

Abstract
Neutrophils promote tumor growth and metastasis at multiple stages of cancer progression. One mechanism through which this occurs is via release of neutrophil extracellular traps (NETs). We have previously shown that NETs trap tumor cells in both the liver and the lung, increasing their adhesion and metastasis following postoperative complications. Multiple studies have since shown that NETs play a role in tumor progression and metastasis. NETs are composed of nuclear DNA-derived web-like structures decorated with neutrophil-derived proteins. However, it is unknown which, if any, of these NET-affiliated proteins is responsible for inducing the metastatic phenotype. In this study, we identify the NET-associated carcinoembryonic Ag cell adhesion molecule 1 (CEACAM1) as an essential element for this interaction. Indeed, blocking CEACAM1 on NETs, or knocking it out in a murine model, leads to a significant decrease in colon carcinoma cell adhesion, migration and metastasis. Thus, this work identifies NET-associated CEACAM1 as a putative therapeutic target to prevent the metastatic progression of colon carcinoma.
Neutrophils are the first lines of defense against infectious insults. Recently, there has been evolving evidence showing that the body’s host defense mechanism can play a protumorigenic role in cancer progression, including neutrophils (1). However, the exact mechanisms remain elusive. The clinical evidence associating neutrophils with cancer progression is strong; elevated neutrophil/lymphocyte ratio correlates with poor prognosis in almost every solid organ malignancy studied to date (2, 3). Neutrophil extracellular traps (NETs) represent a relatively recently described process by which neutrophils responded to an inflammatory insult (4) by releasing a decondensed meshwork of DNA into the surrounding environment (5). Composed of nucleic acids, histones, and cytoplasmic proteins, these web-like structures entrap and kill circulating pathogens (4). Since their discovery, NETs were also implicated in various pathophysiological situations (reviewed in Ref. 6).
To our knowledge, we were the first to demonstrate that NETs are involved in cancer progression (7). NETs have since been further implicated in cancer cell proliferation, growth, progression, metastasis, and cancer-related thrombosis (8, 9). Moreover, we and others have shown that postoperative infection after cancer surgery leads to increased cancer recurrence rates and diminished overall survival in patients (10, 11). We then showed, in a mouse model of postoperative infections (cecal-ligation-puncture [CLP]), that in response to severe infections, NETs are capable of not only capturing circulating tumor cells but, more importantly, also increasing their metastatic potential (7). The precise mechanism for this increased metastatic potential is unknown; however, it is possible that NET-associated proteins may be implicated in this process. Using mass spectrometry, we identified several candidate proteins present in purified NETs that may be involved in capturing cancer cells and promoting NET-facilitated metastasis. One of the more promising candidates identified was carcinoembryonic Ag cell adhesion molecule 1 (CEACAM1), a member of the carcinoembryonic Ag (CEA) family known to be widely expressed in different cell types but especially on human neutrophils (12). CEA family members have been reported to regulate diverse functions including tumor promotion, tumor suppression, angiogenesis and neutrophil activation (13, 14). Expression of host CEACAM1 has also been shown to increase the adhesion of cancer cells and metastasis in the liver, an effect primarily driven by the presence of this adhesion molecule on bone marrow–derived leukocytes, of which neutrophils are by far the most abundant (15). Given these findings linking CEACAM1 and cancer progression, and the fact that CEACAM1 was found to be structurally present on NETs, we investigated the role of CEACAM1 in NET-facilitated cancer cell adhesion and metastasis. We show that NET-associated CEACAM1 promotes adhesion and migration of colon carcinoma cells both in vitro and in vivo and enhances in vivo metastasis. Blocking CEACAM1 decreases adhesion, migration, and metastasis of colon carcinoma cells. This work characterizes an important interaction mechanism between NETs and cancer cells and identifies a promising marker and potential target for novel therapeutics to abrogate these prometastatic interactions between NETs and cancer cells.
Materials and Methods
Cells
Human colon carcinoma cell line (HT-29), murine colon carcinoma subline with low CEACAM1 expression (MC38-CC1−), and murine colon carcinoma subline stably transfected with CEACAM1 long isoform [MC38-CC1L (16)], were obtained from Dr. N. Beauchemin (McGill University, Montreal, QC). A549 were obtained from American Type Culture Collection (Manassas, VA). Cells were maintained in aMEM (HT-29) and DMEM (A549, MC38-CC1−, and MC38-CC1-L) containing 10% FBS and 1% penicillin/streptomycin and incubated at 37°C and 5% CO2. All reagents are from Wisent (St. Bruno, QC).
Human peripheral neutrophil extraction and NET generation
Human neutrophils were isolated from healthy subjects, and NETs were generated as previously described in Najmeh et al. (17). Only neutrophil isolates with >98% purity and viability as determined by methylene blue (Stemcell Technologies) and trypan blue (Wisent) staining, respectively, were used to generate NETs.
Murine bone marrow neutrophil extraction
Neutrophils were isolated from bone marrow of syngeneic mice as described in Mócsai et al. (18). Only neutrophils with >90% pure and 95% viable as determined by methylene blue and trypan blue staining respectively were used.
Murine peripheral neutrophil extraction and NET generation
Peripheral blood from Ceacam 1 knockout (CC1 KO) and C57 BL/6 mice were isolated by heart puncture and lysed with BD cell lysis buffer as per manufacturer’s instructions (BD Biosciences). Neutrophils were sorted following staining with Ly6G-AF647 (1:200) (1A8; BioLegend, San Diego, CA) and then stimulated with 500 nM PMA for 4 h at 37°C, 5% CO2 to induce NETosis.
Mass spectrometry
Mass spectrometry of isolated NETs was performed on samples obtained from neutrophils from triplicate healthy donor controls. Scaffold software (version 4.0.5; Proteome Software, Portland, OR) was used to validate tandem mass spectrometry (MS/MS)-based peptide and protein identifications. Peptide identifications were accepted if they could be established at >95% probability by the Peptide Prophet algorithm (19). Protein identifications were accepted if they could be established at >99% probability and contained at least two identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm as previously described (20). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Immunofluorescence
Human NETs were isolated as described above and used to coat 13 mm glass coverslips (Fisher Scientific, Ottawa, ON) overnight at 4°C. The following day, coverslips were fixed for 10 min with 4% paraformaldehyde (Thermo Fisher Scientific) and gently washed once with PBS (Wisent). Coverslips were blocked with 1% BSA (Wisent) for 1 h at room temperature (RT) prior to staining with anti-human CC1 mAb (5F4, from Dr. R. Blumberg, Harvard) (1:100) for 1 h at RT. After washing once with PBS, isolated NETs were incubated with FITC-conjugated goat-anti mouse Ab (1:200) for 1 h at RT. Coverslips were then gently washed with PBS, and DNA was counterstained with Sytox Orange (1:1000; Life Technologies, Burlington, ON) for 10 min at RT. Coverslips were mounted using Mowiol mounting medium (Sigma-Aldrich).
Mouse neutrophils were isolated as previously described and loaded on a μ-SlideVI chamber (ibidi Biosciences, Fitchburg, WI) in the presence or absence of calcium ionophore (2 μM) (A23187; Sigma-Aldrich) to stimulate NETs release. Neutrophils were stained with Ly6G-AF647 (1:200) (1A8; BioLegend), extracellular DNA was stained with Sytox Orange (5 μM), and CC1 was labeled with anti-mouse CC1 (1:100) (E-1; Santa Cruz Biotechnologies, Dallas, TX) labeled with an Alexa Fluor 350 labeling kit (Life Technologies). The images were acquired for up to 1 h poststimulation.
