

Abstract
Inference of causality between gene expression and complex traits using Mendelian randomization (MR) is confounded by pleiotropy and linkage disequilibrium (LD) of gene-expression quantitative trait loci (eQTL). Here, we propose an MR method, MR-link, that accounts for unobserved pleiotropy and LD by leveraging information from individual-level data, even when only one eQTL variant is present. In simulations, MR-link shows false-positive rates close to expectation (median 0.05) and high power (up to 0.89), outperforming all other tested MR methods and coloc. Application of MR-link to low-density lipoprotein cholesterol (LDL-C) measurements in 12,449 individuals with expression and protein QTL summary statistics from blood and liver identifies 25 genes causally linked to LDL-C. These include the known SORT1 and ApoE genes as well as PVRL2, located in the APOE locus, for which a causal role in liver was not known. Our results showcase the strength of MR-link for transcriptome-wide causal inferences.
Subject terms: Statistical methods, Gene expression, Genomics
Mendelian randomization is a useful tool to infer causal relationships between traits, but can be confounded by the presence of pleiotropy. Here, the authors have developed MR-link, a Mendelian randomization method which accounts for unobserved pleiotropy and linkage disequilibrium between instrumental variables.
Introduction
Mendelian randomization (MR) is a method that can infer causal relationships between two heritable complex traits from observational studies1,2. In recent years, MR has gained popularity in the epidemiological field and its application has provided valuable insights into the risk factors that cause diseases and complex traits1–3. MR studies have, for example, successfully identified causal relationships between low-density lipoprotein cholesterol (LDL-C) and coronary artery disease, in turn informing therapeutic strategies4,5. MR studies have also shown that a causal relationship between high-density lipoprotein cholesterol (HDL-C) and coronary artery disease is unlikely, which is in contrast to previous epidemiological associations6. The same approach has been applied to identify molecular marks that are causal to disease7–10. Since gene expression is one of these marks, investigating its causal role in complex traits is of particular interest given that complex trait loci are enriched for expression quantitative trait loci (eQTLs)11.
MR infers a causal relationship between an exposure (e.g., a risk factor) and an outcome (e.g., a complex trait) by leveraging QTL variants of the exposure as instrumental variables (IVs). The mathematical model behind MR relies on three main assumptions to correctly infer causality: the IVs have to be (i) associated with the exposure, (ii) independent of any confounder of the exposure-outcome association, and (iii) conditionally independent of the outcome given the exposure and confounders. One major challenge of applying MR to gene expression is correcting for deviations from the third assumption, which can occur in the presence of linkage disequilibrium (LD) between the eQTL variants used as IVs, or in the presence of pleiotropy, i.e., when IVs affect the outcome through pathways other than the exposure of interest. Accounting for LD is necessary when gene expression is the exposure trait in MR because, in contrast to the majority of complex traits, the genetic architecture of gene expression is characterized by the presence of strong-acting eQTLs located proximal to their transcript (in cis), which are often correlated through LD12,13. On top of this, the presence of pleiotropy cannot be excluded a priori given that the majority of variants in our genome are likely to affect one or multiple phenotypes14–16. There are MR methods7,17–21 that extend standard MR analysis to correct for LD and pleiotropy, however, the application of these methods is not optimal because they require either the removal of pleiotropic IVs from the statistical model7,19,20, that all sources of pleiotropy are measured and incorporated into the model22,23, or that both the exposure and the outcome are measured in the same cohort21. These constraints limit robust inference of gene-expression traits as there are often only a limited number of IVs (i.e., eQTL variants) available, and subsequent removal of outliers will substantially reduce power. Likewise, it is not always possible to measure all sources of pleiotropy because it could come from expression of a gene in a different tissue or even from other unobserved molecular marks or phenotypes.
Here we introduce MR-link, an MR method that allows for causal inference in the presence of LD and an unobserved pleiotropic effect, without requiring the removal of pleiotropic IVs or measuring all sources of pleiotropy. MR-link uses summary statistics of an exposure combined with individual-level data on the outcome to estimate the causal effect of an exposure from IVs (i.e., eQTLs if the exposure is gene expression), while at the same time correcting for pleiotropic effects using genetic variants that are in LD with these IVs (cis-genetics) (Fig. 1).
Fig. 1. Graphical representation of the study.

The Biobank Integrative Omics Study (BIOS) cohort was used to identify expression quantitative trait loci (eQTLs) and characterize the genetic architecture of gene expression. Dashed outbox: Knowledge used in a simulation scheme that mimicked gene-expression traits, including linkage disequilibrium (LD) between eQTL single nucleotide polymorphism (SNPs). We used this simulation to assess the false positive rates and power for widely used Mendelian randomization (MR) methods. We applied our MR method, MR-link, to both the simulations and to individual-level data of low-density lipoprotein cholesterol (LDL-C) in 12,449 individuals (Lifelines) combined with BIOS and GTEx eQTL as well as protein quantitative trait loci (pQTL) summary statistics to identify gene-expression changes and protein level changes that are causally linked to LDL-C within or outside a genome-wide association study (GWAS) locus.
We assess the performance of MR-link using simulated data in 100 different scenarios that mimic the genetic architecture of gene expression. We derive this information from eQTL association patterns in a large cohort of samples with genetic and transcriptomics data13. Subsequently, we apply MR-link to individual-level data for LDL-C measurements in 12,449 individuals with four different eQTL summary statistic datasets: blood eQTLs identified in the BIOS cohort (Fig. 1) and eQTLs from blood, liver, and cerebellum from the GTEx Consortium24 (Fig. 1). We further explore the performance of MR-link on another molecular layer, protein levels, through the application of MR-link on protein quantitative trait loci (pQTL) summary statistics from Sun et al. combined with our LDL-C measurements25. Our results in simulated and real data show that MR-link can robustly identify causal relationships between molecular traits—such as gene expression and protein levels—and an outcome (e.g., a complex trait), even when the information for causal inference is very limited.
Results
eQTL variants between different genes are often in LD
In a standard MR analysis, IVs need to be independent (not in LD) and have to affect the outcome only through the exposure (absence of pleiotropy). Even in absence of pleiotropy, correlated IVs in the cis locus may negatively influence an MR analysis (Fig. 2a). In the presence of pleiotropy, we distinguish two scenarios: (i) pleiotropic variants that are in LD with an IV (pleiotropy through LD, Fig. 2b) and (ii) when the IV and the pleiotropic variant are the same and affect the outcome through two distinct mechanisms (pleiotropy through overlap Fig. 2c). If pleiotropy through LD is prevalent, genetic variants in the cis-region other than those selected as IVs can be used to explain the pleiotropic effects. Incorporating these variants in an MR model can then account for this pleiotropy through LD (Fig. 2b).
Fig. 2. Typical scenarios of pleiotropy in causal inference of gene expression changes as an exposure.

Typical scenarios to consider when performing causal inference in gene expression: a expression quantitative trait locus (eQTL) single nucleotide polymorphisms (SNPs) used as instrumental variables (IVs) for the same gene (exposure) are in linkage disequilibrium (LD) and pleiotropic effects are absent, b pleiotropy is present through LD between IVs for different exposures (pleiotropy through LD), and c pleiotropy is present through overlap of the IVs (pleiotropy through overlap). In each panel, the left image shows the genomic context while the right image is a schematic diagram of the corresponding causal effects. Please note that the unobserved exposure trait does not necessarily need to be a protein product: it could be any measured or unmeasured phenotype that is regulated by the genetic locus.
We investigated how often pleiotropy through LD occurs in gene expression by looking at how frequently eQTL variants are shared between genes in cis. Using data from the BIOS Consortium, a cohort of 3503 Dutch individuals whose genome and whole-blood transcriptome has been characterized (Fig. 1), we searched for eQTLs located 1.5 megabases (Mb) on both sides of the translated region of 19,960 genes (see “Methods”)13. We then applied a summary statistics-based stepwise linear regression approach (GCTA-COJO) to identify jointly significant variants, e.g., one or more variants that jointly associate significantly with expression changes of a gene26 (“Methods”). We observed that 54% of the genes with an eQTL at p < 5 × 10−8 (13,778 genes) had two or more jointly significant eQTL variants at p < 5 × 10−8 (“Methods”) (Fig. 1 and Fig. 2a). These genetic effects were mostly non-overlapping: only 13.4% of the genes have overlapping (r2 > 0.99) top eQTL variants. In contrast, genetic variants regulating gene expression of a gene were very often in LD with other eQTLs: 40.6% of top variants are in LD (r2 > 0.5) between genes, and this percentage increased to 60.3% if all jointly significant eQTL variants were considered (“Methods”).
To strengthen our inferences on the genetic regulation of gene expression in cis, we performed statistical fine-mapping using FINEMAP v1.3.127 on 13,276 genes (“Methods”). Only 373 (2.8%) genes have full eQTL overlap (all variants in the top configuration of a gene are identical or in high LD (r2 > 0.99)), while 33.2% of the genes have at least one variant in r2 > 0.5 LD with a variant in the top configuration of another gene. These percentages are higher for configurations with larger posterior inclusion probabilities (“Methods”) (Supplementary Data 1), but overall the results are similar to our observations from the GCTA-COJO analysis, i.e., the genetics of gene expression in whole blood is mostly regulated by variants that do not overlap but are in moderate LD with variants associated with gene expression changes of another gene. Based on these results, it seems likely that pleiotropy through LD is more common than pleiotropy through overlap in gene-expression traits.
