

ABSTRACT
The importance of speed to clinic for medicines that may address unmet medical needs puts pressure on product development timelines. Historically, both toxicology and first-in-human clinical materials are generated using the same clonal-derived cells to ensure safety and minimize any development risks. However, cell line development with single cell cloning is time consuming, and aggravated by the time needed to screen for a lead clone based on cell line stability and manufacturability. In order to achieve faster timelines, we have used pools of up to six clones for earlier production of drug substance for regulatory filing-enabling toxicology studies, and then the final single clone was selected for production of clinical materials. This approach was enabled by using platform processes across all stages of early development, including expression vectors, host cell lines, media, and production processes. Through comprehensive cell culture and product quality analysis, we demonstrated that the toxicology material was representative of the clinical material for all six monoclonal antibody programs evaluated. Our extensive development experience further confirmed that using a pool of clones for toxicology material generation is a reliable approach to shorten the early development timeline.
KEYWORDS: Cell line development, stable pool, Chinese Hamster Ovary (CHO), product quality, speed to clinic
Introduction
In biopharmaceutical development, the early process development strategy for biologics production in Chinese hamster ovary (CHO) cells is typically supported by the initial evaluation of biochemical and biophysical characteristics, as well as platform fit assessments, both of which enable manufacturability with reduced timelines.1,2 After the candidates are transitioned from drug discovery to process development, a substantial amount of time in early development is dedicated to cell line development, which starts from transfection of the desired sequence into host cells until single cell cloning (SCC), accompanied by multiple screenings for desired productivity and product quality.3–5 Single cell clones are further evaluated for manufacturability by cell growth/titer, product quality, and cell line stability profiles until a lead clone is selected. The importance of single cell cloning and cell line stability with respect to product quality has been emphasized by regulatory guidelines6 and extensively discussed in the industry.7–9 In essence, the product must be consistently expressed from cell lines with the right sequence identity, and product quality attributes must be controlled, throughout the product’s life cycle. Due to such expectations, the biopharmaceutical industry maintains rigorous efforts to ensure clonality and stability for clones selected for biologics manufacturing.
Traditionally, purified material from the selected lead clone is used for toxicology (Tox) studies, also called safety assessments, which include study designs in rodents and non-rodents to address potential toxicity in humans, repeat dosing effects and safety pharmacology evaluations.10 Purified Tox material is also used for Investigational New Drug (IND)-enabling product stability studies, and serves as the interim reference standard until clinical material is available.11 To minimize risks and to adhere as closely as possible to the final process for clinical manufacturing, the traditional approach in industry is to produce both Tox and clinical materials from the same lead clone with minimum process changes.
To enable the generation of an earlier supply of representative Tox material, we and others12-18 have explored a strategy comprising the use of a pool of clones for which cell line stability data have not yet been generated. This strategy relies on the observation that top-producing clones produce drug substance with very similar product quality profiles. Good comparability between pools and clonal materials have been reported by us and others in the industry.12–14,16-18 This strategy decouples the Tox material production from the final clone selection, and keeps Tox studies off the critical path while allowing parallel data accumulation for a full clone stability study. In the pool of clones strategy, stability studies are carried out for all clones present in the Tox material production and a lead clone is selected for master cell bank (MCB) manufacture for GMP (Good manufacturing practice) clinical manufacturing. The use of pools to generate Tox material is enabled by the use of our platforms, including expression vector, host cell line, media and process, and appropriate scale-up strategies. For each program, representative Tox material is produced using a pool of 6 clones and clinical material is produced using the lead clone selected from the 6 clones.
Here, we report our experience with the use of a pool of clones for Tox strategy in six monoclonal antibody (mAb) programs. Cell culture performance and product quality attributes are compared to demonstrate that Tox material produced via this methodology is representative of the clinical material and supports the continued implementation of the strategy described for future programs.
Results
Workflow
Our workflow and activities at each stage of cell line development are shown in Figure 1. Historically, Tox material was generated using the lead clone to ensure consistency with the first-in-human (FIH) clinical material and to avoid any unexpected safety readouts. With this approach, it takes longer to get to first-in-human trials in our experience, mostly because the IND-enabling Tox study is on the critical path. Recent reports indicate that industry is adapting the “pool of clones” strategy for Tox material generation,12,13 and early development timeline can be shortened by as much as 4 months.12
Figure 1.
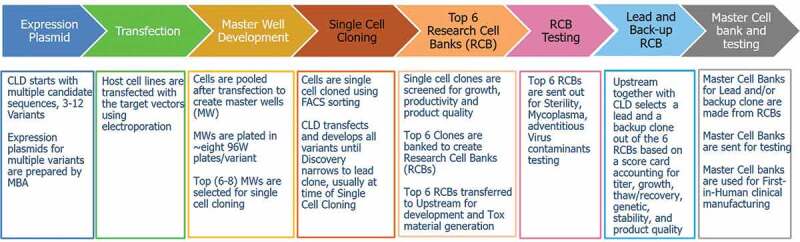
Standard cell line development and process workflow at BMS.