Both immunofluorescence assays were visualized using an LSM 780 laser scanning confocal microscope (Carl Zeiss, Dorval, QC) equipped with a temperature and CO2 controlled chamber.
Western blot
Western blots were performed to verify the presence of CC1 in NETs. To do so, cell-free NET stock was obtained from murine or human blood. A549, HT-29, MC38-CC1L, and MC38-CC1− cell lysates were obtained by incubating cells with 500 μl of RIPA buffer (Pierce, Edmonton, AB, Canada) containing protease inhibitor (Roche Canada, Laval, QC, Canada) for 30 min. Prepared samples were then loaded onto an SDS-PAGE gel and electro-transferred to a nitrocellulose membrane. The membrane was then blocked for an hour with 1% BSA in PBS, then incubated with primary Abs consisting of either human (5F4, 1:100) or mouse (E-1, 1:100) anti-CC1 Abs and β-actin (1:20,000) overnight at 4°C. The following day, the membranes were incubated with HRP-conjugated secondary Ab at 1:10,000 dilution for 1 h at RT. Detection of proteins was performed using 20× LumiGlo image capturing. GAPDH protein levels were used at all time points for positive control.
Animals
Seven- to ten-week-old C57BL/6 (Charles River, St-Constant, QC) and CC1 KO (from Dr. Beauchemin) mice were used for all experiments. Peritonitis was induced by CLP as previously described (21, 22). For mice treated with DNase I, 2.5 mg/kg DNase I (Biomatik, Cambridge, ON) was given daily i.m. starting 24 h prior to CLP until the termination of the experiment. All mice experiments were carried out in strict accordance with the recommendations of the Canadian Council on Animal Care and under the conditions and procedures approved by the Animal Care Committee of McGill University (Animal Use Protocol no. 7724).
In vitro adhesion assay on NETs
A general description of the in vitro adhesion assay of cancerous cells on NETs is described in (17). Briefly, isolated NETs were left to adhere overnight at 4°C. The next day, nonadherent NETs were aspirated, and wells were blocked with 1% BSA for 1 h at RT. After blocking, 200 μg/ml anti-CEACAM1 mAb (5F4) or 200 μg/ml mouse IgG isotype control (34B1, from Dr. Blumberg) were added to some wells and incubated for 1 h at 37°C and 5% CO2. After washing, 2 × 104 HT-29 or A549 cells were stained with CFSE dye for 10 min (Life Technologies) and then washed and added to the NET monolayers and incubated for 90 min at 37°C, 5% CO2. Cells were subsequently gently aspirated, washed once, and fixed in 4% paraformaldehyde. In some experiments, 1000 U of DNase I were added to NET–cell mixtures 10 min prior to fixation and quantification of adhesion. Adhesion was quantified as the number of cells in 4 random high-power fields (hpf) at 20× using a Nikon TE300 microscope (Nikon, Mississauga ON).
In vitro adhesion assay to mouse neutrophils
A total of 5 × 105 neutrophils isolated form C57BL/6 or CC1 KO mice were plated in 24-well plates (Falcon) and incubated for 1 h at 37°C and 5% CO2 in DMEM. A total of 105 MC38-CC1L or MC38-CC1− CFSEstained cells were added to wells in the presence of 500 nM PMA with or without 1000 U DNase I. Unstimulated neutrophils and untreated tumor cells in DMEM served as controls. Following incubation for 4 h at 37°C and 5% CO2, wells were washed with PBS and fixed in 4% paraformaldehyde. Adhesion was quantified as the number of cells in 4 random hpf at 10× and 20× using a Nikon TE300 microscope.
In vitro migration assay
Twenty-four–well Boyden chambers with 5-μm PET membranes (Fisher Scientific, Montreal, QC) were used for migration assays. For the human neutrophil migration, 2.5 × 105 neutrophils from healthy human volunteers were incubated with media alone or with 500 nM PMA for 1 h at 37°C, 5% CO2 and placed in the upper chamber. HT-29 cells from 80% confluent cultures were detached using 0.5% trypsin (Wisent) and stained for 10 min with CFSE, and 2.5 × 105 cells were added to the upper wells. Neutrophil–tumor cell suspensions were treated with either 500 nM PMA or with 500 nM PMA with 200 μg/ml 5F4 mAb or 200 μg/ml IgG isotype control or 10 μM neutrophil elastase inhibitor (NEi). The cell suspensions were incubated for 24 h at 37°C, 5% CO2. After incubation, the contents of the upper chambers were aspirated, washed with PBS, and wiped with a sterile cotton swab. Cells adherent to the undersurface of the membrane were quantified in 5 random hpf, and representative images were taken as described. Human blood was obtained from consented healthy volunteers as per institutional review board protocol no. 2007-856.
For mouse neutrophil migration assays, 2.5 × 105 neutrophils from C57BL/6 or CC1 KO mice were incubated with media alone or with 500 nM PMA for 1 h at 37°C, 5% CO2 and placed in the upper chamber. MC38-CC1L cells from 80% confluent cultures were detached using 0.5% trypsin (Wisent), and 2.5 × 105 cells were added to the upper wells. Neutrophil-tumor cell suspensions were treated with either 500 nM PMA or media alone. The cell suspensions were incubated for 24 h at 37°C, 5% CO2. After incubation, the contents of the upper chambers were aspirated, washed with PBS, and wiped with a cotton swab. Cells adhering to the undersurface of the membrane were fixed with 4% paraformaldehyde, permeabilized with methanol, and stained with crystal violet. Adherent cells were quantified in 4 random hpf, and representative images were taken as described.
Depletion reinfusion experiment and in vivo adhesion assay
Neutrophils were depleted from C57BL/6 mice using i.p. injections of 150 μg of anti-Ly6G/GR1 Ab (Thermo Fisher Scientific, Waltham, MA). The next day, neutrophils were extracted from bone marrow of C57BL/6 or CC1 KO mice as previously described and then incubated with or without 500 nM PMA and/or with or without 100 U DNase I for 1 h at 37°C and 5% CO2 and washed, and then 106 neutrophils were reinfused into the depleted mice. After 20 min, the reinfused mice were intrasplenically injected with CFSE-labeled 1.5 × 105 MC38-CC1L cells per mouse. After 10 min, the livers were visualized in vivo and CFSE-expressing tumor cells were imaged using epifluorescence and cells arrested within unoccluded sinusoids were considered adherent. Cells or tumor islands were quantified as the number of cells/micrometastatic foci in 8–10 hpf, and representative images were recorded.
Gross liver metastasis assay
Liver metastases were quantified 18 d after intrasplenic injection of 1.5 × 105 MC38-CC1L cells per mouse, with a maximum quantifiable number of 400 gross metastases per liver. H&E staining was performed, and representative images of the gross metastases and H&E are shown.
Statistics
One-way ANOVA with post hoc multiple comparison was employed. All data are presented as mean 6 SEM. Statistical significance was set at p < 0.05. GraphPad Prism 6 software was used for all statistical analyses and graphing.