MR-link outperforms other methods in discriminative ability
We have developed an MR method, MR-link, that uses the genetic region surrounding IVs as a covariate to correct for pleiotropic effects (“Methods”, Fig. 2 and Supplementary Note 1). The model underlying MR-link is informed by the observation that the genetic regulation of gene expression is characterized mostly by eQTLs that are in LD, but not overlapping, between genes. This suggests that the variants in the genetic vicinity of the IVs can be used to correct for pleiotropic effects.
MR-link gathers information from all genetic variants in LD with an IV to jointly model the outcome through the IVs and their genetic vicinity (“Methods”). Compared to other MR methods that require summary statistics of both the exposure and the outcome (two-sample MR), our approach adds a requirement of individual-level data for the outcome, but has the advantage that it can perform causal inference even when only a single IV is available. Strictly speaking, MR-link corrects for pleiotropy under the assumption that pleiotropy can be better explained by variants in LD with the IV (pleiotropy through LD) (Fig. 2b) and that pleiotropy through overlap is absent (Fig. 2c). In the case of a single IV, this assumption needs to be fully accounted for, but when multiple IVs are available, this assumption can be relaxed somewhat. Differences in effect sizes between IVs can be used to distinguish the causal effect of interest from a pleiotropic effect in the same way that multivariable MR corrects for pleiotropy22. Of note, MR-link does not require the source of pleiotropy to be specified in the model; MR-link can account for pleiotropic effects arising from, for instance, gene expression in other tissues or from other molecular layers or phenotypes.
We assessed the performance of MR-link under different scenarios and compared it to four other MR methods: Inverse variance weighting (IVW), which assumes the absence of LD and pleiotropy, and the pleiotropy-robust methods MR-Egger, LDA-MR-Egger, and MR-PRESSO (Table 1)17–19,28. In addition, we compared MR-link to the widely used Bayesian colocalization method coloc29, although this is not a formal test for assessing causal relationships, but rather a way to evaluate if two traits share the same causal variant(s) in a locus29.
Table 1.
MR methods assessed in this study.
| MR method | Data required | Pleiotropy correction | LD correction | Minimum no. of IVs required |
|---|---|---|---|---|
| MR-link | Sumstats: E; ILD: O | Yes | Yes | 1 |
| Inverse variance weighted (IVW)28 | Sumstats: O, E | No | No | 1 |
| LDA-MR-Egger17 | Sumstats: O, E, LDR | Yes | Yes | 3 |
| MR-Egger18 | Sumstats: O, E | Yes | No | 3 |
| MR-PRESSOa,19 | Sumstats: O, E | Yes | No | 3a |
MR methods assessed in our simulation with information about the type of data needed to make a causal estimate, the ability to correct for pleiotropy or linkage disequilibrium, and the minimum number of instrumental variables required.
IVW inverse variance weighting, Sumstats summary statistics, ILD individual-level data, O outcome, E exposure, LDR linkage disequilibrium reference panel. For more details, see “Methods”.
aWe refer here to the MR-PRESSO test that reports estimates after identifying and removing outliers, as the test without outliers generalizes to IVW estimates.
We simulated causal relationships between an exposure and an outcome in a 5 Mb region, based on LD structure estimated for 403 European samples from the 1000 Genomes project30 (“Methods”). All tested MR methods were assessed in 1500 simulated datasets for 100 different scenarios that varied with respect to the absence or presence of causality, the absence or presence of pleiotropy, and the number of causal eQTL variants. We initially evaluated two approaches to select QTL variants as IVs: GCTA-COJO (v1.26.0) and p value clumping (“Methods”)26,31. We observed that GCTA-COJO was best suited for IV selection because: (i) the median number of IVs identified by GCTA-COJO better represented the number of simulated causal variants (Supplementary Data 2) and (ii) the false-positive rates (FPRs) in the MR analysis using the IVW method were lower (median FPR was 0.057 using GCTA-COJO versus 0.115 using clumping) (Supplementary Fig. 1 and Supplementary Data 2). We therefore selected IVs for the exposure using the GTCA-COJO approach in subsequent analyses.
When we simulated pleiotropy through LD with no causal effect of the known exposure on the outcome (Figs. 2b, 3a, Supplementary Data 3 and “Methods”), all existing MR-methods showed inflated FPRs (up to 0.71, 0.15, 0.13, and 0.27 for IVW, MR-Egger, LDA-MR-Egger, and MR-PRESSO, respectively), whereas MR-link presented an FPR close to expectation (median: 0.05, maximum: 0.058). In addition, for LDA-MR-Egger, MR-Egger, and MR-PRESSO, the FPR was undesirably dependent on the number of causal SNPs simulated (Fig. 3a).
Fig. 3. Relative performance of different MR methods.

The figure shows performance of MR methods on simulations representing the pleiotropy through linkage disequilibrium (LD) scenario (depicted in Fig. 2b) when 1, 3, 5, or 10 causal expression quantitative trait locus (eQTL) single nucleotide polymorphisms (SNPs) were simulated (“Methods”). a False positive rates (at alpha = 0.05) in scenarios where no causal relationship is simulated. b Power to detect a small causal effect (at alpha = 0.05). c Power to detect a large causal effect (at alpha = 0.05). Note that MR-link is the only MR method that can adjust for pleiotropy when only one or two instrumental variables are available. MR methods that had fewer than 100 out of 1500 estimates in a scenario are not shown (“Methods”). Extended results, including those that are not shown in the figure, can be found in Supplementary Data 3.
In the scenarios of pleiotropy through LD and non-null causal effects (bE = 0.05, bE = 0.1, bE = 0.2, and bE = 0.4), MR-link has high detection power (up to 0.89) and strongly outperforms all other pleiotropy-robust methods (maximum detected power was 0.28 for MR-Egger, 0.26 for LDA-MR-Egger and 0.65 for MR-PRESSO) (Fig. 3b, c, Supplementary Data 3 and “Methods”). Among all the methods tested, including MR-link, and for all scenarios, IVW had the greatest detection power but also an inflated FPR (minimum FPR: 0.63), making this MR method unreliable in such pleiotropic scenarios (“Methods”).
When we simulated increasing levels of pleiotropy through overlap (Fig. 2c and “Methods”), a situation we expect to be rare in real-world scenarios based on our observation in the BIOS cohort, we observed that all methods including MR-link have increased FPRs (up to 0.22 for MR-link, 0.77 for IVW, 0.10 for LDA-MR-Egger, 0.13 for MR-Egger, and 0.30 for MR-PRESSO) (Supplementary Data 4). Nonetheless, MR-link remains a powerful method when a causal effect is simulated: maximum power was 0.79 for MR-link, 0.98 for IVW, 0.29 for MR-Egger, 0.28 for LDA-MR-Egger, and 0.65 for MR-PRESSO (Supplementary Data 4). Although IVW again had the highest power (0.98) here, the FPR was likewise highly inflated (0.77).
Finally, we compared MR-link to the coloc package using the area under the receiver operator characteristic curve (AUC) metric as well as FPRs and power (calculated using coloc PP4 > 0.9 as a threshold) (“Methods”). We used the AUC metric because coloc provides posterior probabilities of causal variant sharing and not p values (“Methods”). As coloc assumes that the exposure and the outcome share only one causal variant, we also included the recently implemented coloc variations (coloc-cond and coloc-masked) in our comparison. These variations are expected to perform better in scenarios with multiple causal variants32. When comparing MR-link to the coloc variations through the AUC metric, we find that MR-link consistently outperforms coloc and coloc-masked in all scenarios, and coloc-cond in pleiotropic scenarios. In non-pleiotropic scenarios, MR-link and coloc-cond have approximately the same performance (Supplementary Fig. 2 and Supplementary Data 5). As expected, coloc-cond has better discriminative performance compared to the original coloc when multiple causal variants are simulated (Supplementary Fig. 2 and Supplementary Data 5).
To illustrate detection rates in standard coloc settings as they may be used in a real-world analysis, we determined power and FPR for all coloc variations at a PP4 threshold of > 0.9 (Supplementary Fig. 3 and Supplementary Data 6). In the non-pleiotropic case, coloc and coloc-cond have the best detection power (up to 0.79 for coloc and 0.76 for coloc-cond), combined with near zero FPRs (max: 0 for coloc and 0.0006 for coloc-cond) while coloc-masked has lower power (up to 0.40) with a zero FPR (Supplementary Fig. 3a–c) (Supplementary Data 6). In simulations of pleiotropy through LD, all coloc methods have increased FPRs (medians: 0.026 for coloc, 0.142 for coloc-cond, and 0.0037 for coloc-masked) with a decrease in power relative to the non-pleiotropic simulations (max: 0.37 for coloc, 0.43 for coloc-cond, and 0.14 for coloc-masked) (Supplementary Fig. 3d–f and Supplementary Data 6). These patterns were even more apparent in cases of pleiotropy through overlap (Supplementary Fig. 3g–i and Supplementary Data 6). This comparison through FPRs and power indicates again that MR-link has superior discriminative ability over coloc variations, especially in the presence of pleiotropy.
MR-link identifies gene expression causal to LDL-C levels
We applied MR-link to four separate summary statistics-based eQTL datasets combined with individual-level genotype data and LDL-C measurements in 12,449 individuals from the Lifelines cohort33 (Fig. 1). We assessed the causal effect of gene expression changes in (i) whole blood (using eQTLs from BIOS (n = 3503) and GTEx (n = 369)), (ii) liver as the main tissue important for cholesterol metabolism (using eQTLs from GTEx, n = 153), and (iii) cerebellum tissue (using eQTLs from GTEx, n = 154) as a tissue not involved in cholesterol metabolism but with similar sample size (and thus power) to liver tissue24,34.