Given its potential to accelerate the start of Tox studies, a “pool of clones” strategy, enabled by using a platform approach for cell line, media, and process ranges in standard upstream procedures, was implemented in our early development workflow. Bristol Myers Squibb (BMS) upstream platform process was used for Tox production and proper scale-down models were used at all stages to predict the process performance at large scale.19 The top 6 clones were selected based on cell growth/productivity and product quality, and were verified to have an absence of sequence variants.20 The analysis of sequence variants was accomplished through a combination of next-generation sequencing and mass spectroscopy, where clones with any detectable sequence variants were eliminated at an early stage of clone evaluation to ensure that the top 6 clones selected were free of detectable sequence variants. During cell line development, the top 6 clones were pooled for Tox material production. In parallel, clones were screened in either ambr®250 or 5 L bioreactors to ensure proper scale-translations. Of the clones pooled for Tox, the lead clone was selected based on criteria such as thaw recovery, doubling time, cell growth/viability during passaging, platform suitability, genetic stability, and importantly, quality similarity to those of the Tox material. Genetic stability includes analysis of changes in copy number, structural integrity, loss/addition of bands and splice variants. MCB was prepared from the lead clone and used for GMP campaigns.
Acceptable cell culture performance from a pool of 6 clones for Tox
To support the strategy of using a pool of clones for Tox production, we analyzed data from six different mAb programs to evaluate the process performances between using a pool of clones vs. individual clones. Two expression systems were used: mAb-A, B, C with a CHO DG44 system, mAb-D, E, F with a CHO K1 system. Tox with a pool of six clones at 200 L scale was compared to individual clones at 5 L scale and lead clone performance at 1,000 L scale (Figures 2 and 3). Though using the exact process was desired for both Tox and FIH materials, process changes have been made after Tox production for some programs (Table 1). For mAb-A, D, and F, the same BMS platform process was used to produce both Tox and FIH materials. For mAb-B, C, and E, the FIH process for the lead clone was optimized to improve titer and/or to modulate critical quality attributes after Tox material was made. Main process changes included production media/feed and parameter setpoints. Those changes are well-accepted by regulatory agencies.
Figure 2.
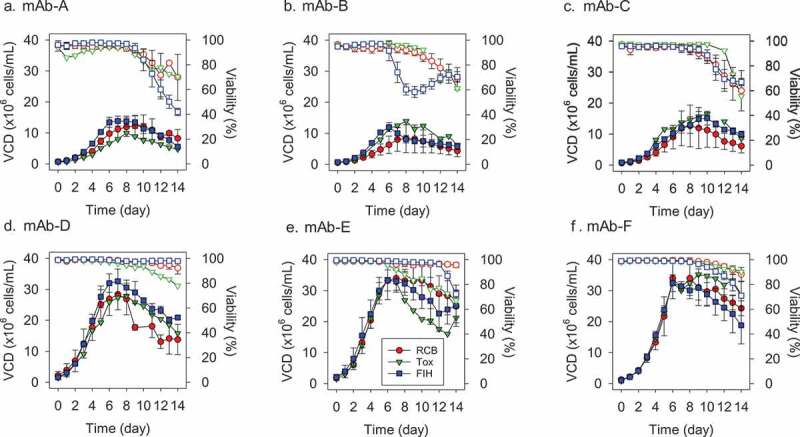
Viable cell density (VCD) and viability profiles for six monoclonal antibodies (A-F: mAb-A to mAb-F) between RCB, Tox, and FIH stages. RCB was at 5 L scale with individual 6 clones (RCBs), Tox was at 200 L scale with a pool of 6 clones (RCBs), and FIH was at 1,000 L scale with the lead clone MCB. Two expression systems were used: mAb-A, B, C with a CHO DG44 system, mAb-D, E, F with a CHO K1 system. RCB: Research Cell Bank; MCB: Master Cell Bank; FIH: First-in-Human. Solid symbols represent VCD profiles and open symbols represent viability profiles. For RCB, error bars represent the spread of all 6 clones (n = 6). For FIH, error bars represent one standard deviation of multiple FIH batches (n = 3–6). Only one batch was produced for Tox (n = 1).
Figure 3.
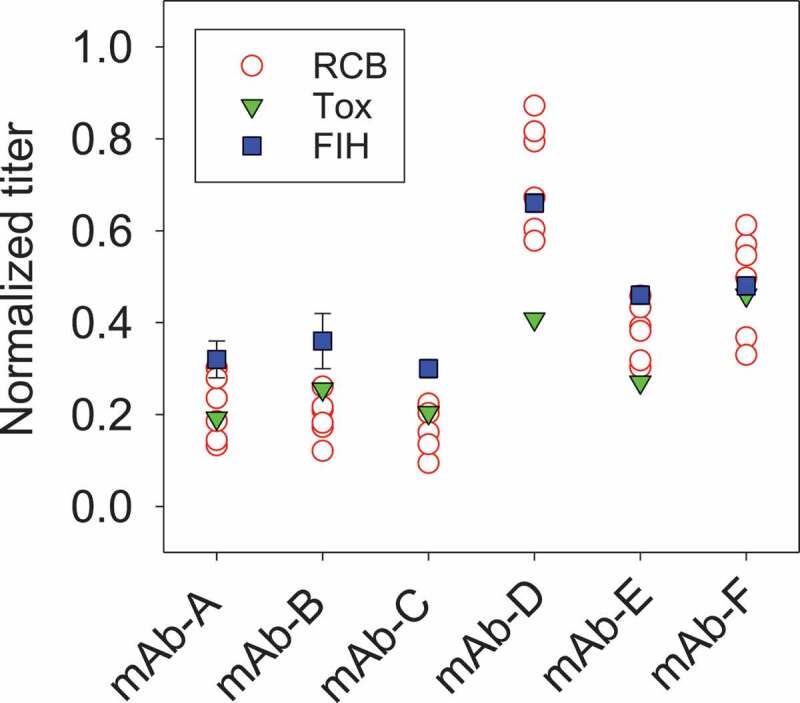
Final titer profiles for six monoclonal antibodies (mAb-A to mAb-F) between RCB, Tox, and FIH stages. RCB was at 5 L scale with individual 6 clones (RCBs), Tox was at 200 L scale with a pool of 6 clones (RCBs), and FIH was at 1,000 L scale with the lead clone. RCB: Research Cell Bank; FIH: First-in-Human. For FIH, error bars represent one standard deviation of multiple FIH batches (n = 3–6).