Results
CEACAM1 is present on murine and human NETs
To identify potential molecular mediators of the tumor–NET interaction, we performed tandem mass spectrometric analysis on NETs isolated from the blood of three healthy volunteers. This led to the identification of 583 distinct proteins (Supplemental Table 1). Of interest, we decided to focus on CEACAM1 (Fig. 1A), given its widely studied implication in various cellular signaling interactions involved in the growth and differentiation of cancer cells and modulation of various types of cancer (14-16, 23). Moreover, CEACAM1 was shown to be present on the surface of activated neutrophils (24, 25), and high expression of CEACAM1 on neutrophils was shown to drive increased infiltration in the tumor microenvironment and correlate with poor prognosis (reviewed in Refs. 26, 27). These studies implicated neutrophil CEACAM1 in cancer progression.
FIGURE 1.

CEACAM1 is present on NETs. (A) Mass spectrometry profile of CEACAM1 in three NET samples obtained from three healthy individuals. (B) Confocal fluorescence imaging of human isolated NETs on glass coverslips stained with anti–H3-Citrulline Ab (red), Sytox Green (green), and anti-human CEACAM1 (CC1) Ab (5F4, blue) (left panels) or IgG isotype instead of 5F4 Ab (blue) (right panels). (C) Confocal fluorescence imaging of bone marrow–derived murine neutrophils stimulated with calcium ionophore (A23187) on glass coverslips stained with Ly6G (blue), Sytox Orange (red), and anti-murine CEACAM1 (CC1) Ab (green). Top panels, Non-NETosed neutrophil; middle panels, NETosing neutrophil; bottom panels, NETosed neutrophil. (D) Western blot on isolated cell-free human NETs probed with an anti-human CEACAM1 Ab and β-actin (loading control) for A549, HT29, and human NETs. (E) Western blot on isolated cell-free murine NETs probed with an anti-murine CEACAM1 Ab and β-actin (loading control) for MC38-CC1−, MC38-CC1L, and murine NETs.
We first validated the findings from the tandem mass spectrometry analysis by coating glass coverslips with isolated human NETs and then staining for NETs and CEACAM1. Extensive colocalization between extracellular DNA (SYTOX, red), NETs (H3-citrulline, blue), and CEACAM1 (5F4 mAb, green) was observed in PMA-stimulated neutrophils, confirming that CEACAM1 is found on human NETs (Fig. 1B). We also sought to determine if CEACAM1 is expressed on murine NETs isolated from C57BL/6 bone marrow–derived neutrophils and stimulated to generate NETs with calcium ionophore (A23187; Sigma-Aldrich), another validated NETosis agent (28). Using live cell imaging, we captured live images of neutrophils (Ly6G, in blue) generating NETs (SYTOX, red) displaying extensive colocalization with CEACAM1 (E-1 mAb, green) (Fig. 1C). The presence of CEACAM1 on isolated cell-free NETs from human and mouse neutrophils was also confirmed by Western blotting (Fig. 1D, 1E, respectively). HT-29 (high CC1 expresser, Supplemental Fig. 1A) and A549 [low CC1 expressor (29)] were used as positive and negative controls, respectively, for the human NETs Western blot (Fig. 1D). MC38-CC1L (high CC1 expressor, Supplemental Fig. 1B) and MC38-CC1− [low expressor of CC1 (30)] were used as positive and negative controls, respectively, for the murine NETs Western blot (Fig. 1E). Moreover, we observed colocalization of NETs and CEACAM1 in vivo (Supplemental Fig. 2) in a mouse model of sepsis (CLP), known to generate NETs (7). These results show that CEACAM1 is released during NETosis and is a component of NETs.
CEACAM1 on NETs is important in adhesion of colon carcinoma cells
To identify the intrinsic function of NET-associated CEACAM1, we performed an in vitro adhesion assay of human colon carcinoma cell line HT-29 with isolated human NETs pretreated with the 5F4 anti-CEACAM1 mAb or isotype control and treated with DNase I (1000 U) or vehicle control (water). The NET monolayer is formed when a high concentration of NETs, obtained from stimulated neutrophils, are collected and plated onto a dish (17). This process allows us to study pure NETs in isolation. We observed a significant 5-fold reduction of adhesion of HT-29 cells to cell-free purified NETs in the presence of 5F4 (p < 0.05), but not isotype or vehicle control (p > 0.05), an adhesion effect equal to NET degradation with DNase I (p < 0.05, Fig. 2A, 2B). This was not the case for the adhesion of the human lung carcinoma cell line A549, which is a low CEACAM 1 expressing cell line (29) (Fig. 1D), whereby 5F4 addition did not decrease adhesion of A549 cells to the huNETs as compared with vehicle and isotype control (Fig. 2C, 2D); this adhesion only decreased following the addition of DNase I (Fig. 2C, 2D).
FIGURE 2.
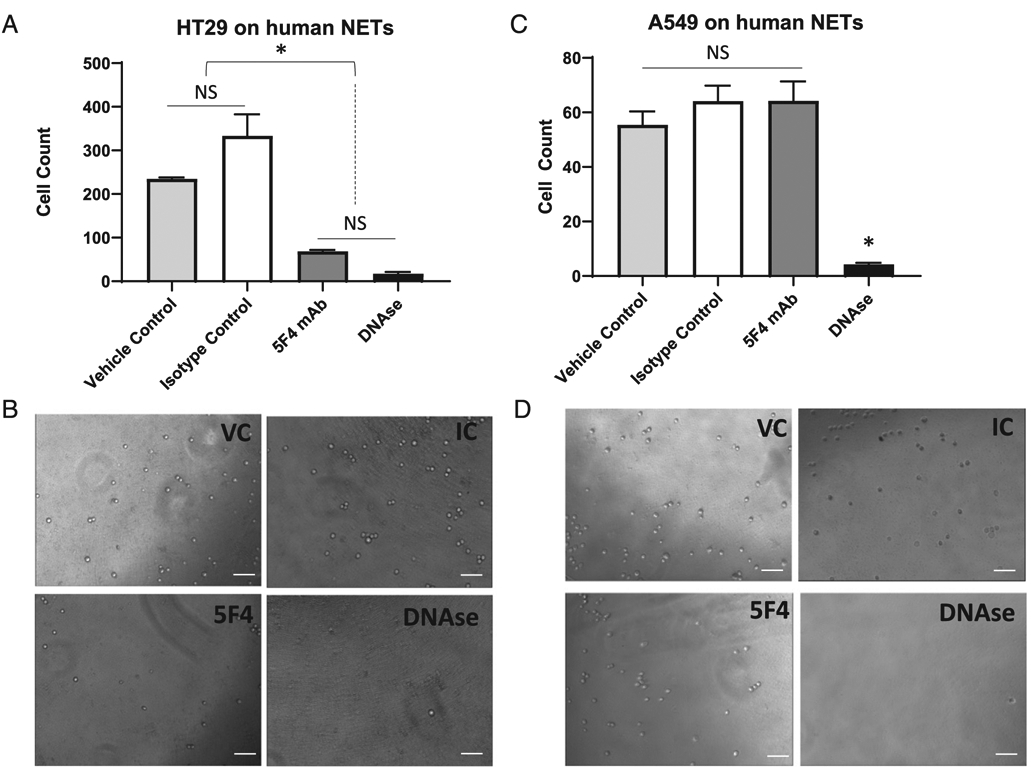
CEACAM1 on NETs is important for adhesion of human colon carcinoma cells. (A) In vitro adhesion assay of CFSE-labeled HT-29 cells on isolated human NETs in the presence of water (vehicle control for DNase I, light gray bar), 200 μg/ml isotype control of the CEACAM1 Ab (white bar), 200 μg/ml CEACAM1 functional blocking Ab (5F4 mAb, dark gray bar), and 1000 U DNase I (black bar). Shown are the mean and mean SE of three experiments. *p < 0.05. (B) Representative bright field images of images analyzed in (A). Scale bar, 100 μm. (C) In vitro adhesion assay of CFSE-labeled A549 cells on isolated human NETs in the presence of water (vehicle control for DNase I, light gray bar), 200 μg/ml isotype control of the CEACAM1 Ab (white bar), 200 μg/ml CEACAM1 functional blocking Ab (5F4 mAb, dark gray bar), and 1000 U DNase I (black bar). *p < 0.05. (D) Representative bright field images of images analyzed in (C). Scale bar, 100 μm.