Transcriptome-wide application of MR-link to these eQTL datasets identified 24 significant genes whose variation in blood (18 using BIOS eQTLs, 2 using GTEx eQTLs) or liver (4 genes) was causally related to LDL-C (Tables 2, 3, Supplementary Tables 1 and 2). No significant genes were found in the cerebellum (Supplementary Table 2).
Table 2.
MR-link results using BIOS blood eQTLs.
| Gene name | Causal effect | p Value | #IVs | Biological function and link to LDL-C |
|---|---|---|---|---|
| IGLC5 | −0.05313 | 2.08E−09 | 1 | Immunoglobulin lambda constant 5 (IGLC5) is a pseudogene; three other genes in the same locus and belonging to the same family appear in this table (IGLV-70, IGLV4-69, and IGLC6). IGLC5 does not have a known function in LDL-C metabolism |
| KB-1460A1.5 | 0.15354 | 1.35E−08 | 1 | KB-1460A1.5 is an RNA gene with unknown function in LDL-C metabolism |
| ABO | −0.08224 | 4.84E−08 | 4 | Alpha 1-3-N-Acetylgalactosaminyltransferase And Alpha 1-3-Galactosyltransferase (ABO) is a protein-coding blood group gene. ABO is located in LDL-C GWAS locus35,36,72. ABO is part of the KEGG pathway Glycosphingolipid biosynthesis although the direct link between ABO and LDL-C remains unclear. |
| UNC5B | −0.01235 | 4.87E−08 | 1 | Unc-5 Netrin Receptor B (UNC5B) is a protein-coding gene and a receptor for the NETRIN1 protein. It is associated to the disease Hyperinsulinemic Hypoglycemia familial 3. UNC5B is localized in lipid rafts, membrane compartments that contain high levels of cholesterol and lipids73. The direct link of this gene to LDL-C remains unclear. |
| TMEM176B | −0.0287 | 1.11E−07 | 4 | Transmembrane protein 176B (TMEM176B) is a protein-coding gene located in the TMEM176B-TMEM176A-AOC1 GWAS locus for HDL-C35. Two IVs for TMEM176B are overlapping with IVs for TMEM176A. |
| REEP1 | −0.02183 | 1.25E−07 | 1 | Receptor Accessory Protein 1 (REEP1) is a protein-coding gene. Mutations of the N-terminal of REEP1 lead to accumulation in lipid droplets in the endoplasmic reticulum74. REEP1 does not have a known function in LDL-C metabolism. |
| KRT79 | −0.05904 | 1.54E−07 | 2 | Keratin 79 (KRT79) is a protein-coding gene, that promotes sebaceous gland maintenance in mice hair follicles75. The sebaceous gland produces up to 90% of the lipids present in the epidermis. Although a direct link to LDL-C levels remains unclear. |
| IGLC6 | −0.07662 | 2.21E−07 | 3 | Immunoglobulin lambda constant 6 (IGLC6) is a pseudogene; three other genes in the same locus belonging to the same family appear in this table (IGLV-70, IGLV4-69, and IGLC5). IGLC6 does not have a known function in LDL-C metabolism. |
| MAP1LC3A | 0.037236 | 2.32E−07 | 1 | Microtubule Associated Protein 1 Light Chain 3 Alpha (MAP1LC3A) is a protein-coding gene. MAP1LC3A has no known function in cholesterol metabolism. |
| AOC1 | −0.00861 | 2.48E−07 | 1 | Amine Oxidase, Copper Containing 1 (AOC1) is a protein-coding gene located in the TMEM176A-TMEM176B-AOC1 GWAS locus for HDL-C35. |
| IGLV4-69 | 0.08767 | 3.1E−07 | 1 | Immunoglobulin Lambda Variable 4-69 (IGLV4-69) is a protein-coding gene; three other genes in the same locus and belonging to the same family appear in this table (IGLV-70, IGLC5, and IGLC6). IGLV4-69 does not have a known function in cholesterol metabolism |
| SYCP2L | −0.01941 | 4.09E−07 | 2 | Synaptonemal Complex Protein 2 Like (SYCP2L) is a protein-coding gene, located in a GWAS locus for antiphospholipid syndrome and fatty acid measurements. SYCP2L does not have a known function in LDL-C metabolism38,76. |
| C10orf10/DEPP1 | −0.06893 | 4.74E−07 | 1 | DEPP1 (also known as C10orf10) is an autophagy regulator highly expressed in adipose tissue. DEPP1 overexpression in mice reduces glucose and triglyceride levels77, although a direct link of this gene to LDL-C metabolism remains unclear. |
| TMEM176A | −0.02242 | 4.77E−07 | 3 | Transmembrane protein 176A (TMEM176A) is a protein-coding gene located in the TMEM176B-TMEM176A-AOC1 GWAS locus for HDL-C35. Two IVs for TMEM176A are overlapping with TMEM176B. |
| RP11-18H21.1 | 0.029575 | 5.02E−07 | 2 | RP11-18H21.1 is a non-coding RNA gene without a known function in LDL-C metabolism. |
| TACSTD2 | −0.01865 | 8.65E−07 | 2 | Tumor Associated Calcium Signal Transducer 2 (TACSTD2) is a protein-coding gene located on the cell membrane involved in the superpathway Ca, cAMP, and Lipid Signaling. The function of TACSTD2 in LDL-C metabolism is unclear. |
| MSLN | −0.02566 | 1.39E−06 | 4 | Mesothelin (MSLN) is a protein-coding gene, its link to LDL-C is unclear. |
| IGLVI-70 | 0.112435 | 3.49E−06 | 2 | Immunoglobulin Lambda Variable (I)-70 (IGLVI-70) is a pseudogene; three other genes in the same locus and belonging to the same family appear in this table (IGLV4-69, IGLC5, and IGLC6). IGLVI-70 does not have a known function in cholesterol metabolism |
This table shows 18 Bonferroni-significant genes identified by MR-link as causal for LDL-C levels in the analysis that included eQTLs from the BIOS cohort. Gene names are according to ENSEMBL GENES 96 database (human Genome build 37). The causal effect estimate represents the changes in LDL-C (mg per dL) per standard deviation increase in gene expression. p values listed in this table are not adjusted for multiple testing (MR-link calibrated two-sided p value, see Supplementary Note 1). Full summary statistics of the genes are shown in Supplementary Table 1.
LDL-C low-density lipoprotein cholesterol, IV instrumental variable, HDL-C high-density lipoprotein cholesterol, GWAS genome-wide association study, siRNA small interfering RNA.
Table 3.
MR-link results using GTEx liver eQTLs.
| Gene name | Causal effect | p Value | #IVs | Biological function and link to LDL-C |
|---|---|---|---|---|
| PVRL2 | 0.3177 | 3.0E−14 | 1 | Poliovirus receptor-related 2 (PVRL2) is a protein-coding gene also known as NECTIN2. It is a cell-membrane protein located in the LDL-C GWAS locus APOE. siRNA experiments show that LDL-C uptake is increased in cells upon its downregulation48. PVRL2 knockout mice also had less atherosclerosis46. Both studies indicate a reduction in LDL-C upon downregulation of PVRL2. |
| PSRC1 | −0.0847 | 3.99E−09 | 1 | Proline and serine rich coiled-coil 1 (PSRC1) is a protein-coding gene located in an LDL-C GWAS locus35. PSRC1 has not been found to have an effect on cholesterol despite being targeted in a specific functional study43. The IV for PSRC1 is overlapping with the IV for CELSR2 and SORT1. |
| SORT1 | −0.0865 | 5.89E−09 | 1 | Sortilin (SORT1) is a protein-coding gene located in an LDL-C GWAS locus35. siRNA and knockdown experiments have functionally validated that SORT1 has a negative effect on LDL-C levels41,42. The IV for SORT1 is overlapping with the IV for PSRC1 and CELSR2. |
| CELSR2 | −0.0993 | 6.8E−08 | 1 | Cadherin EGF LAG Seven-Pass G-Type Receptor 2 (CELSR2) is a protein-coding gene located in an LDL-C GWAS locus35. CELSR2 has not been found to have an effect on cholesterol despite being targeted by a specific functional study43. The IV for CELSR2 is overlapping with the IV for PSRC1 and SORT1. |
This table lists four Bonferroni-significant genes that were identified using GTEx liver eQTLs. Gene names are according to ENSEMBL GENES 96 database (human Genome build 37). The causal effect estimate represents changes in LDL-C (mg per dL) per standard deviation increase in gene expression. p Values listed in this table are not adjusted for multiple testing (MR-link calibrated two-sided p values, see Supplementary Note 1). Full summary statistics of these genes are shown in Supplementary Table 2. LDL-C low-density lipoprotein cholesterol, GWAS genome-wide association study, siRNA small interfering RNA, IV instrumental variable.
MR analysis that used whole-blood eQTLs from GTEx was, as expected, underpowered compared to the analysis using BIOS eQTLs. Only two genes were found to be significant here, but they were not significant in the analysis that used BIOS eQTLs, where a more robust estimate could be made thanks to higher number of IVs identified (Supplementary Fig. 4a). Despite the limited power, we observed high concordance between effect sizes from the two analyses for all genes that showed nominal significance (p < 0.05) in the analysis that used BIOS eQTLs, with 94.8% of genes showing the same effect direction (Supplementary Fig. 4b).