Table 1.
Process changes between Tox and FIH batches a.
| Molecule | Media Changes b | Process Setpoint Changes |
|---|---|---|
| mAb-A |
|
|
| mAb-B |
|
|
| mAb-C |
|
|
| mAb-D |
|
|
| mAb-E |
|
|
| mAb-F |
|
|
a. Process changes were for productivity and/or quality considerations.
b. Media changes were limited to implementation of different versions of BMS platform media, in which main components were the same.
Variability in viability profiles have been observed between Tox and FIH material production: mAb-A and -F had lower final viability and mAb-D and -E had higher final viability in FIH batches, while mAb-B and -C had comparable final viability between Tox and FIH batches. This suggested that process changes did not necessarily lead to improved viability for all mAbs. In general, such variations were acceptable and within our typical experience where most FIH batches were harvested at >60% viability except mAb-A. The minimum impact from viability differences was confirmed by evaluating the process impurity levels: residual host cell protein (HCP) and DNA levels for the FIH clinical materials were well within our platform specifications for all six mAb programs (Table 2). As residual HCP and DNA levels in Tox materials were not analyzed using the same qualified methods, a direct comparison between Tox and FIH materials cannot be made in this study. Still, the residual HCP and DNA levels from the FIH materials suggest that the viability levels experienced in FIH batches is acceptable. Cell growth was also comparable between Tox and FIH batches, and the variations were within the distribution of six individual clones. Change of process conditions in mAb-B, -C and -E did not affect the initial growth and peak cell density.
Table 2.
Residual HCP and residual DNA levels in FIH drug substance batches a.
| Molecule/Attribute | Residual HCP (ELISA) (ng/mg protein) b |
Residual DNA (qPCR) (pg/mg protein) c |
|---|---|---|
| mAb-A (n = 6) | <9 – <10 | <1 |
| mAb-B (n = 3) | 6–18 | <2.1 – <2.2 |
| mAb-C (n = 6) | 18–39 | <0.03 |
| mAb-D (n = 4) | <3 – 5 | <0.21 |
| mAb-E (n = 3) | <14 – 22 | <0.40 – <0.42 |
| mAb-F (n = 4) | <2 – 2 | <0.05 |
a. Only results from FIH drug substance batches were included as they were the only ones tested using qualified methods.
b. Residual HCP: platform specification for early phase development is not greater than 100 ng/mg. Values indicate range of experience.
c. Residual DNA: specification varied between programs as maximum doses differed; none of the specs exceeded the WHO exposure limit of 10 ng/dose. Values indicate range of experience.
Distributions of process productivity for all six mAb programs were evaluated (Figure 3). For mAb-A, B, C, E, F, Tox titer were within the distribution of 6 research cell banks (RCBs). For mAb-A and -F, the Tox batch from a pool of 6 clones had a titer approximately equivalent to the average titer of the individual clones (<15% difference). Since individual clones were pooled at the N-1 stage, a passage before production, equal numbers of cells of each clone were in the Tox production bioreactor, and therefore the individual clones average titer approximately equals the Tox batch titer. For mAb-B and -C, Tox had slightly higher titer than the average titer of the individual clones (>15% higher) because the high-producing clones were also faster growers in those cases (data not shown). For mAb-D and -E, the Tox batch had a lower titer than the average titer of the individual clones (>30% lower) because some of the clones had titer instability. Especially for mAb-D, Tox titer was outside of the distribution of the 6 RCBs. As part of the workflow, a short-term stability assessment was performed to ensure that there was no productivity loss or genetic rearrangements with cell age and the most genetically-stable clone with acceptable quality profiles was chosen as the lead clone. For mAb-B, -C and -E, process improvements post-Tox led to improved titers in FIH batches. Regardless of the changes made between Tox and FIH batches, cell culture performance was in general within expectations. Importantly, pool of clones for Tox was able to meet all material requirements in non-clinical studies.
Acceptable product quality from using a pool of 6 clones for Tox
It is critical to ensure product quality consistency throughout the development life cycle. We have previously shown that antibody products produced from masterwell cells and clonal-derived lines from the same masterwell were highly similar.18 Here, quality profiles, including charge variants, N-linked glycans, and product- and process- related impurities, between different conditions were compared (Figure 4 and Table 2, 3).