When neutrophils extracted from CC1 KO mice were stimulated with PMA, the same percentage of NET release was observed as compared with those extracted from C57BL/6 mice (Fig. 3A, 3B). This indicated that CC1 KO neutrophils have the same capability to release NETs as C57BL/6 neutrophils. We then performed an in vitro adhesion assay of a murine colon carcinoma cell line overexpressing CEACAM1-L (CC1L) on isolated bone marrow neutrophils from either C57BL/6 or CC1 KO mice. These neutrophils were stimulated or not with 500 nM of PMA for 1 h to undergo NETosis (ex vivo) prior to addition of the MC38-CC1L. We observed that adhesion of MC38-CC1L to PMA-stimulated C57BL/6 mouse neutrophils (black bars) was increased by ~3-fold compared with unstimulated neutrophils (p < 0.05), an effect that was completely attenuated by DNase I treatment (p < 0.05) but not its vehicle control (Fig. 3C, 3D). When a neutralizing Ab for CEACAM1 was added, the adhesion of MC38-CC1L was significantly decreased by 2-fold (Fig. 3C, 3D). This is was not the case for the isotype control of the CEACAM1 Ab (Fig. 3C, 3D). PMA stimulation of neutrophils isolated from CC1 KO mice (gray bars) did not increase the adhesion to MC38-CC1L cells as compared with C57BL/6 neutrophils (p > 0.05) and was same as the adhesion observed using C57BL/6 and CC1 KO neutrophils treated with DNase I (Fig. 3C, 3D). Moreover, addition of the neutralizing Ab had no effect on the adhesion of MC38-CC1L on CC1 KO neutrophils, same as with its isotype control (Fig. 3C, 3D). Based on these results from both human and murine adhesion assays, we conclude that, in vitro, CEACAM1 on NETs is an important adhesion molecule mediating colon carcinoma-NET interaction.
FIGURE 3.
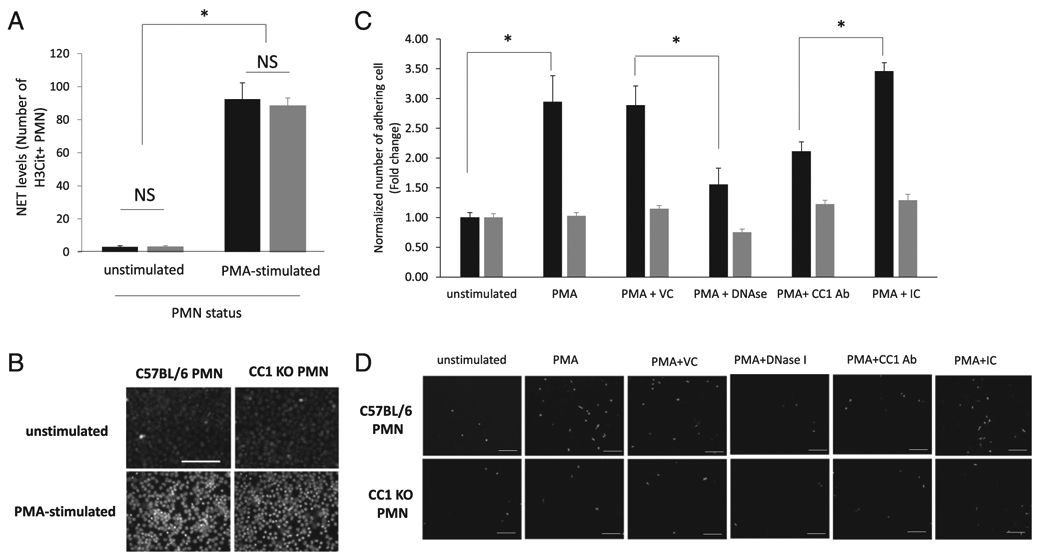
CEACAM1 on NETs is important for adhesion of murine colon carcinoma cells. (A) Bar graph showing number of H3-Citrulline–positive neutrophil (obtained by counting under a fluorescent microscope) in unstimulated neutrophils and in neutrophils stimulated with 500 nM PMA for 4 h (n = 5). *p < 0.05. (B) Representative images of each condition in (A) acquired using fluorescent microscopy. Scale bar, 100 μm. (C) In vitro adhesion assay of CFSE-labeled MC38-CC1L cells on C57BL/6 (WT, black bars) or CC1 KO (gray bars) murine neutrophils in the presence or absence of 500 nM PMA, water (the vehicle control [VC] to DNase I), 1000 U DNase I, a murine CEACAM1 neutralizing Ab (CC1 Ab), and its isotype control (IC). Shown are the mean and mean SE of three experiments. *p < 0.05. (D) Representative fluorescent images of data presented in (C). Scale bar, 100 μm.
CEACAM1 on NETs is important in migration of colon carcinoma cells
To investigate whether CEACAM1 on NETs plays a postadhesive role in tumor progression, we performed conventional Boyden chamber migration assays. We show that PMA-stimulated neutrophils from healthy individuals treated with 5F4 (CEACAM1 mAb, second bar) or an NEi (Sivelestat) (fourth bar) significantly decreased (by 4-fold) the migration of human HT-29 colon carcinoma cells compared with their migration on PMA-stimulated neutrophils (first bar) and on IgG control treated PMA-stimulated neutrophils (third bar) (p < 0.05, Fig. 4A, 4B). Moreover, we show that PMA addition does not affect tumor cell migration in the absence of neutrophils (p > 0.05, Supplemental Fig. 3A), an indication that the enhanced migration observed following PMA stimulation of neutrophils is due to NETs and not PMA.
FIGURE 4.