Several genes located in genome-wide association study (GWAS) loci for cholesterol metabolism were found significant in the MR analysis that used blood eQTLs from BIOS, using a Bonferroni threshold that accounted for 13,778 genes being tested (0.05/13778 = 3.6 × 10−6). These include ABO, located in a LDL-C locus, AOC1, TMEM176A, and TMEM176B, which are all located in the same HDL-C-associated locus35,36, and SYCP2L, which is located in a GWAS locus for polyunsaturated fatty acids and related to LDL-C levels37,38. For the other genes identified, there was no evidence in the literature for a direct role in cholesterol metabolism, although some interesting patterns were evident. For example, we observed multiple genes involved in immunoglobulin production (IGLC5, IGLC6, IGLV4-69, and IGLVI-70) and insulin metabolism (UNC5B, DEPP1), mechanisms that are consistent with the role of cholesterol in inflammation and insulin resistance39,40. For all 18 genes, the effect direction estimated by MR-link was concordant with the direction estimated by other MR-methods when they were available, except in the case of MSLN, where only LDA-MR-Egger gave discordant results compared to all other methods (Table 1, Supplementary Fig. 5, and Supplementary Table 3). Interestingly, 17 of the 18 genes did not pass significance after multiple testing correction using the other tested methods: only ABO passed Bonferroni significance and only when using the IVW method (Table 1, Supplementary Fig. 5, and Supplementary Table 3). In 13 genes, a causal effect could not be estimated by MR-Egger, LDA-MR-Egger, and MR-PRESSO because there were too few IVs. Furthermore, MR-PRESSO did not make a causal estimate in the remaining 5 genes as it identified too many outliers (Table 1, Supplementary Fig. 5, and Supplementary Table 3).
In the MR analysis using eQTLs from liver, all the genes identified at the Bonferroni significance level of 3.2 × 10−5 (0.05/1557) fall within LDL-C GWAS loci. Among these, we found a negative causal effect for the well-known SORT1 gene (MR-link calibrated two-sided p = 5.9 × 10−9). Multiple functional studies have shown that this gene encodes the protein Sortilin (encoded by SORT1) and that it affects plasma LDL-C levels by acting on clearance of LDL-C and on secretion of very-LDL (VLDL) by the liver41–43 (Table 3 and Supplementary Table 2). We also found two other genes in the same GWAS locus, PSRC1, and CELSR2, but the IV (only one was found) for these genes was identical to that of SORT1 due to the high correlation between expression levels of these genes. Full overlap of a single IV in this locus makes it is impossible to discern causal from pleiotropic genes using MR-methods, including MR-link. The fourth gene found to be significant using liver eQTLs is PVRL2 (MR-link calibrated two-sided p = 3 × 10−14), which is located in the APOE locus associated to LDL-C (Table 3)35,36. For PVRL2, we estimated a positive causal effect; higher expression of PVRL2 is causally related to higher LDL-C (Table 3). PVRL2 is 17.5 kb downstream of the APOE gene, and two common missense polymorphisms in APOE account for a large fraction of the association signal36,44. Interestingly, in the most recent GWAS meta-analysis for lipids, 19 jointly significant LDL-C variants were found spanning a 162 kb region that encompasses PVRL236. This indicates that, while missense mutations in APOE play a major role, other genes in this locus are also likely involved in LDL-C regulation and that pleiotropic effects are to be expected. Our analyses indicate that PVRL2 is one of the causal genes at this locus. The positive effect of PVRL2 on LDL-C was also seen in the analysis that used blood eQTLs from BIOS (MR-link calibrated two-sided p = 4.3 × 10−5), although it did not pass our significance threshold in that analysis. Likewise, variation in gene expression of PVRL2 in blood has been found to be associated with LDL-C in a transcriptome-wide association analysis carried out in a very large genetic association study36. Of note, since the LD between IVs used in the analysis of blood and liver eQTLs was low (r2 < 0.2), the results potentially indicate a dual causal role for PVRL2 across these two tissues.
PVRL2 has mostly been studied in the context of atherosclerosis, where it has been shown to act as cholesterol-responsive gene involved in trans-endothelial migration of leukocytes in vascular endothelial cells, a key feature in atherosclerosis development45–47. Our results indicate a role for PVRL2 in modulating plasma levels of LDL-C via its expression variation in the liver. Biologically the role in liver could be explained by increased production of very-LDL or decreased LDL-C uptake (Fig. 4). In line with this hypothesis, a siRNA screen in hepatic cell lines of genes in the APOE locus showed that downregulation of PVRL2 gene expression promotes LDL-C uptake48 (Fig. 4). Overall, our results and existing functional evidence support that PVRL2 expression is correlated with LDL-C levels and show a causal effect in liver (Fig. 4).
Fig. 4. Biological interpretation of PVRL2.
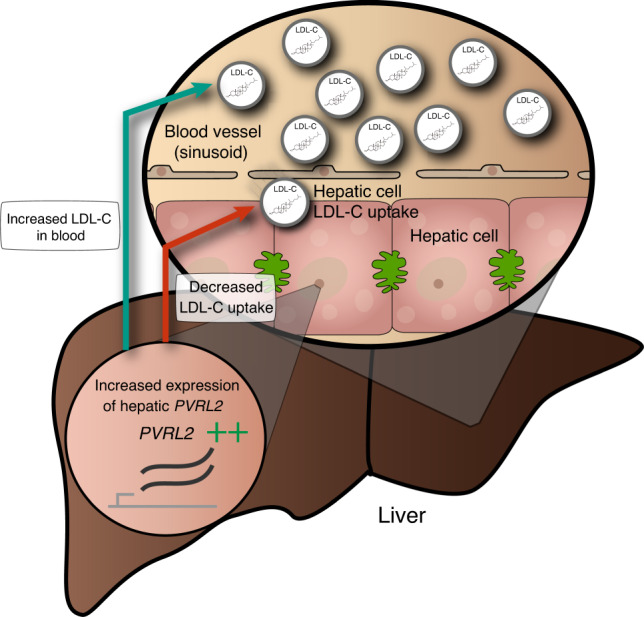
Functional and statistical evidence for the causal effect of PVRL2 on low-density lipoprotein cholesterol (LDL-C) levels. The teal arrow indicates a positive causal relationship between PVRL2 expression in liver and LDL-C levels in plasma—this relationship was detected in our MR analysis. The red arrow indicates a negative causal relationship between PVRL2 expression and LDL-C uptake in hepatic cells—this relationship was detected in small interfering RNA (si) experiments described in Blattman et al.48.
MR-link confirms ApoE changes affect LDL-C levels
To assess the effectiveness of MR-link in proteomics measurements, we combined the aforementioned LDL-C measurements in the Lifelines cohort with cis-pQTL summary statistics of 471 plasma protein measurements (measured using the SOMAscan platform in a cohort of 3301 individuals) (“Methods”)25,49. One protein passes the Bonferroni multiple testing threshold (p < 1.05 × 10−4): ApoE3, an isoform of ApoE (causal effect: 0.40 (+/−0.13 s.e.), MR-link calibrated two-sided p = 4.65 × 10−5, SOMAmer ID: APOE.2937.10.2). pQTLs were also available for ApoE2 (SOMAmer ID: APOE.5312.49.3), another isoform of ApoE but the causal effect was weaker and did not pass the Bonferroni threshold (causal effect = 0.56 (+/−0.24 s.e.), MR-link calibrated two-sided p = 0.002)44. These results are in line with the well-known causal relationship between increased ApoE plasma levels and LDL-C, and the widely described stronger impact of the E3 isoform compared to the E2 isoform44. Interestingly, MR-link did not estimate BGAT, the protein product of ABO, to be significant in this dataset (SOMAmer ID: ABO.9253.52.3, MR-link calibrated two-sided p = 0.18) We compared the IVs identified for BGAT (rs9411463 and rs72775494) with those used in the ABO blood eQTL analysis and found that only one IV for the BGAT protein was in LD (rs9411463) with any of the four IVs for ABO expression in BIOS. This scenario is in line with the overall patterns observed in the proteomics study—only a small fraction of eQTLs in blood also affect protein levels, but our results could also reflect targeting of the SOMAmer to a specific ABO protein isoform25. Unfortunately, further isoform information for BGAT was not available in the original study.
Discussion
Identification of genes whose changes in expression are causally linked to a phenotype is crucial for understanding the mechanisms behind complex traits. While several methods exist that infer causal relationships between two phenotypes, these rely on a set of assumptions that are often violated when gene expression is the exposure. Specifically, the presence of LD and pleiotropy between the genetic variants chosen as IVs are the main cause of violations of such assumptions17–19,28. Here we interrogated a large gene-expression dataset and showed that the eQTLs of a gene, which can be used as IVs, are very likely to be in LD, but not overlapping, with eQTLs of other genes, indicating that potential sources of pleiotropy in transcriptome-wide MR analyses are likely to come from variants in LD with the IVs.
We therefore developed MR-link, a causal inference method that is robust to unobserved pleiotropy. Our in silico results show that MR-link has the best discriminative ability compared to all other MR methods we tested, as well as to the Bayesian colocalization method coloc. MR-link jointly models the outcome using jointly significant eQTLs as IVs, combined with variants in LD, to correct for all potential sources of pleiotropy. To our knowledge, this approach has never been used in a causal inference method.
We applied MR-link to real data by applying it to LDL-C cholesterol measurements and eQTLs derived from blood, cerebellum and liver. This identified known and previously unknown causal genes within and outside GWAS loci. For example, in liver we identified the well-known negative causal relationship between expression of SORT1 in liver and LDL-C41–43. In liver, and suggestively in blood, we detected a causal effect for PVRL2, a gene located in the APOE locus. While a role for this gene is mostly known for immune and endothelial cells and in the context of atherosclerosis45,47, our results indicate that regulation of expression of this gene in both blood and liver causally affects LDL-C levels. Given its established role in atherogenesis, PVRL2 has been proposed as a potential therapeutic target for atherosclerosis. Our study indicates that such strategies should not only take into account the effect on atherosclerotic plaques, but also consider the hepatic function of PVRL2 in regulating plasma LDL-C levels in humans.