Figure 4.
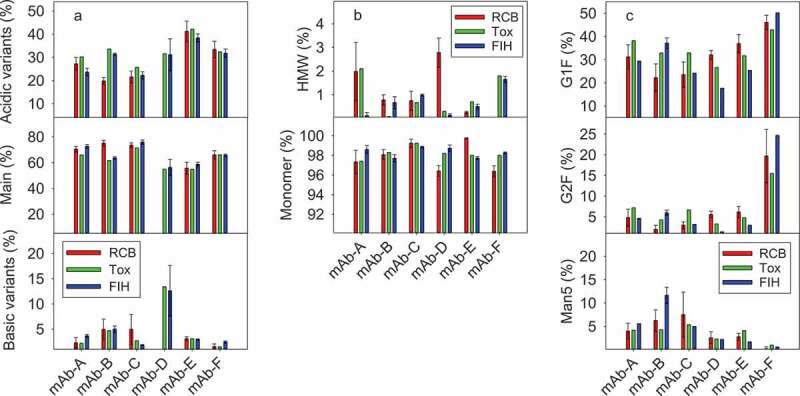
Quality attributes for six mAbs (mAb-A to mAb-F) between RCB, Tox, and FIH stages. RCB was at 5 L scale with individual 6 clones (RCBs), Tox was at 200 L scale with a pool of 6 clones (RCBs), and FIH was at 1,000 L scale with the lead clone. (a) Charge variants: acidic variants, main, and basic variants; (b) HMW and Monomer; (c) Major N-linked glycans: G1F, G2F, and Man5. RCB: Research Cell Bank; FIH: First-in-Human; HMW: High molecular weight. For RCB, error bars represent the spread of all 6 clones (n = 6). For FIH, error bars represent one standard deviation of multiple batches (n = 3–6).
Table 3.
Comparison of purity and potency profiles between Tox and FIH drug substance batches a.
| Molecule/Attribute | Reduced CE-SDS Purity (%) | Non-Reduced CE-SDS Purity (%) |
ELISA Potency (%) |
|
|---|---|---|---|---|
| mAb-A | Tox | 97.4 | 97.5 | 100 |
| FIH (n = 6) | 98.6 ± 0.4 | 98.8 ± 0.4 | 102 ± 14 | |
| mAb-B | Tox | 98.7 | 98.5 | 110 |
| FIH (n = 3) | 97.1 ± 0.9 | 97.9 ± 0.8 | 89 ± 8 | |
| mAb-C | Tox | 99.1 | 98.6 | 113 |
| FIH (n = 6) | 99.3 ± 0.1 | 98.5 ± 0.1 | 110 ± 12 | |
| mAb-D | Tox | 98.8 | 97.3 | 107 |
| FIH (n = 4) | 98.7 ± 0.4 | 94.8 ± 0.4 | 103 ± 4 | |
| mAb-E | Tox | 99 | 96 | 100 |
| FIH (n = 3) | 98.2 ± 0.2 | 97.6 ± 0.6 | 99 ± 4 | |
| mAb-F | Tox | 98.9 | 98 | 100 |
| FIH (n = 4) | 99.2 ± 0.1 | 97.8 ± 0.1 | 104 ± 5 |
a. Data presented as average ± one standard deviation.
Charge variant distributions between individual clones, Tox, and FIH materials were generally comparable (Figure 4(a)). Though minor differences were observed by visual assessment, those differences were typically <5% in absolute values and were considered as part of practical process variations. For mAb-D, RCB charge variants were not included for comparison because a different method was used for analysis. Additionally, mAb-D acidic and basic variants had larger variations in FIH batches, and efforts are ongoing to further understand the reason for this result. For all six mAbs, acidic variants were in the range of 20–40% and main peak was in the range of 50–70%. As charge variants could relate to some of the degradation pathways (e.g., deamidation, oxidation, isomerization) and IND-enabling stability studies typically use Tox drug substance/drug product materials, similar charge variants between Tox and FIH materials ensure that IND-enabling stability studies can be confidently used to support GMP stability and regulatory filing activities. Additionally, modulation of charge variants can be realized if needed in the final process via different culture (e.g., pH and temperature) and media levers,21 though it was not required in the cases covered here.
Tox material from a pool of clones was in general representative of FIH materials with a single clone for N-linked glycan profiles (Figure 4(c)). For most mAbs, the N-glycan core was fucosylated, with G0F and G1F being the major glycoforms. In general, the G1F and G2F glycoforms were quite similar: G1F at 20–40%, and G2F at <10%. The exception was mAb-F, where much higher G2F glycoform (15–25%) was generated than other mAbs. Still, the overall G1F and G2F levels are similar to those routinely observed in standard mAbs. Furthermore, modulation of galactosylation (G0F, G1F, and G2F) could be readily achieved if needed in the final process through media efforts, such as optimizing the uridine, manganese chloride, and galactose concentrations.22 For mAb-B, there was a significant increase in Man5 glycoform to 10–15% in the FIH material. This likely was due to the changes in media and process conditions done post-Tox to improve titer (Table 1). Antibodies with high mannose glycoforms (Man5 and higher) are cleared from the bloodstream at a faster rate than those with more mature glycoforms.23-25 Levels of high mannose glycoforms at <10% are generally considered as a low risk for standard mAbs, and there is little emphasis on its control unless it is for biosimilar development.26 In our case, higher than 10% Man5 glycoform raised development concerns. Potency enzyme-linked immunosorbent assay (ELISA) and Fcγ receptor binding kinetics verification using a Biacore assay were performed, and it was shown that there was no major impact due to process changes and the associated Man5 difference on relative potency and FcγRIIIa (CD16a) binding activity (data not shown). Overall, the increased Man5 level in mAb-B FIH materials was deemed as a low development risk and the material was introduced in clinical trials.