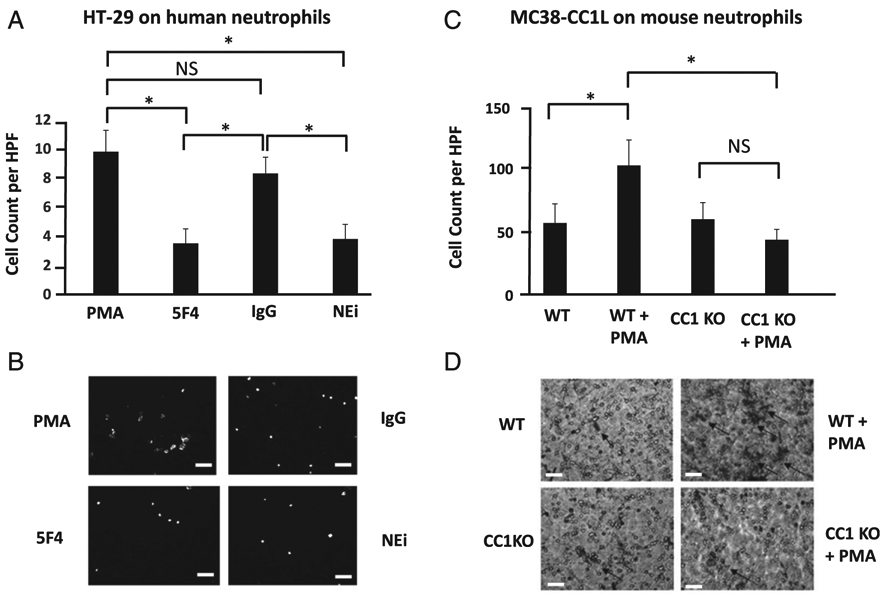
CEACAM1 on NETs is important in migration of colon carcinoma cells. (A) Boyden chamber assays of HT-29 cells on NET monolayer isolated from human neutrophils stimulated with 500 nM PMA and treated with 200 μg/ml 5F4 mAb (5F4), 200 μg/ml isotype control of the 5F4 mAb (IgG), or 10 μM Sivelestat (NEi). Shown are the mean and mean SE of five fields per condition done in triplicate. *p < 0.05, NS compared with stimulated neutrophils. (B) Representative fluorescent images of the migrated CFSE-HT-29 cells of the four conditions presented in (A). Scale bar, 20 μm. (C) Boyden chamber assays of MC38-CC1L cells on PMA-stimulated (light gray bar) or non-stimulated (black bar) neutrophils from C57BL/6 (WT) or PMA-stimulated (white bar) or non-stimulated neutrophils (dark gray bar) from CC1 KO mice. Shown are the mean and mean SE of four fields per condition done in triplicate. *p < 0.05. (D) Representative bright field images of crystal violet stained MC38-CC1L migrated cells of the four conditions depicted in (C). Scale bar, 20 μm.
We also observed that PMA-stimulated neutrophils from C57BL/6 mice (second bar) significantly increased (by 50%) MC38-CC1L cancer cell migration compared with their migration on unstimulated neutrophils (first bar, p < 0.05, Fig. 4C, 4D). However, PMA-stimulated neutrophils isolated from CC1 KO mice (fourth bar) did not increase the migration of MC38-CC1L cells compared with their migration on unstimulated neutrophils from CC1 KO mice (third bar, p > 0.05, Fig. 4C, 4D). In fact, MC38-CC1L cells exposed to PMA-stimulated CC1 KO neutrophils had a significant (by 50%) reduction in migration compared with those exposed to PMA-stimulated neutrophils from C57BL/6 mice (p < 0.05, Fig. 4C, 4D). PMA addition does not affect tumor cell migration in the absence of neutrophils (p > 0.05, Supplemental Fig. 3B), again an indication that the enhanced migration observed following PMA-stimulation of neutrophils is due to NETs and not PMA.
These results suggest that CEACAM1 on NETs help facilitate murine and human colon carcinoma cell migration in vitro.
CEACAM1 on NETs is important in adhesion of murine colon carcinoma cells to liver sinusoids in vivo
To validate these findings in vivo, we performed intravital hepatic microscopy on C57BL/6 mice intrasplenically injected with MC38-CC1L. Using this same model, and through a series of neutrophil depletion–reinfusion experiments, we have previously demonstrated that in vivo adhesion of cancer cells increased in the presence of circulating NETs (7). Using the same experimental design, we compared in this study the adhesion of MC38-CC1L murine colon carcinoma cells in C57BL/6 mice by intravital microscopy following neutrophil depletion (using a single i.p. injections of 150 μg of anti-Ly6G/GR1 Ab) and reinfusion with either C57BL/6 or CC1 KO neutrophils (experimental design schematically depicted in Fig. 5A). Reinfusion of PMA-stimulated neutrophils from C57BL/6 mice (second bar) significantly increased the adhesion of MC38-CC1L cells by 4-fold compared with reinfusion with unstimulated C57BL/6 neutrophils (first bar) (p < 0.05, Fig. 5B, 5C). Adhesion levels of MC38-CC1L cells in mice depleted and reinfused with PMA-stimulated neutrophils treated with DNase I from C57BL/6 mice (third bar) were comparable to those seen for unstimulated neutrophils (Fig. 5B, 5C). Mice that were neutrophil depleted and reinfused with CC1 KO neutrophils, unstimulated (fourth bar), PMA stimulated (fifth bar) and PMA stimulated and treated with DNase I (six bar) all had similar low adhesion levels of MC38-CC1L cells (Fig. 5B, 5C). MC38-CC1L adhesion levels in neutrophil-depleted mice reinfused with PMA-stimulated CC1 KO neutrophils (fifth bar) are significantly lower than those of neutrophil-depleted mice reinfused with PMA-stimulated C57BL/6 neutrophils (second bar) (p < 0.05, Fig. 5B, 5C). This experiment demonstrates not only that NETs themselves seem to enhance tumor cell arrest in liver sinusoids, as we have previously demonstrated (7), but that in the absence of CEACAM1 on NETs, the effect mimics that of unstimulated neutrophils. Therefore, the proarrest activity of NETs is dependent on CC1 expression.
FIGURE 5.

CEACAM1 on NETs is important in adhesion of murine colon carcinoma cells to liver sinusoids in vivo. (A) Schematic depiction of the depletion reinfusion experiment. IVM, intravital microscopy; PMN, polymorphonuclear neutrophils. (B) Hepatic intravital video microscopy of CFSE-labeled MC38-CC1L in the liver sinusoids of neutrophil-depleted C57BL/6 mice (WT) reinfused with neutrophils from C57BL/6 mouse neutrophils either nonstimulated (first bar) or stimulated with 500 nM PMA (second bar) or with PMA and DNase 1 (1000 U) (third bar) or from CC1 KO mouse neutrophils either nonstimulated (fourth bar) or stimulated with PMA (fifth bar) or with PMA and DNase 1 (sixth bar). Shown are the mean and mean SE of five to seven experiments. *p < 0.05. (C) Representative images of the six conditions presented above with CFSE-labeled MC38-CC1L cells shown in green. Scale bar, 10 μm.
Tumor cells have significantly reduced metastatic potential in CC1 KO mice even in the presence of widespread NET deposition
As we have demonstrated above, CEACAM1 on NETs is able to adhere to and capture colon carcinoma cell lines both in vitro and in vivo. As we have previously shown that CLP results in widespread intravascular NET deposition (7), we sought to employ this clinically relevant model of postoperative infection to determine the influence of NET-associated CEACAM1 on cancer metastasis. We first show that CC1 KO and C57BL/6 mice have the same number of neutrophils in circulation (around 9–10% of all circulating WBCs; Supplemental Fig. 4). Thus, induction of NETosis in the two mouse models will result in similar number of NETs being released because we have previously shown that CC1 KO neutrophils have the same capability to release NETs as C57BL/6 neutrophils (Fig. 3A, 3B). MC38-CC1L colon cancer cells were intrasplenically injected into C57BL/6 and CC1 KO mice 24 h following CLP (to induce NET formation), and gross hepatic metastases were quantified 18 d following tumor injection. In C57BL/6, gross hepatic metastases were significantly (p < 0.05) increased in CLP-treated versus sham mice (median of 188 nodules versus 4.5 nodules, respectively), and this increase in gross hepatic metastases was completely attenuated by degrading NETs with daily DNase I injection (median of four nodules, p < 0.05) (Fig. 6). In CC1 KO mice, gross hepatic metastases were not significantly increased in CLP-treated mice compared with sham or to CLP-treated mice treated with DNase I (median of three nodules compared with 11 nodules and 9.5 nodules respectively, p > 0.05) (Fig. 6). CC1 KO CLP-treated mice had significantly less hepatic metastasis than C57BL/6 CLP-treated mice (median of 11 nodules compared with 188 nodules respectively, p < 0.05) (Fig. 6).