All the genes identified in the analyses that used eQTLs from blood were different from those identified using eQTLs from liver. While this is partly due to statistical power, as the BIOS cohort is more than 20 times larger than the GTEx cohort used to derive eQTLs in liver, this may also be related to tissue-specific mechanisms. We expect that causal genes found in whole blood will affect LDL-C through pathways that signal for lipid changes or regulate lipid binding to erythrocytes, as hypothesized for the ABO gene, whereas genes found in liver are more likely to be involved in lipid metabolism50,51.
MR-link has several advantages over other recent MR methods developed to overcome bias from LD and pleiotropy17,23. First, MR-link can model unobserved pleiotropy, whereas sources of pleiotropy need to be specified in multivariate MR methods. This is particularly important because sources of pleiotropy may be context-dependent and may arise from a phenotype other than those being measured in a cohort14,34. Second, MR-link can derive robust causal estimates even when only one or two IVs are available. The majority of genes tested in our large eQTL dataset have fewer than three IVs (68%), which makes it impossible for MR-PRESSO, MR-Egger, and LDA-MR-Egger to make causal estimates17–19.
One of the MR-link assumptions is that the IVs affect the outcome only through the exposure, conditional on the unmeasured pleiotropic effect. This assumption is violated when the IVs of the exposure and of the pleiotropic effect are fully overlapping. This assumption must not be violated when a single IV is available, but can be relaxed when multiple IVs are used in the model, as the relative effects of the IVs help to discriminate between a true causal effect and a pleiotropic effect, similar to multivariable Mendelian randomization methods22. In the case of multiple IVs that are fully overlapping, we have shown that MR-link has an increased FPR, yet still maintains higher power compared to other MR-methods and superior discriminative ability compared to coloc.
The application of MR-link is not restricted to gene expression or proteomics datasets; it can also be applied to other molecular layers that are known to have a similar genetic architecture to gene expression, such as metabolites. Given the increases in sharing of summary statistics from functional genomics QTL studies, coupled with the development of very large biobanks such as the UK biobank, the Estonian Biobank, the Lifelines cohort study, and the Million Veteran Program cohort33,52–54, we foresee many opportunities for applications of MR-link to individual-level data for the identification of the molecular mechanisms underlying complex traits. Of note, while we have limited our simulations to quantitative traits as an outcome in this paper, MR-link could be applied to binary traits such as human diseases. However, we have not investigated its performance in detail for binary outcome phenotypes. Furthermore, as for all MR studies, our method can be applied to populations of any ethnicity, provided that the summary statistics of the exposure are derived from a population that is ethnically-matched with the outcome cohort.
We foresee that many causal relationships will be discovered if highly powered causal inference methods such as MR-link are applied to many human traits. This could make it possible to build extensive causal networks similar in size and complexity to metabolic networks of small molecules, which would provide valuable insights into the mechanisms behind human traits and diseases.
Methods
BIOS consortium cohort genotype and expression analysis
We used genotype and expression measurements on 3746 Dutch individuals from the Biobank-based Integrative Omics Study (BIOS; http://www.bbmri.nl/acquisition-use-analyze/bios/), a collection of six different data cohorts: Lifelines DEEP55, Prospective ALS Study Netherlands56, Leiden Longevity Study57, Netherlands Twin Registry58, The Cohort on Diabetes and Atherosclerosis Maastricht59, and the Rotterdam Study60. All cohorts from the BIOS consortium were approved by their ethical committees, as follows: the LLDEEP was approved by the medical ethics committee of the University Medical Center Groningen; the Prospective ALS Study Netherlands was conducted with the approval of the institutional review board of the University Medical Centre Utrecht; the Leiden Longevity Study was approved by the Medical Ethical Committee of the Leiden University Medical Center; the Netherlands Twin Registry was approved by Central Ethics Committee on Research Involving Human Subjects of the VU University Medical Center, Amsterdam, an Institutional Review Board certified by the US Office of Human Research Protections (IRB number IRB-2991 under Federal-wide Assurance-3703; IRB/institute codes, NTR 03-180); the Rotterdam Study was approved by the institutional review board (Medical Ethics Committee) of the Erasmus Medical Center and by the review board of The Netherlands Ministry of Health, Welfare and Sports; the CODAM study was approved by the medical ethics committee of Maastricht University. An informed consent form was obtained from all the participants. Genotyping was performed separately per cohort (see references). All combined genotypes were imputed to the Haplotype reference consortium dataset61 using the Michigan imputation server62. We retained only biallelic SNPs and confined our analyses to variants with minor allele frequency (MAF) > 0.01, Hardy–Weinberg equilibrium (HWE) p value >10−6 and an imputation quality RSQR > 0.8. A genetic relationship matrix (GRM) was derived based on LD-pruned genotypes using the Plink 1.9 command --indep 50 5 2, and one individual was kept from all pairs of individuals that had a GRM value > 0.1 using the --rel-cutoff Plink 1.9 command31. Population outliers were identified using a principal component analysis of the GRM, and individuals more distant than three standard deviations from the mean of principal component 1 and principal component 2 were removed.
RNA-seq gene-expression quality control and processing are the same as those of Zhernakova et al.13. RNA extracted from whole blood was paired-end sequenced using the Illumina HiSeq 2000 instrument. RNA-seq read alignment was performed using STAR (version 2.3.0e)63. During alignment, variants with MAF < 0.01 from the Genome of the Netherlands were masked64. Gene expression was quantified using HTSeq (version v0.6.1p1)65. Samples with < 80% of reads mapping to exons were considered of low quality and removed. Samples were also removed if they had < 85% of mapped reads, or if they had a median 3′ bias larger than 70% or smaller than 45%. To further account for unobserved confounders, the expression matrix was corrected for the first 25 principal components as well as 5′ bias, 3′ bias, GC content, intron base-pair percentage, and sex following the procedure of Zhernakova et al.13. After genotype and expression quality control filters, 3503 individuals with expression data of 19,960 transcripts and genotype information of 7,838,327 SNPs were available for analyses. In this set, 57% were female and the average age was 52.8 years (±16.0 Stand. Dev.). eQTL association analysis was performed for SNPs located ±1.5 Mb of the transcript using Plink 1.9 and the --assoc command31. For 13,778 genes, at least one eQTL at p < 5 × 10−8 was identified, and those genes were used for all the analyses described in this manuscript.
We quantified how many genetic variants are necessary to explain gene expression using a conditional joint analysis approach. We identified jointly significant eQTLs by applying GCTA-COJO (v1.26.0)26 to eQTL summary statistics, using the BIOS cohort as LD reference panel, and selecting jointly significant variants that showed a p < 5 × 10−8 in this analysis step. To infer how often eQTLs are shared between genes, we assessed the percentage of genes with top eQTLs (or jointly significant variants) that have LD r2 > 0.99. We used the r2 > 0.5 threshold to see how often eQTL variants were in LD with each other.
We performed statistical fine-mapping of all genes using the FINEMAP v1.3.1 program27. First, we searched for associated eQTL variants (p < 5 × 10−8) in the cis-associated region. We then padded the associated regions with 100 kb and only looked for variants in this extended region. FINEMAP requires the same number of individuals across all variants, therefore we analyzed only the genes with the associated variants available in all subcohorts. We ran FINEMAP on these genes with the --sss option, using LD computed with Plink v1.9, with the --r command. Furthermore, genes were not run if they had less than 25 variants available in the region, or if a combination of variants led to an invalid posterior probability, leaving 13,276 genes which were successfully fine-mapped.
FINEMAP provides several configurations of statistically fine-mapped variants, along with their posterior probability of being causal. Studies that identify causal variants usually use a high posterior inclusion probability of multiple causal variant configurations to make sure the causal variant is captured in analysis. In MR studies it is not necessary to identify true causal variants, as the IV only needs to explain the exposure signal the best. In our analysis of LD between FINEMAP variants, we have therefore only considered the most likely configuration identified by FINEMAP, as these variants better explain the exposure variation.
Lifelines cohort genotype data and LDL-C levels
Lifelines is a multi-generational cohort study of 167,000 individuals from the north of The Netherlands. It was approved by the medical ethics committee of the University Medical Center Groningen and conducted in accordance with Helsinki Declaration Guidelines. All participants signed an informed consent form prior to enrollment. A subset of 13,436 Lifelines samples were genotyped with the cytoSNP array and underwent the quality control steps described in Scholtens et al.33: Genotyped variants were retained based on three criteria: MAF > 0.001, HWE p > 10−4, and a genotyping call rate > 0.95. After genotype quality control, samples were imputed using the Genome of the Netherlands reference panel64 and Minimac version 2012.10.366. Variants were further excluded if they were of bad imputation quality (RSQR < 0.3), showed deviation from HWE (p < 10−6), or if they were absent in the set of quality controlled genotyped and imputed variants of the BIOS cohort.
