google Scholar]
- Moore DT, Nygren P, Jo H, Boesze‐Battaglia K, Bennett JS, DeGrado WF (2012) Affinity of talin‐1 for the beta3‐integrin cytosolic domain is modulated by its phospholipid bilayer environment. Proc Natl Acad Sci USA 109: 793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Keely PJ (2009) The role of focal adhesion kinase in tumor initiation and progression. Cell Adh Migr 3: 347–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA (2017) cryoSPARC: algorithms for rapid unsupervised cryo‐EM structure determination. Nat Methods 14: 290–296 [DOI] [PubMed] [Google Scholar]
- Ransom RC, Carter AC, Salhotra A, Leavitt T, Marecic O, Murphy MP, Lopez ML, Wei Y, Marshall CD, Shen EZ et al (2018) Mechanoresponsive stem cells acquire neural crest fate in jaw regeneration. Nature 563: 514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Righetto RD, Biyani N, Kowal J, Chami M, Stahlberg H (2019) Retrieving high‐resolution information from disordered 2D crystals by single‐particle cryo‐EM. Nat Commun 10: 1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N (2015) CTFFIND4: fast and accurate defocus estimation from electron micrographs. J Struct Biol 192: 216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PB, Henderson R (2003) Optimal determination of particle orientation, absolute hand, and contrast loss in single‐particle electron cryomicroscopy. J Mol Biol 333: 721–745 [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815 [DOI] [PubMed] [Google Scholar]
- Saltel F, Mortier E, Hytonen VP, Jacquier MC, Zimmermann P, Vogel V, Liu W, Wehrle‐Haller B (2009) New PI(4,5)P2‐ and membrane proximal integrin‐binding motifs in the talin head control beta3‐integrin clustering. J Cell Biol 187: 715–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K, Stevens C, Brunton VG, Langdon WY, Vidal M et al (2011) Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol 14: 51–60 [DOI] [PubMed] [Google Scholar]
- Seeliger MA, Young M, Henderson MN, Pellicena P, King DS, Falick AM, Kuriyan J (2005) High yield bacterial expression of active c‐Abl and c‐Src tyrosine kinases. Protein Sci 14: 3135–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong J, Tajik A, Sun J, Guan JL, Humphries MJ, Craig SE, Shekaran A, Garcia AJ, Lu S, Lin MZ et al (2013) Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc Natl Acad Sci USA 110: 19372–19377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrels B, Sandilands E, Serrels A, Baillie G, Houslay MD, Brunton VG, Canel M, Machesky LM, Anderson KI, Frame MC (2010) A complex between FAK, RACK1, and PDE4D5 controls spreading initiation and cancer cell polarity. Curr Biol 20: 1086–1092 [DOI] [PubMed] [Google Scholar]
- Shaiken TE, Opekun AR (2014) Dissecting the cell to nucleus, perinucleus and cytosol. Sci Rep 4: 4923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzmaier FJ, Jean C, Schlaepfer DD (2014) FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer 14: 598–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Guo SS, Fassler R (2016) Integrin‐mediated mechanotransduction. J Cell Biol 215: 445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavora B, Reynolds LE, Batista S, Demircioglu F, Fernandez I, Lechertier T, Lees DM, Wong PP, Alexopoulou A, Elia G et al (2014) Endothelial‐cell FAK targeting sensitizes tumours to DNA‐damaging therapy. Nature 514: 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro‐Tapia G, Villaseca S, Beyer A, Roycroft A, Marcellini S, Mayor R, Torrejon M (2018) The Ric‐8A/Galpha13/FAK signalling cascade controls focal adhesion formation during neural crest cell migration in Xenopus. Development 145: dev164269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torsoni AS, Constancio SS, Nadruz W Jr, Hanks SK, Franchini KG (2003) Focal adhesion kinase is activated and mediates the early hypertrophic response to stretch in cardiac myocytes. Circ Res 93: 140–147 [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, flexible, and free. J Comput Chem 26: 1701–1718 [DOI] [PubMed] [Google Scholar]
- Walkiewicz KW, Girault JA, Arold ST (2015) How to awaken your nanomachines: site‐specific activation of focal adhesion kinases through ligand interactions. Prog Biophys Mol Biol 119: 60–71 [DOI] [PubMed] [Google Scholar]
- Wang HB, Dembo M, Hanks SK, Wang Y (2001) Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc Natl Acad Sci USA 98: 11295–11300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L et al (2018) SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46: W296–W303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrle‐Haller B (2012) Assembly and disassembly of cell matrix adhesions. Curr Opin Cell Biol 24: 569–581 [DOI] [PubMed] [Google Scholar]
- Winograd‐Katz SE, Fassler R, Geiger B, Legate KR (2014) The integrin adhesome: from genes and proteins to human disease. Nat Rev Mol Cell Biol 15: 273–288 [DOI] [PubMed] [Google Scholar]
- Wong VW, Rustad KC, Akaishi S, Sorkin M, Glotzbach JP, Januszyk M, Nelson ER, Levi K, Paterno J, Vial IN et al (2011) Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat Med 18: 148–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Subramaniam S, Case DA, Wu KW, Brooks BR (2013) Targeted conformational search with map‐restrained self‐guided Langevin dynamics: application to flexible fitting into electron microscopic density maps. J Struct Biol 183: 429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila‐Contreras EM, Qi Y, Lee J, Monje‐Galvan V, Venable RM et al (2014) CHARMM‐GUI membrane builder toward realistic biological membrane simulations. J Comput Chem 35: 1997–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel‐Bar R, Itzkovitz S, Ma'ayan A, Iyengar R, Geiger B (2007) Functional atlas of the integrin adhesome. Nat Cell Biol 9: 858–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Cordon‐Barris L, Zurashvili T, Bayascas JR (2014) Fine‐tuning the intensity of the PKB/Akt signal enables diverse physiological responses. Cell Cycle 13: 3164–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The final structures and cryo‐EM maps as well as raw data images are available in the following databases:
Protein Data Bank accession codes 6TY3 (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TY3; Apo) and 6TY4 (http://www.rcsb.org/pdb/explore/explore.do?structureId=6TY4; AMP‐PNP)
Electron Microscopy Data Bank accession codes EMD‐10615 (http://www.ebi.ac.uk/pdbe/entry/EMD-10615; apo) and EMD‐10616 (http://www.ebi.ac.uk/pdbe/entry/EMD-10616; AMP‐PNP)
Electron Microscopy Public Image Archive accession code EMPIAR‐10347 https://www.ebi.ac.uk/pdbe/emdb/empiar/entry/10347/)