FIGURE 6.

CC1 KO mice have significantly less metastatic potential in the presence of widespread NET deposition in a sepsis mouse model. (A) In vivo hepatic metastasis assay was performed on C57BL/6 (WT) and CC1 KO mice under three conditions each: sham, CLP, and CLP followed by daily i.m. DNase 1 treatment. Shown are average number of visible hepatic nodules (with SEs) of the six groups above mentioned with the number of mice shown below as n. *p < 0.05. (B) Representative images of the livers of the six groups of mice above mentioned at necropsy, 18 d after MC38-CC1L tumor injection. (C) H&E staining on representative liver sections from the six groups of mice mentioned above. Scale bar, 100 μm.
These results prove that tumor cells have significantly reduced metastatic potential in CC1 KO mice even in the presence of widespread NET deposition.
Discussion
Neutrophils are the most abundant WBCs in the body and the first responders to tissue injury and infection. Many studies have reported an association between higher number of neutrophils and/or high neutrophil/lymphocyte ratios in circulation and poor prognosis, adverse outcome and decreased survival in multiple cancer types (reviewed in Refs. 2, 3). Indeed, neutrophils can enhance adhesion of circulating tumor cells, thereby promoting metastatic progression via direct interactions (31), secreted factors (32), and NETs (7).
NETs are linked to several pathological conditions (reviewed in Ref. 6) and were shown to drive tumor progression via multiple mechanisms (reviewed in Refs. 8, 9). We have previously shown that one modality through which NETs enhance tumor progression is by trapping circulating tumor cells (7). This was shown in the context of postoperative complications (sepsis) and could be reversed by treating NETs with DNase I or Sivelestat, a known NET degrader and inhibitor, respectively (7). This prompted us to identify the adhesion molecules present on NETs that enhance adhesion and subsequently metastasis of colon cancer cells to secondary organs. In this study, we identified 283 proteins present on NETs. Many of these were adhesion molecules belonging to the integrin (ITGAM, ITGB2, ITGAIIb, ITGAL) and the CEACAM family (CEACAM1, CEACAM6, CECAM8). Identifying α2 integrin on NETs further validates our previous work that identified β1 integrin on tumor cells as an important interacting partner with NETs (33), because integrin α2 and β1 are known to form heterodimers to promote tumor migration and metastasis (34).
In this study, we focused our work on CEACAM1 because this adhesion molecule is extensively studied in multiple cancer types and is shown to play important roles in tumor–immune cell interaction (reviewed in Refs. 14, 26). Indeed, CEACAM1 is now used as a biomarker for diagnosis of melanoma as well as breast, pancreatic, and bladder cancers (35-38) and is involved in multiple essential cancer-related phenomena including proliferation, metastasis, apoptosis, inflammation, and angiogenesis, among others (reviewed in Ref. 14). Moreover, CEACAM1 is expressed by several immune cell types (T cells, B cells, NK cells, dendritic cells, and neutrophils) and has been associated with development and progression of various cancers and in boosting metastasis (reviewed in Ref. 14). Indeed, nonstimulated neutrophils express little surface CEACAM1; only following activation is CEACAM1 transferred from the granules to the surface (24). Moreover, one group has reported that neutrophil infiltration and CEACAM1 overexpression on tumor cells are associated with poor clinical outcomes in tongue squamous cell carcinoma (39). However, to date, how CEACAM1 expression on neutrophils and on their NETs affects tumor progression remains unclear.
We first confirmed that CEACAM1 is present on both murine and human NETs. We then show that, by blocking CEACAM1 on human NETs or knocking it down in mice, we decrease adhesion and migration of tumor cells in vitro by more than 50%. Although adhesion and migration require different sets of activation patterns, we believe that they do occur in this case sequentially. Our hypothesis is that once cancer cells are trapped in the NET complexes, the cells are exposed to a variety of proteins that are present on the NETs (Supplemental Table 1). The NETs thus create a “micro-microenvironment” where tumor cells are near a concentrated area of proteins that can induce signaling in the tumor cells leading to adhesion, proliferation, migration among others. Therefore, it is possible that tumor cells get activated in the NET complex, leading to their release from the NET, and then subsequently those cells can adhere, intravasate, migrate and seed and grow in the secondary organ.
In vivo, PMA-stimulated C57BL/6 neutrophils reinfused in C57BL/6 neutrophil-depleted mice significantly increased tumor cell adhesion to the hepatic sinusoids compared with reinfusion of nonstimulated neutrophils or controls or PMA-stimulated CEACAM1 KO neutrophils. Finally, using an experimental liver metastasis model, intrasplenic injection, we show that tumor cells have significantly reduced metastatic potential in CC1 KO mice even in the presence of widespread NET deposition. To note, the intrasplenic injection does not encompass all the steps of the metastatic cascade such as EMT transition and extravasation for example; however, it does cover a substantial step from adhesion to intravasation to seeding, survival, and proliferation. This strongly suggests that CEACAM1 on NETs is a major mediator of tumor cell adhesion at metastatic sites.
CEACAM1 is known to have both homophilic interactions (40) and heterophilic interactions (41) with other members of the CEACAM family (reviewed in Ref. 42). Therefore, what is attracting about a CEACAM1 targeted therapy, in case of a homophilic interaction, is that targeting CEACAM1 will block it on both tumor cells and NETs creating a stronger double inhibition. Indeed, we do show that the colon carcinoma cell lines used in this study, MC38 and HT-29, have high CEACAM1 levels (~80 and 100% respectively), which can be involved in the homophilic interaction with the CEACAM1 on NETs (Fig. 1D, 1E, Supplemental Fig. 1). However, as mentioned above, CEACAM1 is also capable of forming heterodimers with CEA and CEACAM6 (43). It has been previously shown that HT-29 and MC38 do express high levels of CEA and CEACAM6, which can be involved in their heterophilic interaction with the NETs CEACAM1 (44, 45).
In addition, we show that in the context of sepsis, using a CLP model, we observe a significant decrease in circulating tumor cell metastasis to the hepatic sinusoids in CEACAM1 KO versus wild-type (WT) mice. We have previously shown that CLP induces massive NET deposition in the liver (7), which, based on our results in this study, is unequivocally covered with CEACAM1. Moreover, CEACAM1 on neutrophils was shown to prolong their survival by delaying apoptosis (46). Therefore, blocking or knocking out CEACAM1 from neutrophil will not only decrease available CEACAM1 sites for tumor cells to attach to but will also increase neutrophil apoptosis. Because tumor-associated neutrophils were shown to have the capacity to be immunosuppressive (47), this should relieve this immunosuppression and thus play an antitumor role. In addition, CEACAM1 was shown to play an immunosuppressive role by inhibiting T cells and NK cells cytotoxic activities (reviewed in Ref. 14). These findings lead to the speculation that CEACAM1 immunotherapy (alone or in combination with available immunotherapies such as CTLA-4 or PD-1) could soon become a reality. Indeed, CEACAM1 mAbs were successfully tested as immunotherapy drugs for both melanoma and colorectal cancers (48, 49). To note, we have used in this study an intrasplenic injection of colon carcinoma cells in our in vivo experimental metastasis assay. This commonly used inducible liver metastasis model (50) is a very robust and reproducible approach to investigating the seeding, adhesion, and growth of tumor cells in liver. Looking at the role of CEACAM1 in other metastasis models is important to corroborate our results in other organ systems.