Low-density lipoprotein cholesterol (LDL-C) was estimated using the Friedewald equation67, based on triglycerides, high-density lipoprotein, and total cholesterol levels33. Total cholesterol levels of individuals who were prescribed cholesterol-lowering medication were divided by 0.8 prior to calculating LDL-C. Individuals with >4.52 mmol per liter total triglycerides were removed67. In addition, LDL-C levels were corrected for age, age squared, and sex. After genotype and LDL-C quality control, 12,449 individuals (of which 58.8% were female and the average age was 48.7 years (±11.5 Stand. Dev.)) and 7,336,374 variants remained for analyses. Association analysis for additive effects on LDL-C was performed using linear regression on standardized genotypes, e.g., transforming genotypes into a distribution with mean 0 and variance 1. Summary statistics of this analysis were used to perform MR analyses using the existing MR methods listed in Table 1.
GTEx download and analysis
We downloaded GTEx version 7 eQTL summary statistics, including non-significant results, from the GTEx website (https://gtexportal.org/home/datasets/)24. For every gene with at least one eQTL at p < 5 × 10−8, conditional analysis using GCTA-COJO was performed to select secondary variants at the same threshold, using the BIOS cohort as an LD reference. This resulted in 4028, 1557, and 1726 genes with at least one jointly significant eQTL for whole blood, liver, and brain (cerebellum) tissues, respectively.
pQTL summary statistics download and analysis
We downloaded the proteomics summary statistics of Sun et al.25 from the GWAS catalog (ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/SunBB_29875488_GCST005806). We isolated cis-regions by selecting variants within +/−1.5 Mb from each transcript. These variants already passed the quality control steps of Sun et al.25: (i) INFO score > = 0.7; (ii) minor allele count > = 8; (iii) Hardy–Weinberg equilibrium p > = 5 × 10−6. For all these variants we used UK10K minor allele frequencies (ftp://ngs.sanger.ac.uk/production/uk10k/UK10K_COHORT/REL-2012-06-02/UK10K_COHORT.20160215.sites.vcf.gz) as this information was not provided in the summary statistics but it is required for GCTA-COJO IV selection. We selected IVs using Lifelines genotypes as an LD reference33. To run MR-link, we first selected proteins with significantly (p < 5 × 10−8) associated variants that were shared between the cis summary statistics and the Lifelines cohort. This resulted in 471 proteins with significantly associated variants (p < 5 × 10−8) that are overlapping with the variants in the Lifelines cohort and for which GCTA-COJO was able to identify IVs.
Simulation of genotypes
Four hundred and three non-Finnish European individuals were isolated from the 1000 Genomes phase 3 release and used as a starting point for genotype simulation30. We simulated genotype data for 25,000 individuals in a chromosomal region (Chromosome 2, 100–105 Mb, human genome build 37) using the HAPGEN2 program (v.2.2.0), combined with interpolated HAPMAP3 recombination rates68. The region was then reduced to 1 Mb in length: between 102 Mbp and 103 Mb. Only biallelic SNPs with MAF < 0.01 were retained from simulated genotypes, leaving 3101 variants in this region. Simulated individuals were separated into an outcome cohort of 15,000 individuals, and into an exposure cohort and an LD reference cohort of 5000 individuals each. These cohort sizes were chosen to roughly represent the sizes of BIOS and Lifelines cohorts.
Simulation of phenotypes
We simulated quantitative phenotypes representing the exposures by randomly selecting SNPs from the simulated genetic region, and subsequently assigning these an effect. Causal SNPs were selected to represent both pleiotropy through LD (Fig. 2b) and pleiotropy through overlap (Fig. 2c). For the scenario of pleiotropy through LD (Fig. 2b), one to ten causal SNPs (subset sE) for the exposure were randomly selected from the entire simulated genetic region, and the same number of causal SNPs (subset sU) for the unobserved (pleiotropic) exposure was randomly selected from all SNPs in moderate LD (0.25 < r2 < 0.95) with SNPs in sE.
When pleiotropy through overlap was simulated (Fig. 2c), the causal SNPs for the observed and unobserved exposure were selected to be identical: sE = sU. A combination of pleiotropy through overlap and pleiotropy through linkage was simulated by choosing some or all of the SNPs of the unobserved exposure (subset sU) to be overlapping and some being in LD (0.25 < r2 < 0.95) with SNPs in sE.
The mathematical framework for the simulation of phenotypes is as follows. For each selected causal SNP of the exposure (subset sE), we simulated an effect-size from the uniform distribution U(−0.5,0.5) and then simulated the observed exposure yE as:
| 1 |
where X is a genotype matrix of size n × m, with n being the number of individuals (5000) and m the number of variants in the region (3101 in the simulated data), βE is the vector of effects
, and C ~ N(0,0.5)n is an n-vector of independent scalar draws from N(0,0.5), representing a cohort-specific confounder value per individual. Finally, is an n-vector of the measurement error of the exposure. Similarly, the unobserved exposure yU was simulated as:
| 2 |
where βU is the vector of effects defined as: , sU is the selection of SNPs for the unobserved exposure and are measurement errors distributed as . The outcome phenotype yo was then simulated as a linear combination of the observed and unobserved exposures:
| 3 |
where the causal effect of interest is parameterized per simulation run as and the (unknown) pleiotropic effect is the parameter reflecting absence and presence of a pleiotropic effect in a locus. Again, the measurement error is drawn from N(0,1)n.
The genetic variants of the exposures (sE, sU) and their effect sizes βE, βU were drawn and used in both cohorts (exposure and outcome), while the other random variables C, were randomly drawn in a cohort-specific manner. Since our model was built to account for unobserved pleiotropy, the observed and unobserved exposure were used to generate the outcome phenotype as in Eq. (3), but only the outcome phenotypes and the summary statistics of the (observed) exposure phenotype were used in the causal inference analysis.
Simulation parameters and scenarios
We simulated 1500 runs per scenario, each with a unique outcome (O) and two exposures (E and U). The scenarios differed in the number of causal SNPs (which varied from one to ten for both the observed and unobserved exposure), the strength of the causal relationship of interest (varied from no causal effect up to a large effect () and the presence (bU = 0.4) or absence ((bU = 0.0) of the pleiotropic effect. This resulted in 10 × 5 × 2 = 100 different scenarios.
In certain cases, an estimate cannot be made by an MR method, for instance when insufficient IVs are identified or a solution is not found in the estimation method. As a result, there are sometimes fewer estimates than expected in the final results. To ensure the stability of our FPR and power estimates, we have only reported results for a MR method in a specific scenario if we had more than 100 estimates out of the 1500 simulated runs.
Instrumental variable selection
IV selection can be difficult when there is LD between association signals. In simulations, we used two IV selection techniques: GCTA-COJO26 and p value clumping, using standard settings of Plink 1.9 except for the r2 threshold, which was set to 0.131. Both selection methods used a p value threshold of p < 5 × 10−8. When selecting IVs for BIOS and GTEX, we only used the GCTA-COJO technique.
MR-link
MR-link is a method for causal inference that is robust to the presence of LD and unobserved pleiotropy. It is an MR approach that requires individual-level data from the outcome cohort and summary statistics (effect sizes, standard errors and MAFs) from an exposure. Conceptually, MR-link jointly models a known exposure with SNPs that are in LD with the exposure IVs (tag-SNPs). Tag-SNPs are used to account for the unobserved pleiotropic effect present in a locus.
We defined our model in the following manner. Let X be a genotype matrix of n × m where n is the number of individuals in the outcome study and m are all the SNPs in a cis-region around the transcript (±1.5 Mb of the transcript), in which SNPs at indices sE are the causal genetic variants (IVs) for the exposure E. If we define the exposure E and the unobserved (pleiotropic) exposure U as in Eqs. (1) and (2), then the outcome phenotype yo from Eq. (3) can be represented as a function of E and U with the following equation:
| 4 |
where bE is the causal effect of interest of the exposure on the outcome, bU is the causal effect of the unobserved exposure, Co is a n-vector of independent scalars representing specific confounder per individual and is the measurement error of the outcome. In the hypothetical case that the genetic effects for both the exposure E and the pleiotropic exposure U are known, we can estimate bE by solving Eq. (4) in an analysis that is similar to multivariate MR22. In a real-world scenario, only the IV(s) for the exposure are known, while the variants that contribute to the unobserved (pleiotropic) exposure and their effect on the outcome are unknown.
Under Eq. (4), MR-link relies on the assumption that SNPs on sE influence the outcome yO only through their effect on yE, when conditioning on sU.
MR-link uses the following procedure to estimate causal effects:
A selection of IVs for the exposure and conditional effect sizes for these IVs are determined using the GCTA-COJO method26. A vector of effect sizes for all SNPs in the region is thus defined as: .
All SNPs in LD 0.1 < r2 < 0.99 with the exposure IVs are potential tag-SNPs. These variants are iteratively pruned for high LD so that tag-SNPs, sT, are always r2 < 0.95 with each other in order to reduce collinearity and computation time.
-
The following equation is solved for bE using ridge regression:
where XT is the genotype matrix of the outcome containing only tagging variants as defined in step (2), mT is the number of tagging variants and is used to normalize for the number of tags in the region, and mE represents the number of IVs selected by the selection method and is a parameter used to remove the dependency of the model on the number of IVs. The resulting coefficient vector contains the causal effect of interest bE, and the vector βUbU of length mT is a nuisance parameter that captures pleiotropic effects.5 Because individual-level data of the outcome is modeled by MR-link, MR-link does not use any summary statistics of the outcome.
We also considered solving the Eq. (5) using ordinary least squares (OLS). However, due to the multicollinear nature of the matrix, this approach leads to very low detection power (Supplementary Figs. 6–9; Supplementary Data 2–4, 7, and Supplementary Note 1). We therefore applied ridge regression to solve the equation and determined a T statistic and subsequent Wald test two-sided p value for ridge regression69. Due to the over-conservative nature of the resulting p value in simulations and real data (Supplementary Figs. 6–8, 10; Supplementary Data 2–4, 7, and Supplementary Note 1), we calibrated the p value distribution of each different scenario by fitting a beta distribution to null estimates to derive the final p values (Supplementary Note 1). When we report results for MR-link, it is these calibrated p values that we are referring to.