Presumed product-related impurity levels were also highly similar between Tox and FIH materials. High molecular weight (HMW) and monomer profiles were comparable for all individual clones vs. pool of clones (Figure 4(b)). In mAb-A, there was a significant reduction in HMW when moving from the Tox to FIH process, while a slight increase was observed for mAb-B. Nevertheless, all Tox and FIH materials had relatively modest HMW levels at <2%, and further HMW reduction could be achieved through purification unit operations. Similarly, antibody purity levels from the measurement of reduced and non-reduced capillary electrophoresis-sodium dodecyl sulfate (CE-SDS) were similar for both Tox material from a pool of clones and FIH material from a single clone (Table 3). As noted in the previous section, process-related impurities including residual HCP and DNA from FIH materials were well within our platform specifications (Table 2). The ELISA-based potency results were also highly comparable between Tox and FIH materials for all six mAbs (Table 3). In a typical biologics development program, Tox material is used as the interim reference standard until the GMP material is available. All analytical methods and formulations went through phase appropriate validation using the Tox material, which was also used to help define the specifications. In general, Tox material attributes were informative for setting FIH specifications.
Discussion
In this report, we described our experience using a pool of 6 clones for Tox material generation, followed by use of the lead clone for first-in-human GMP production. This approach was implemented on the foundation of robust platforms built in BMS’s biologics development organization. We systematically compared the cell culture performance and product quality attributes between individual clones and pools of clones. Six mAb programs, three derived from a CHO DG44 host and three derived from a CHO K1 host, were included in our assessment.
Overall process performance and quality attributes were acceptable for all six mAb programs. The process performance variations exhibited between individual clones at 5 L scale, Tox materials at 200 L scale, and clinical materials at 1,000 L scale were within our typical process experience27 and that seen during cell culture scale-up reported by others.28,29 In general, we showed good cell culture scalability from 5 L to 1,000 L; and process performance at 200 L was also indicative of 1,000 L scale characteristics, even though one used a pool of clones and the other used a single clone (Figure 2). For some mAb programs, process changes were introduced between Tox and FIH processes (Table 1), but they were not big enough to cause any significant changes in antibody quality attributes, with some exceptions such as the high Man5 glycoform in mAb-B FIH materials. In that particular case, FIH materials were analyzed by ELISA potency and Biacore binding kinetics to ensure there was no impact on binding and effector functions. Additional extensive analytical testing could be done to ensure any changes/differences will not pose concerns regarding safety and potency. In an ideal scenario, the Tox and FIH processes should be the same, which would reduce development risks. Continued platform process improvement focusing on productivity and product quality can lead to a more streamlined development workflow and eliminate the need for any process changes. We expect such efforts will further reduce the potential differences between Tox and FIH materials to a minimal level.
Use of stable pools or a pool of clones to produce Tox materials has been recently described in the literature.12–15 There are, however, few published reports of real-world application of such a strategy using non-clonal-derived proteins for IND-enabling toxicology studies.12,13 To the best of our knowledge, this is the first report on systematic implementation of this strategy in standard biologics development workflows, supported by comprehensive comparison in cell culture performance and quality attributes. Compared with the use of bulk or mini-pools to generate IND-enabling Tox material, use a pool of 6 clones naturally narrows down the selection to only top 6 clones from cell line development and reduces the possibility of drastic quality attributes differences. Including more than 6 clones in the pool of clones strategy, though possible, does not save additional time within the overall FIH development timeline in our current workflow.
It is worthwhile to note that using a pool of clones for Tox strategy also adds pressure to process development because the development timeline is reduced. In our case, clone selection becomes a parallel activity with Tox production and process optimization happens after Tox material is made. Thus, further improved conditions are only introduced in the FIH process if deemed necessary, as shown in the process changes made from Tox to FIH processes (Table 1). However, the generally acceptable mAb quality attributes between clones and extensive cell culture knowledge for standard mAbs makes the benefits in timesaving from using a pool of clones to generate Tox material outweigh the potential differences in quality attributes. Our study indicates that using a pool of clones to generate materials for toxicology studies is a reliable approach to produce representative material, especially for mAbs, and has been successfully applied as part of our early development strategy.