Finally, a key reason to identify molecular mediators of NET-tumor cell interactions is to specifically target these to avoid the side effects of degrading NETs in general. Indeed, systemic degradation of NETs (e.g., by using DNase I) could lead to increased chances of sepsis, because NETs play an important role in clearing out microorganisms (4). Thus, the identification of β1 integrin (33) and CEACAM1 on NETs opens the door to establishing therapeutic targets of tumor cell adhesion and metastasis that warrant testing by blocking them both.
Supplementary Material
Acknowledgments
We thank Dr. Min Fu and the staff of the molecular imaging platform of the Research Institute of the McGill University Health Centre for help with the fluorescent confocal images acquired in this manuscript.
This work was supported by Canadian Institutes of Health Research Grant MOP-133567 (to L.E.F.).
Abbreviations used in this article:
- CC1 KO
Ceacam 1 knockout
- CEA
carcinoembryonic Ag
- CEACAM1
carcinoembryonic Ag cell adhesion molecule 1
- CLP
cecal-ligation-puncture
- Hpf
high-power field
- MS/MS
tandem mass spectrometry
- NEi
neutrophil elastase inhibitor
- NET
neutrophil extracellular trap
- RT
room temperature
- WT
wild-type
Footnotes
Disclosures
The authors have no financial conflicts of interest.
The online version of this article contains supplemental material.
References
- 1.Tüting T, and de Visser KE. 2016. CANCER. How neutrophils promote metastasis. Science 352: 145–146. [DOI] [PubMed] [Google Scholar]
- 2.Shen M, Hu P, Donskov F, Wang G, Liu Q, and Du J. 2014. Tumor-associated neutrophils as a new prognostic factor in cancer: a systematic review and meta-analysis. PLoS One 9: e98259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Templeton AJ, McNamara MG, Šeruga B, Vera-Badillo FE, Aneja P, Ocaña A, Leibowitz-Amit R, Sonpavde G, Knox JJ, Tran B, et al. 2014. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J. Natl. Cancer Inst 106: dju124. [DOI] [PubMed] [Google Scholar]
- 4.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, and Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535. [DOI] [PubMed] [Google Scholar]
- 5.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, and Zychlinsky A. 2007. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol 176: 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sprensen OE, and Borregaard N. 2016. Neutrophil extracellular traps - the dark side of neutrophils. J. Clin. Invest 126: 1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, Bourdeau F, Kubes P, and Ferri L. 2013. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Invest DOI: 10.1172/JCI67484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cedervall J, and Olsson AK. 2016. Immunity gone astray - NETs in cancer. Trends Cancer 2: 633–634. [DOI] [PubMed] [Google Scholar]
- 9.Cools-Lartigue J, Spicer J, Najmeh S, and Ferri L. 2014. Neutrophil extracellular traps in cancer progression. Cell. Mol. Life Sci 71: 4179–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andalib A, Ramana-Kumar AV, Bartlett G, Franco EL, and Ferri LE. 2013. Influence of postoperative infectious complications on long-term survival of lung cancer patients: a population-based cohort study. J. Thorac. Oncol 8: 554–561. [DOI] [PubMed] [Google Scholar]
- 11.Nojiri T, Hamasaki T, Inoue M, Shintani Y, Takeuchi Y, Maeda H, and Okumura M. 2017. Long-term impact of postoperative complications on cancer recurrence following lung cancer surgery. Ann. Surg. Oncol 24: 1135–1142. [DOI] [PubMed] [Google Scholar]
- 12.Gu A, Zhang Z, Zhang N, Tsark W, and Shively JE. 2010. Generation of human CEACAM1 transgenic mice and binding of Neisseria Opa protein to their neutrophils. PLoS One 5: e10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skubitz KM, and Skubitz AP. 2008. Interdependency of CEACAM-1, −3, −6, and −8 induced human neutrophil adhesion to endothelial cells. J. Transl. Med 6: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beauchemin N, and Arabzadeh A. 2013. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 32: 643–671. [DOI] [PubMed] [Google Scholar]
- 15.Arabzadeh A, Chan C, Nouvion AL, Breton V, Benlolo S, DeMarte L, Turbide C, Brodt P, Ferri L, and Beauchemin N. 2013. Host-related carcinoembryonic antigen cell adhesion molecule 1 promotes metastasis of colorectal cancer. Oncogene 32: 849–860. [DOI] [PubMed] [Google Scholar]
- 16.Arabzadeh A, Dupaul-Chicoine J, Breton V, Haftchenary S, Yumeen S, Turbide C, Saleh M, McGregor K, Greenwood CM, Akavia UD, et al. Carcinoembryonic antigen cell adhesion molecule 1 long isoform modu-lates malignancy of poorly differentiated colon cancer cells. Gut 65: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Najmeh S, Cools-Lartigue J, Giannias B, Spicer J, and Ferri LE. 2015. Simplified human neutrophil extracellular traps (NETs) isolation and handling. J. Vis. Exp Available at: https://www.jove.com/video/52687/simplified-humanneutrophil-extracellular-traps-nets-isolation. Accessed: March 16, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macsai A, Zhang H, Jakus Z, Kitaura J, Kawakami T, and Lowell CA. 2003. G-protein-coupled receptor signaling in Syk-deficient neutrophils and mast cells. Blood 101: 4155–4163. [DOI] [PubMed] [Google Scholar]
- 19.Keller A, Nesvizhskii AI, Kolker E, and Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem 74: 5383–5392. [DOI] [PubMed] [Google Scholar]
- 20.Nesvizhskii AI, Keller A, Kolker E, and Aebersold R. 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem 75: 4646–4658. [DOI] [PubMed] [Google Scholar]
- 21.Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moldawer LL, and Efron PA. 2010. Cecal ligation and puncture Curr. Protoc. Immunol Chapter 19: Unit 19.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gommerman JL, Oh DY, Zhou X, Tedder TF, Maurer M, Galli SJ, and Carroll MC. 2000. A role for CD21/CD35 and CD19 in responses to acute septic peritonitis: a potential mechanism for mast cell activation. J. Immunol 165: 6915–6921. [DOI] [PubMed] [Google Scholar]
- 23.Fiori V, Magnani M, and Cianfriglia M. 2012. The expression and modulation of CEACAM1 and tumor cell transformation. Ann. Ist. Super. Sanita 48: 161–171. [DOI] [PubMed] [Google Scholar]
- 24.Ducker TP, and Skubitz KM. 1992. Subcellular localization of CD66, CD67, and NCA in human neutrophils. J. Leukoc. Biol 52: 11–16. [DOI] [PubMed] [Google Scholar]