Mendelian randomization analyses
Causal relationships were estimated with MR-link and four other existing methods: Inverse variance weighting (IVW)28, LDA-MR-Egger regression17, MR-Egger regression18, and MR-PRESSO19. All methods were (re-)implemented in Python and compared to present equal results when compared with their original implementation. The corresponding code is available at https://github.com/adriaan-vd-graaf/genome_integration.
The IVW method is a weighted meta-analysis of causal estimates from single IVs. Specifically, a causal estimate bi for an IV i is estimated as , where β′O,i is the marginal effect of SNP i on the outcome and β′E,i is the marginal effect of the exposure. For the estimation of the causal effect, single IV causal estimates are combined using weights proportional to the inverse variance of such estimates using the two-terms definition of standard error: as following Burgess and Thompson70.
MR-Egger regression adjusts for average pleiotropy by fitting a weighted linear regression between the exposure SNP-effects and the outcome SNP-effects18. It assumes that <50% of the variants have a pleiotropic effect. MR-Egger can be applied when three or more instruments are available.
LDA-MR-Egger is similar to MR-Egger but also recognizes LD. LDA-MR-Egger can only be used when LD information between the IVs is available17,18.
MR-PRESSO is a method of causal inference that implements an approach to identify and remove outliers from the IVW framework19. It assumes that <50% of the variants have a pleiotropic effect. MR-PRESSO is unable to adjust for the presence of pleiotropy if fewer than three IVs are available, of if fewer than two IVs are left after outlier correction.
We applied these four methods to both simulated and real data. For real data, we used the LDL-C full GWAS summary statistics derived from the association carried out in the Lifelines study, as described above.
Prior to MR analyses, for each IV, we select the allele with positive effect on the exposure.
Colocalization analyses
We have run colocalization analyses on the simulated data using the R package coloc v4, git commit 6f3cbb1e5e90f07de772339d6e4af362140affc3, specifically its coloc.abf() function for the original coloc functionality and the coloc.signals() function for the masked (coloc-masked) and conditional (coloc-cond) estimates29,32. We used marginal effect sizes, standard errors and the MAFs as input that were calculated separately for the exposure and outcome. The LD for the conditional and masked coloc analysis was derived from the simulated reference cohort. For original coloc, we used the H4 test statistic of the coloc.abf() function as our result metric, which provides the posterior probability of sharing of the causal variants between the two traits being tested. For the coloc-cond and coloc-masked results, we have used the maximum PP4 reported by the coloc.signals() function, as this represents the largest posterior probability that a causal variant is shared between traits. We compared the discriminative ability of all coloc variations with that of MR-link using (i) false-positive rate and power when using a PP4 > 0.9 to declare colocalization and (ii) an area under the curve (AUC) statistic of the receiver operator curve, where scenarios with bE = 0 (null causal effect of the exposure) were considered true negative observations and were considered the true positive observations. We determined the AUC using the sklearn library71.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We are very grateful for the altruistic donation of biological materials and questionnaire data by our generous study participants, without them this study would not be possible. In addition, we thank the UMCG Genomics Coordination center and the UG Center for Information Technology, and their sponsors BBMRI-NL & TarGet, for storage and computing infrastructure. We thank BBMRI-NL for providing the transcriptome and genotyped data for the BIOS cohort. We thank P. Visscher, N. Wray, J. Yang, E. Lopera-Maya, O. Bakker, and N. de Klein for valuable advice during the development and writing of this work. We also thank K. McIntyre for editorial assistance and C. Benner for support on running FINEMAP. This work is financed by the Netherlands Organization for Scientific Research (NWO): NWO Spinoza Prize SPI 92-266 (to C.W.), by Fondation Lefoulon-Delalande (to A.R.), and by Radboud University Medical Centre Hypatia Grant 2018 (to Y.L.).
Author contributions
A.v.d.G. conceived and designed MR-link with critical input from S.S.; A.v.d.G. performed all simulations and data analyses on the datasets used in this study; A.v.d.G. and A.C. performed quality control analyses on the BIOS and Lifelines cohorts; B.C. and C.W. provided access to the datasets used in this study; A.v.d.G. and S.S. wrote the paper with critical inputs from Y.L., H.J.W., A.R., and A.C.; Y.L., S.S., and C.W. supervised the study; C.W. provided funding for this study. All authors read and approved the paper.
Data availability
Individual-level data of Lifelines cohorts are available to all bona-fide researchers upon request to the Lifelines biobank (https://www.lifelines.nl/researcher). Individual-level data (genotypes and RNA-seq data) of the BIOS Consortium cohorts can be downloaded by researchers of Dutch Institutes, or analyzed (but not downloaded) by any non-Dutch researcher in a Cloud environment (https://www.bbmri.nl/acquisition-use-analyze/bios). GTEx summary statistics can be downloaded from the GTEx website (https://gtexportal.org/home/datasets). pQTLs summary statistics can be downloaded from GWAS Catalog (ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/SunBB_29875488_GCST005806/). Simulated data can be recreated using the code at the link provided in Code availability statement. Imputation with the Haplotype Reference Consortium dataset can be done at the following link: https://imputationserver.sph.umich.edu/index.html#!. Raw data used to draw Fig. 3 can be found in Supplementary Data 3.
Code availability
An implementation of MR-link, the methods to recreate the simulated data, and instructions on usage can be found at https://github.com/adriaan-vd-graaf/genome_integration. This repository also includes implementation of the other MR methods and the coloc method used in this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Carlo Berzuini, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors jointly supervised this work: Yang Li, Cisca Wijmenga, Serena Sanna.
A list of authors and their affiliations appears at the end of this paper.
Contributor Information
Serena Sanna, Email: serena.sanna@irgb.cnr.it.
BIOS Consortium:
Bastiaan T. Heijmans, Peter A. C.’t Hoen, Joyce B. J. van Meurs, Rick Jansen, and Lude Franke
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-020-18716-x.
References
- 1.Burgess S, Foley CN, Zuber V. Inferring causal relationships between risk factors and outcomes from genome-wide association study data. Annu. Rev. Genomics Hum. Genet. 2018;19:303–327. doi: 10.1146/annurev-genom-083117-021731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pingault JB, et al. Using genetic data to strengthen causal inference in observational research. Nat. Rev. Genet. 2018;19:566–580. doi: 10.1038/s41576-018-0020-3. [DOI] [PubMed] [Google Scholar]
- 3.Evans DM, Davey Smith G. Mendelian randomization: new applications in the coming age of hypothesis-free causality. Annu. Rev. Genomics Hum. Genet. 2015;16:327–350. doi: 10.1146/annurev-genom-090314-050016. [DOI] [PubMed] [Google Scholar]
- 4.Ference BA, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. Ration. Pharmacother. Cardiol. 2013;9:90–98. doi: 10.1016/j.jacc.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 5.Ference BA, et al. Association of genetic variants related to CETP inhibitors and statins with lipoprotein levels and cardiovascular risk. JAMA - J. Am. Med. Assoc. 2017;318:947–956. doi: 10.1001/jama.2017.11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voight BF, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu Z, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016;48:481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 8.Gusev A, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016;48:245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luijk R, et al. Genome-wide identification of directed gene networks using large-scale population genomics data. Nat. Commun. 2018;9:3097. doi: 10.1038/s41467-018-05452-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gamazon ER, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015;47:1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li YI, et al. RNA splicing is a primary link between genetic variation and disease. Science. 2016;352:600–604. doi: 10.1126/science.aad9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobbyn A, et al. Landscape of conditional eQTL in dorsolateral prefrontal cortex and co-localization with schizophrenia GWAS. Am. J. Hum. Genet. 2018;102:1169–1184. doi: 10.1016/j.ajhg.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhernakova DV, et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nat. Genet. 2017;49:139–145. doi: 10.1038/ng.3737. [DOI] [PubMed] [Google Scholar]
- 14.Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell. 2017;169:1177–1186. doi: 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu B, Gloudemans MJ, Rao AS, Ingelsson E, Montgomery SB. Abundant associations with gene expression complicate GWAS follow-up. Nat. Genet. 2019;51:768–769. doi: 10.1038/s41588-019-0404-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Li YI, Pritchard JK. Trans effects on gene expression can drive omnigenic inheritance. Cell. 2019;177:1022–1034.e6. doi: 10.1016/j.cell.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barfield R, et al. Transcriptome-wide association studies accounting for colocalization using Egger regression. Genet. Epidemiol. 2018;42:418–433. doi: 10.1002/gepi.22131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowden J, Smith GD, Burgess S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015;44:512–525. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018;50:693–698. doi: 10.1038/s41588-018-0099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu Z, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 2018;9:224. doi: 10.1038/s41467-017-02317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berzuini C, Guo H, Burgess S, Bernardinelli L. A Bayesian approach to Mendelian randomization with multiple pleiotropic variants. Biostatistics. 2020;21:86–101. doi: 10.1093/biostatistics/kxy027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am. J. Epidemiol. 2015;181:251–260. doi: 10.1093/aje/kwu283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porcu E, et al. Mendelian randomization integrating GWAS and eQTL data reveals genetic determinants of complex and clinical traits. Nat. Commun. 2019;10:377267. doi: 10.1038/s41467-019-10936-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aguet F, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun BB, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–79. doi: 10.1038/s41586-018-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 2012;44:369–375. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benner C, et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 2016;32:1493–1501. doi: 10.1093/bioinformatics/btw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013;37:658–665. doi: 10.1002/gepi.21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giambartolomei C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Auton A, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wallace C. Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses. PLoS Genet. 2020;16:e1008720. doi: 10.1371/journal.pgen.1008720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scholtens S, et al. Cohort Profile: LifeLines, a three-generation cohort study and biobank. Int. J. Epidemiol. 2015;44:1172–1180. doi: 10.1093/ije/dyu229. [DOI] [PubMed] [Google Scholar]