Materials and methods
Cell line development
In our standard workflow, cell line activities started prior to the final identification of desired protein sequence. Cells were transfected with multiple variants. Two host systems were used: mAb-A, B, C used a CHO DG44 system, and mAb-D, E, F used a CHO K1 system. Once the sequence was identified, cells with the target sequence were plated in 96-well plates. They were non-homogeneous and non-clonal. These master wells were screened based on titer, and then proceeded to single cell cloning using fluorescence-activated cell sorting (BD FACSJazz™, BD Biosciences, San Jose, CA). Following single cell cloning, 24 single cell clones of 384 clones were selected based on titer. These 24 clones were further narrowed down to the top 6 clones (evaluation of 6 clones was a BMS internal practice to maximize the probability of identifying high-producing clones with given resources) based on cell growth, titer, and quality attributes profiles using procedures detailed previously.18 The 6 high-producing clones were banked to create individual RCBs. The RCB vials were tested for sterility, identity, bacteriostasis, fugastasis, mycoplasma and mycoplasmastasis; and were evaluated for manufacturability, phenotypic stability, and genetic stability. Of the 6 clones, a lead and a backup clone were selected for the preparation of MCBs. The lead clone MCB is tested and released for the first-in-human clinical manufacturing.
Cell expansion and production
RCB vials were thawed and cells were expanded in shake flasks followed by a Wave bioreactor (BIOSTAT® RM 20/50, Sartorius Stedim, Göttingen, Germany). For Tox material generation, cells from 6 clones were expanded separately and pooled at the N-1 stage into two 50 L Wave bioreactors. This culture was then used to inoculate a 200 L single-use bioreactor (SUB; Xcellerex XDR200, GE Healthcare, Marlborough, MA). All 6 RCBs were evaluated in 5 L glass bioreactors (Sartorius Stedim) with Finesse controllers (Finesse Solutions, Santa Clara, CA). GMP clinical material was produced using the lead clone in a 1,000 L SUB (Xcellerex XDR1000, GE Healthcare). A platform 14-day fed-batch process was used. Bioreactor operating conditions used were platform conditions for all mAbs unless specified otherwise. Target inoculation density, temperature, dissolved oxygen, and pH controls were the same across all bioreactor scales. BMS-proprietary platform basal and feed media were used for expansion and production steps.
Cell culture sample analysis
Bioreactor samples were taken daily and analyzed immediately. Viable cell density (VCD) and viability were measured using the trypan blue dye exclusion method on a Vi-CELL XR cell counter (Beckman Coulter, Fullerton, CA). Offline pH, pO2, pCO2, and nutrients/metabolites were measured using a BioProfile 400 or a FLEX analyzer (Nova Biomedical, Waltham, MA).
Analytical methods and data analysis
Supernatant titer was measured using an Acquity H-Class ultra-performance liquid chromatography (UPLC, Waters, Milford, MA) with a Protein A column (POROSTM A 20, Thermo Fisher Scientific, Bedford, MA). Harvested cell culture fluids were purified using a TECAN-based high-throughput method (Tecan Systems, San Jose, CA) with Protein A resin, and purified samples were analyzed for quality attributes.
Monomer and HMW species were determined using an Alliance high-performance liquid chromatography (HPLC, Waters) with a Tosoh TSK G3000SWXL column. Charge variants were determined with either an imaged capillary isoelectric focusing method using an iCE3 instrument (Protein Simple, San Jose, CA) or a cation exchange chromatography (CEX) method using an Alliance HPLC (Waters) with a ProPac® WCX-10 analytical column (Dionex, Sunnyvale, CA). Purity levels were determined with reduced and non-reduced CE-SDS methods using a PA800 Plus system (Sciex, Framingham, MA). N-Linked glycosylation was analyzed using an Acquity UPLC (Waters) or a LabChip GXII instrument (Caliper, Hopkinton, MA). Glycans were released enzymatically using PNGase F (ProZyme, Hayward, CA), then fluorescent-labeled prior to measurements. The same methods were used for analyzing samples across different bioreactor scales for the same mAb.
Residual host cell DNA was analyzed using a quantitative polymerase chain reaction (qPCR) method using a ViiA 7 Real-Time PCR system (Applied Biosystems, Carlsbad, CA). Residual HCP was analyzed using an ELISA method, with a commercial CHO-host cell protein 3rd generation ELISA kit (Cat# F550, Cygnus Technologies, Southport, NC) used. Samples were reacted with anti-CHO horseradish peroxidase (anti-CHO:HRP, Cat# F551, Cygnus) in a 96-well microtiter plate consisting of wells coated with an affinity-purified anti-CHO-HCP capture antibody (Cat# F552, Cygnus). The plate was washed to remove any unbound conjugated antibodies. 3,3′,5,5′-tetramethylbenzidine (TMB) substrate was then added to yield a colorimetric reaction. The reaction was terminated with a stop solution and the absorbance was measured using a SpectraMax microplate reader (Molecular Devices, Sunnyvale, CA) at 450 nm. Sample residual HCP concentrations were determined by interpolation from a standard curve.
Relative potency was analyzed using an ELISA method. The assay was performed on a 96-well microtiter plate coated with the specific recombinant human protein, which captures the target antibody in the sample. Drug substance samples were diluted and added to the coated plate. Antibody bound to the target protein is detected with a HRP-conjugated goat anti-human IgG, F(ab’)2 fragment-specific antibody (Cat#109-036-097, Jackson ImmunoResearch Laboratories, West Grove, PA). Addition of a TMB substrate solution initiates a reaction with HRP that results in absorbance at 450 nm. The measured absorbance is directly proportional to the amount of antibody bound on the plate. The potency of the sample was calculated relative to the reference standard.