- 25.Jantscheff P, Nagel G, Thompson J, Kleist SV, Embleton MJ, Price MR, and Grunert F. 1996. A CD66a-specific, activation-dependent epitope detected by recombinant human single chain fragments (scFvs) on CHO transfectants and activated granulocytes. J. Leukoc. Biol 59: 891–901. [DOI] [PubMed] [Google Scholar]
- 26.Dankner M, Gray-Owen SD, Huang YH, Blumberg RS, and Beauchemin N. 2017. CEACAM1 as a multi-purpose target for cancer immunotherapy. OncoImmunology 6: e1328336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang W, and Ferrara N. 2016. The complex role of neutrophils in tumor angiogenesis and metastasis. Cancer Immunol. Res 4: 83–91. [DOI] [PubMed] [Google Scholar]
- 28.Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, Brinkmann V, Bernuth HV, and Zychlinsky A. 2017. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife 6: e24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singer BB, Scheffrahn I, Kammerer R, Suttorp N, Ergun S, and Slevogt H. 2010. Deregulation of the CEACAM expression pattern causes undifferentiated cell growth in human lung adenocarcinoma cells. PLoS One 5: e8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Z, Chen L, Baker K, Olszak T, Zeissig S, Huang YH, Kuo TT, Mandelboim O, Beauchemin N, Lanier LL, and Blumberg RS. 2011. CEACAM1 dampens antitumor immunity by down-regulating NKG2D ligand expression on tumor cells. J. Exp. Med 208: 2633–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spicer JD, McDonald B, Cools-Lartigue JJ, Chow SC, Giannias B, Kubes P, and Ferri LE. 2012. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer Res. 72: 3919–3927. [DOI] [PubMed] [Google Scholar]
- 32.Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, Stolz DB, Land SR, Marconcini LA, Kliment CR, et al. 2010. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med 16: 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Najmeh S, Cools-Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F, Giannias B, Berube J, Rousseau S, Ferri LE, and Spicer JD. 2017. Neutrophil extracellular traps sequester circulating tumor cells via b1-integrin mediated interactions. Int. J. Cancer 140: 2321–2330. [DOI] [PubMed] [Google Scholar]
- 34.Naci D, Vuori K, and Aoudjit F. 2015. Alpha2beta1 integrin in cancer development and chemoresistance. Semin. Cancer Biol 35: 145–153. [DOI] [PubMed] [Google Scholar]
- 35.Markel G, Ortenberg R, Seidman R, Sapoznik S, Koren-Morag N, Besser MJ, Bar J, Shapira R, Kubi A, Nardini G, et al. 2010. Systemic dysregulation of CEACAM1 in melanoma patients. Cancer Immunol. Immunother 59: 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simeone DM, Ji B, Banerjee M, Arumugam T, Li D, Anderson MA, Bamberger AM, Greenson J, Brand RE, Ramachandran V, and Logsdon CD. 2007. CEACAM1, a novel serum biomarker for pancreatic cancer. Pancreas 34: 436–443. [DOI] [PubMed] [Google Scholar]
- 37.Tilki D, Singer BB, Shariat SF, Behrend A, Fernando M, Irmak S, Buchner A, Hooper AT, Stief CG, Reich O, and Ergün S. 2010. CEACAM1: a novel urinary marker for bladder cancer detection. Eur. Urol 57: 648–654. [DOI] [PubMed] [Google Scholar]
- 38.Yang C, He P, Liu Y, He Y, Yang C, Du Y, Zhou M, Wang W, Zhang G, Wu M, and Gao F. 2015. Assay of serum CEACAM1 as a potential biomarker for breast cancer. Clin. Chim. Acta 450: 277–281. [DOI] [PubMed] [Google Scholar]
- 39.Wang N, Feng Y, Wang Q, Liu S, Xiang L, Sun M, Zhang X, Liu G, Qu X, and Wei F. 2014. Neutrophils infiltration in the tongue squamous cell carcinoma and its correlation with CEACAM1 expression on tumor cells. PLoS One 9: e89991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watt SM, Teixeira AM, Zhou GQ, Doyonnas R, Zhang Y, Grunert F, Blumberg RS, Kuroki M, Skubitz KM, and Bates PA. 2001. Homophilic adhesion of human CEACAM1 involves N-terminal domain interactions: structural analysis of the binding site. Blood 98: 1469–1479. [DOI] [PubMed] [Google Scholar]
- 41.Klaile E, Vorontsova O, Sigmundsson K, Müller MM, Singer BB, Ofverstedt LG, Svensson S, Skoglund U, and Obrink B. 2009. The CEACAM1 N-terminal Ig domain mediates cis- and trans-binding and is essential for allosteric rearrangements of CEACAM1 microclusters. J. Cell Biol 187: 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gray-Owen SD, and Blumberg RS. 2006. CEACAM1: contact-dependent control of immunity. Nat. Rev. Immunol 6: 433–446. [DOI] [PubMed] [Google Scholar]
- 43.Oikawa S, Kuroki M, Matsuoka Y, Kosaki G, and Nakazato H. 1992. Homotypic and heterotypic Ca(++)-independent cell adhesion activities of biliary glycoprotein, a member of carcinoembryonic antigen family, expressed on CHO cell surface. Biochem. Biophys. Res. Commun 186: 881–887. [DOI] [PubMed] [Google Scholar]
- 44.Guadagni F, Witt PL, Robbins PF, Schlom J, and Greiner JW. 1990. Regulation of carcinoembryonic antigen expression in different human colorectal tumor cells by interferon-gamma. Cancer Res. 50: 6248–6255. [PubMed] [Google Scholar]
- 45.Holmer R, Wätzig GH, Tiwari S, Rose-John S, and Kalthoff H. 2015. Interleukin-6 trans-signaling increases the expression of carcinoembryonic antigen-related cell adhesion molecules 5 and 6 in colorectal cancer cells. BMC Cancer 15: 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singer BB, Klaile E, Scheffrahn I, Müller MM, Kammerer R, Reutter W, Obrink B, and Lucka L. 2005. CEACAM1 (CD66a) mediates delay of spontaneous and Fas ligand-induced apoptosis in granulocytes. Eur. J. Immunol 35: 1949–1959. [DOI] [PubMed] [Google Scholar]
- 47.Fridlender ZG, and Albelda SM. 2012. Tumor-associated neutrophils: friend or foe? Carcinogenesis 33: 949–955. [DOI] [PubMed] [Google Scholar]
- 48.Liu J, Di G, Wu CT, Hu X, and Duan H. 2013. Development and evaluation of a novel anti-colorectal cancer monoclonal antibody, WL5. Biochem. Biophys. Res. Commun 432: 370–377. [DOI] [PubMed] [Google Scholar]
- 49.Ortenberg R, Sapir Y, Raz L, Hershkovitz L, Ben Arav A, Sapoznik S, Barshack I, Avivi C, Berkun Y, Besser MJ, et al. 2012. Novel immunotherapy for malignant melanoma with a monoclonal antibody that blocks CEACAM1 homophilic interactions. Mol. Cancer Ther 11: 1300–1310. [DOI] [PubMed] [Google Scholar]
- 50.Evans JP, Sutton PA, Winiarski BK, Fenwick SW, Malik HZ, Vimalachandran D, Tweedle EM, Costello E, Palmer DH, Park BK, and Kitteringham NR. 2016. From mice to men: murine models of colorectal cancer for use in translational research. Crit. Rev. Oncol. Hematol 98: 94–105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.