- 34.Ongen H, et al. Estimating the causal tissues for complex traits and diseases. Nat. Genet. 2017;49:1676–1683. doi: 10.1038/ng.3981. [DOI] [PubMed] [Google Scholar]
- 35.Willer CJ, et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013;45:1274–1285. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klarin D, et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet. 2018;50:1514–1523. doi: 10.1038/s41588-018-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ander BP, Dupasquier CMC, Prociuk MA, Pierce GN. Polyunsaturated fatty acids and their effects on cardiovascular disease. Exp. Clin. Cardiol. 2003;8:164–172. [PMC free article] [PubMed] [Google Scholar]
- 38.Lemaitre RN, et al. Genetic loci associated with plasma phospholipid N-3 fatty acids: a meta-analysis of genome-wide association studies from the charge consortium. PLoS Genet. 2011;7:e1002193. doi: 10.1371/journal.pgen.1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barchetta, I. et al. Neurotensin is a lipid-induced gastrointestinal peptide associated with visceral adipose tissue inflammation in obesity. Nutrients10, 526 (2018). [DOI] [PMC free article] [PubMed]
- 40.Earnest CP, Jordan AN, Safir M, Weaver E, Church TS. Cholesterol-lowering effects of bovine serum immunoglobulin in participants with mild hypercholesterolemia. Am. J. Clin. Nutr. 2005;81:792–798. doi: 10.1093/ajcn/81.4.792. [DOI] [PubMed] [Google Scholar]
- 41.Kjolby M, et al. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab. 2010;12:213–223. doi: 10.1016/j.cmet.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 42.Musunuru K, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, et al. Interrogation of the atherosclerosis-associated SORT1 (sortilin 1) locus with primary human hepatocytes, induced pluripotent stem cell-hepatocytes, and locus-humanized mice. Arterioscler. Thromb. Vasc. Biol. 2018;38:76–82. doi: 10.1161/ATVBAHA.117.310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phillips MC. Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life. 2014;66:616–623. doi: 10.1002/iub.1314. [DOI] [PubMed] [Google Scholar]
- 45.Erbilgin A, et al. Gene expression analyses of mouse aortic endothelium in response to atherogenic stimuli. Arterioscler. Thromb. Vasc. Biol. 2013;33:2509–2517. doi: 10.1161/ATVBAHA.113.301989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rossignoli A, et al. Poliovirus receptor-related 2: a cholesterol-responsive gene affecting atherosclerosis development by modulating leukocyte migration. Arterioscler. Thromb. Vasc. Biol. 2017;37:534–542. doi: 10.1161/ATVBAHA.116.308715. [DOI] [PubMed] [Google Scholar]
- 47.Skogsberg J, et al. Transcriptional profiling uncovers a network of cholesterol-responsive atherosclerosis target genes. PLoS Genet. 2008;4:e1000036. doi: 10.1371/journal.pgen.1000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blattmann P, Schuberth C, Pepperkok R, Runz H. RNAi-based functional profiling of loci from blood lipid genome-wide association studies identifies genes with cholesterol-regulatory function. PLoS Genet. 2013;9:e1003338. doi: 10.1371/journal.pgen.1003338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Candia J, et al. Assessment of variability in the SOMAscan assay. Sci. Rep. 2017;7:1–13. doi: 10.1038/s41598-017-14755-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klop B, et al. Erythrocyte-bound apolipoprotein B in relation to atherosclerosis, serum lipids and ABO blood group. PLoS ONE. 2013;8:e75573. doi: 10.1371/journal.pone.0075573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McLachlan, S. et al. Replication and characterization of association between ABO SNPs and red blood cell traits by meta-analysis in Europeans. PLoS ONE11, e0156914 (2016). [DOI] [PMC free article] [PubMed]
- 52.Gaziano JM, et al. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 2016;70:214–223. doi: 10.1016/j.jclinepi.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 53.Leitsalu L, et al. Cohort profile: Estonian Biobank of the Estonian Genome Center, University of Tartu. Int. J. Epidemiol. 2015;44:1137–1147. doi: 10.1093/ije/dyt268. [DOI] [PubMed] [Google Scholar]
- 54.Sudlow C, et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tigchelaar EF, et al. Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: study design and baseline characteristics. BMJ Open. 2015;5:e006772. doi: 10.1136/bmjopen-2014-006772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huisman MHB, et al. Population based epidemiology of amyotrophic lateral sclerosis using capture-recapture methodology. J. Neurol. Neurosurg. Psychiatry. 2011;82:1165–1170. doi: 10.1136/jnnp.2011.244939. [DOI] [PubMed] [Google Scholar]
- 57.Deelen J, et al. Employing biomarkers of healthy ageing for leveraging genetic studies into human longevity. Exp. Gerontol. 2016;82:166–174. doi: 10.1016/j.exger.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 58.Lin BD, et al. The genetic overlap between hair and eye color. Twin Res. Hum. Genet. 2016;19:595–599. doi: 10.1017/thg.2016.85. [DOI] [PubMed] [Google Scholar]
- 59.van Greevenbroek MMJ, et al. The cross-sectional association between insulin resistance and circulating complement C3 is partly explained by plasma alanine aminotransferase, independent of central obesity and general inflammation (the CODAM study) Eur. J. Clin. Invest. 2011;41:372–379. doi: 10.1111/j.1365-2362.2010.02418.x. [DOI] [PubMed] [Google Scholar]
- 60.Hofman A, et al. The Rotterdam Study: 2016 objectives and design update. Eur. J. Epidemiol. 2015;30:661–708. doi: 10.1007/s10654-015-0082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCarthy S, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016;48:1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Das S, et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boomsma DI, et al. The genome of the Netherlands: design, and project goals. Eur. J. Hum. Genet. 2014;22:221–227. doi: 10.1038/ejhg.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anders S, Pyl PT, Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972;18:499–502. doi: 10.1093/clinchem/18.6.499. [DOI] [PubMed] [Google Scholar]
- 68.Su Z, Marchini J, Donnelly P. HAPGEN2: simulation of multiple disease SNPs. Bioinformatics. 2011;27:2304–2305. doi: 10.1093/bioinformatics/btr341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cule E, Vineis P, De Iorio M. Significance testing in ridge regression for genetic data. BMC Bioinforma. 2011;12:372. doi: 10.1186/1471-2105-12-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Burgess, S. & Thompson, S. G. Mendelian randomization: methods for using genetic variants in causal estimation. Mendelian Randomization: Methods for Using Genetic Variants in Causal Estimation, 10.1201/b18084 (CRC Press, 2015).
- 71.Pedregosa F, et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- 72.Hoffmann TJ, et al. A large electronic-health-record-based genome-wide study of serum lipids. Nat. Genet. 2018;50:401–413. doi: 10.1038/s41588-018-0064-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maisse C, et al. Lipid raft localization and palmitoylation: Identification of two requirements for cell death induction by the tumor suppressors UNC5H. Exp. Cell Res. 2008;314:2544–2552. doi: 10.1016/j.yexcr.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 74.Falk J, et al. Functional mutation analysis provides evidence for a role of REEP1 in lipid droplet biology. Hum. Mutat. 2014;35:497–504. doi: 10.1002/humu.22521. [DOI] [PubMed] [Google Scholar]
- 75.Veniaminova NA, et al. Niche-specific factors dynamically regulate sebaceous gland stem cells in the skin. Dev. Cell. 2019;51:326–340. doi: 10.1016/j.devcel.2019.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sugiura-Ogasawara M, et al. The first genome-wide association study identifying new susceptibility loci for obstetric antiphospholipid syndrome. J. Hum. Genet. 2017;62:831–838. doi: 10.1038/jhg.2017.46. [DOI] [PubMed] [Google Scholar]
- 77.Li W, et al. DEPP/DEPP1/C10ORF10 regulates hepatic glucose andm fat metabolism partly via ROS-induced FGF21. FASEB J. 2018;32:5459–5469. doi: 10.1096/fj.201800357R. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Individual-level data of Lifelines cohorts are available to all bona-fide researchers upon request to the Lifelines biobank (https://www.lifelines.nl/researcher). Individual-level data (genotypes and RNA-seq data) of the BIOS Consortium cohorts can be downloaded by researchers of Dutch Institutes, or analyzed (but not downloaded) by any non-Dutch researcher in a Cloud environment (https://www.bbmri.nl/acquisition-use-analyze/bios). GTEx summary statistics can be downloaded from the GTEx website (https://gtexportal.org/home/datasets). pQTLs summary statistics can be downloaded from GWAS Catalog (ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/SunBB_29875488_GCST005806/). Simulated data can be recreated using the code at the link provided in Code availability statement. Imputation with the Haplotype Reference Consortium dataset can be done at the following link: https://imputationserver.sph.umich.edu/index.html#!. Raw data used to draw Fig. 3 can be found in Supplementary Data 3.
An implementation of MR-link, the methods to recreate the simulated data, and instructions on usage can be found at https://github.com/adriaan-vd-graaf/genome_integration. This repository also includes implementation of the other MR methods and the coloc method used in this paper.