Fcγ receptor binding interactions of antibodies were characterized using the surface plasmon resonance method on a Biacore T200 instrument (GE Healthcare, Uppsala, Sweden). Commercially available recombinant human FcγRs were used (Cat# 4325-FC, 1330-CD/CF, 1875-CD, 1257-FC, R&D Systems, Minneapolis, MN); CD16a, CD32a, CD32b/c, and CD64 were immobilized on a sensor chip CM5. Binding of various dilutions of mAbs were measured from triplicate injections. The complex of the captured receptor and the bound mAb was stripped from the immobilized surface with the use of regeneration solution to prepare the surface for subsequent injections. The raw sensorgrams were double-reference subtracted (signal from the blank flow cell and from the 0 nM sample) during the data evaluation process, and the adjusted sensorgrams were processed using Biacore evaluation software to determine association (ka) and dissociation (kd) rate constants as well as equilibrium dissociation constants (KD).
Acknowledgments
The authors thank the Process Development and Clinical Manufacturing groups in New Jersey for supporting the studies, BMS Biologics Development organization for network support, and Henrik Andersen for helpful suggestions.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Yang X, Xu W, Dukleska S, Benchaar S, Mengisen S, Antochshuk V, Cheung J, Mann L, Babadjanova Z, Rowand J, et al. Developability studies before initiation of process development: improving manufacturability of monoclonal antibodies. MAbs. 2013;5(5):787–8. doi: 10.4161/mabs.25269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jarasch A, Koll H, Regula JT, Bader M, Papadimitriou A, Kettenberger H.. Developability assessment during the selection of novel therapeutic antibodies. J Pharm Sci. 2015;104(6):1885–98. doi: 10.1002/jps.24430. [DOI] [PubMed] [Google Scholar]
- 3.Butler M, Meneses-Acosta A. Recent advances in technology supporting biopharmaceutical production from mammalian cells. Appl Microbiol Biotechnol. 2012;96(4):885–94. doi: 10.1007/s00253-012-4451-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lai T, Yang Y, Ng SK. Advances in mammalian cell line development technologies for recombinant protein production. Pharm. 2013;6(5):579–603. doi: 10.3390/ph6050579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le H, Vishwanathan N, Jacob NM, Gadgil M, Hu W-S. Cell line development for biomanufacturing processes: recent advances and an outlook. Biotechnol Lett. 2015;37(8):1553–64. doi: 10.1007/s10529-015-1843-z. [DOI] [PubMed] [Google Scholar]
- 6.EMEA. ICH Q5D: derivation and characterisation of cell substrates used for production of biotechnological/biological products. 1998. [PubMed]
- 7.Frye C, Deshpande R, Estes S, Francissen K, Joly J, Lubiniecki A, Munro T, Russell R, Wang T, Anderson K. Industry view on the relative importance of “clonality” of biopharmaceutical-producing cell lines. Biol. 2016;44(2):117–22. doi: 10.1016/j.biologicals.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Wurm FM, Wurm MJ. Cloning of CHO Cells, productivity and genetic stability-a discussion. Processes. 2017;5(2):20. doi: 10.3390/pr5020020. [DOI] [Google Scholar]
- 9.Welch JT, Arden NS. Considering “clonality”: A regulatory perspective on the importance of the clonal derivation of mammalian cell banks in biopharmaceutical development. Biol. 2019;62(July):16–21. doi: 10.1016/j.biologicals.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Prior H, Baldrick P, de Haan L, Downes N, Jones K, Mortimer-Cassen E, Kimber I. Reviewing the utility of two species in general toxicology related to drug development. Int J Toxicol. 2018;37(2):121–24. doi: 10.1177/1091581818760564. [DOI] [Google Scholar]
- 11.EMEA. ICB Q6B specifications: test procedures and acceptance criteria for biotechnological/biological products. 1999.
- 12.Hu Z, Hsu W, Pynn A, Ng D, Quicho D, Adem Y, Kwong Z, Mauger B, Joly J, Snedecor B, et al. A strategy to accelerate protein production from a pool of clones in Chinese hamster ovary cells for toxicology studies. Biotechnol Prog. 2017;33(6):1449–55. doi: 10.1002/btpr.2467. [DOI] [PubMed] [Google Scholar]
- 13.Munro TP, Le K, Le H, Zhang L, Stevens J, Soice N, Benchaar SA, Hong RW, Goudar CT. Accelerating patient access to novel biologics using stable pool-derived product for non-clinical studies and single clone-derived product for clinical studies. Biotechnol Prog. 2017;33(6):1476–82. doi: 10.1002/btpr.2572. [DOI] [PubMed] [Google Scholar]
- 14.Rajendra Y, Balasubramanian S, McCracken NA, Norris DL, Lian Z, Schmitt MG, Frye CC, Barnard GC. Evaluation of piggyBac-mediated CHO pools to enable material generation to support GLP toxicology studies. Biotechnol Prog. 2017;33(6):1436–48. doi: 10.1002/btpr.2495. [DOI] [PubMed] [Google Scholar]
- 15.Stuible M, van Lier F, Croughan MS, Durocher Y. Beyond preclinical research: production of CHO-derived biotherapeutics for toxicology and early-phase trials by transient gene expression or stable pools. Curr Opin Chem Eng. 2018;22:145–51. doi: 10.1016/j.coche.2018.09.010. [DOI] [Google Scholar]
- 16.Wright C, Alves C, Estes S, Pieracci J, Kshirsagar R. Leveraging a CHO cell line toolkit to accelerate biotherapeutics into the clinic. Biotechnol Prog. 2017:33(6):1468–75. doi: 10.1002/btpr.2548. [DOI] [PubMed] [Google Scholar]
- 17.Scarcelli JJ, Shang TQ, Iskra T, Allen MJ, Zhang L. Strategic deployment of CHO expression platforms to deliver pfizer’s monoclonal antibody portfolio. Biotechnol Prog. 2017:33(6):1463–67. doi: 10.1002/btpr.2493. [DOI] [PubMed] [Google Scholar]
- 18.Fan L, Rizzi G, Bierilo K, Tian J, Yee JC, Russell R, Das TK. Comparative study of therapeutic antibody candidates derived from mini-pool and clonal cell lines. Biotechnol Prog. 2017;33(6):1456–62. doi: 10.1002/btpr.2477. [DOI] [PubMed] [Google Scholar]
- 19.Xu P, Clark C, Ryder T, Sparks C, Zhou J, Wang M, Russell R, Scott C. Characterization of TAP Ambr 250 disposable bioreactors, as a reliable scale‐down model for biologics process development. Biotechnol Prog. 2017;33(2):478–89. doi: 10.1002/btpr.2417. [DOI] [PubMed] [Google Scholar]
- 20.Valliere-Douglass J, Marzilli L, Deora A, Du Z, He L, Kumar SR, Liu Y-H, Mueller H-M, Nwosu C, Stults J, et al. Biopharmaceutical industry practices for sequence variant analyses of recombinant protein therapeutics. PDA J Pharm Sci Technol. 2019;73(6):622–34. doi: 10.5731/pdajpst.2019.010009. [DOI] [PubMed] [Google Scholar]
- 21.Chung S, Tian J, Tan Z, Chen J, Lee J, Borys M, Li ZJ. Industrial bioprocessing perspectives on managing therapeutic protein charge variant profiles. Biotechnol Bioeng. 2018;115(7):1646–65. doi: 10.1002/bit.26587. [DOI] [PubMed] [Google Scholar]
- 22.Gramer MJ, Eckblad JJ, Donahue R, Brown J, Shultz C, Vickerman K, Priem P, van den Bremer ETJ, Gerritsen J, van Berkel PHC. Modulation of antibody galactosylation through feeding of uridine, manganese chloride, and galactose. Biotechnol Bioeng. 2011;108(7):1591–602. doi: 10.1002/bit.23075. [DOI] [PubMed] [Google Scholar]
- 23.Yu M, Brown D, Reed C, Chung S, Lutman J, Stefanich E, Wong A, Stephan J-P BR. Production, characterization and pharmacokinetic properties of antibodies with N-linked Mannose-5 glycans. MAbs. 2012;4(4):475–87. doi: 10.4161/mabs.20737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanda Y, Yamada T, Mori K, Okazaki A, Inoue M, Kitajima-Miyama K, Kuni-Kamochi R, Nakano R, Yano K, Kakita S, et al. Comparison of biological activity among nonfucosylated therapeutic IgG1 antibodies with three different N-linked Fc oligosaccharides: the high-mannose, hybrid, and complex types. Glycobiol. 2007;17(1):104–18. doi: 10.1093/glycob/cwl057. [DOI] [PubMed] [Google Scholar]
- 25.Goetze AM, Liu YD, Zhang Z, Shah B, Lee E, Bondarenko PV, Flynn GC. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiol. 2011;21(7):949–59. doi: 10.1093/glycob/cwr027. [DOI] [PubMed] [Google Scholar]
- 26.Shi HH, Goudar CT. Recent advances in the understanding of biological implications and modulation methodologies of monoclonal antibody N-linked high mannose glycans. Biotechnol Bioeng. 2014;111(10):1907–19. doi: 10.1002/bit.25318. [DOI] [PubMed] [Google Scholar]
- 27.Yongky A, Xu J, Tian J, Oliveira C, Zhao J, McFarland K, Borys MC, Li ZJ. Process intensification in fed-batch production bioreactors using non-perfusion seed cultures. MAbs. 2019;11(8):1502–14. doi: 10.1080/19420862.2019.1652075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu S, Hoshan L, Jiang R, Gupta B, Brodean E, O’Neill K, Seamans TC, Bowers J, Chen H. A practical approach in bioreactor scale-up and process transfer using a combination of constant P/V and vvm as the criterion. Biotechnol Prog. 2017;33(4):1146–59. doi: 10.1002/btpr.2489. [DOI] [PubMed] [Google Scholar]
- 29.Xu S, Jiang R, Mueller R, Hoesli N, Kretz T, Bowers J, Chen H. Probing lactate metabolism variations in large-scale bioreactors. Biotechnol Prog. 2018;34(3):756–66. doi: 10.1002/btpr.2620. [DOI] [PubMed] [Google Scholar]