

Abstract
Genomes have complex three-dimensional architectures. The recent convergence of genetic, biochemical, biophysical and cell biological methods has uncovered several fundamental principles of genome organization. They highlight that genome function is a major driver of genome architecture and that structural features of chromatin act as modulators, rather than binary determinants, of genome activity. The interplay of these principles in the context of self-organization accounts for the emergence of structural chromatin features, the diversity and single-cell heterogeneity of nuclear architecture in cell-types and tissues, and evolutionarily conserved functional features of genomes, including plasticity and robustness.
Keywords: genome organization, chromatin, nuclear architecture, phase-separation, dynamics, gene expression, self-organization
eTOC blurb Misteli
Tom Misteli offers an update on how our view of genome organization has changed since his classic 2007 Review. Principles of heterogeneity and stochasticity, polymer-polymer interactions, chromatin dynamics, phase separation, and architectural elements frame the updated model.
Introduction
The vast majority of an organism’s hereditary information in eukaryotes is stored, transcribed and replicated in the cell nucleus. To accommodate the genome in the confined space of the nucleus, the genetic material – ~2m of DNA in a human cell – must be organized and compacted at multiple levels. Beyond solving this challenging topological problem, the organization of the genome must be such that the correct gene expression programs can be executed at the right time and in the right tissue and cell type.
Deep insights into genome organization have come from microscopy approaches that have documented large-scale subnuclear features, such as morphologically recognizable blocks of heterochromatin, the existence of subnuclear bodies, and the organization of DNA into chromosome territories (Misteli, 2007; Rowley and Corces, 2018). Complementary biochemical approaches, based on chemical crosslinking of chromatin, have provided a comprehensive view of genome topology and have demonstrated the existence of chromatin loops and domains as ubiquitous features in genome organization (Dekker and Mirny, 2016; Pombo and Dillon, 2015). Combined, these approaches have generated a framework for how genomes are organized in space and time.
An important realization from these studies has been that the organization of genomes is characterized by a high degree of order and non-randomness (Misteli, 2007; Rowley and Corces, 2018). An overt example is the physical segregation of transcriptionally active euchromatin from repressed heterochromatin into distinct regions in the cell nucleus of most eukaryotic cells (Fawcett, 1966). Other non-random features of genomes include the formation of chromatin domains and the positioning of genes to preferred location within the nuclear space (Fraser and Bickmore, 2007; Rowley and Corces, 2018). In addition to the genetic material, many proteins are non-randomly distributed in the nucleus and are concentrated in sub-nuclear bodies, such as the nucleolus or splicing factor speckles (Spector and Lamond, 2011; Stanek and Fox, 2017). These observations highlight a considerable degree of order and non-randomness in genome organization.
Elucidating the prominent organizational patterns that are evident at many levels of genome organization has, not surprisingly, been the focus of most efforts in the field. More recently, an appreciation has grown that all genome features are also characterized by a high degree of variability and heterogeneity (Finn and Misteli, 2019). For example, the two alleles of a gene often differ in their 3D position and functional status in the same nucleus, many chromatin-chromatin interactions only occur with low frequency in individual cells in a population, and the shape and number of nuclear bodies fluctuates considerably amongst individual cells (Cattoni et al., 2017; Finn and Misteli, 2019; Rodriguez et al., 2019).
The dichotomy of non-randomness yet high variability presents a paradox for our understanding of the mechanisms that shape genome organization. On the one hand, the presence of non-random patterns points to the existence of specific molecular and physical mechanisms that determine genome architecture. On the other, the high variability suggests that these features are not hard-wired nor strictly required for faithful genome function. The presence of heterogeneity in the spatial organization of genomes raises the question of how the observed non-random structural features and morphological patterns of genome architecture come about. Above all, it provokes the question of whether the organization of the genome is important for its function.
The convergence of genetic, biochemical, biophysical and cell biological observations has provided unprecedented insight into how genomes are organized in 3D space. Here I summarize several major principles that shape genome organization and function. This review focuses on overarching concepts rather than on the detailed role of individual genome regulators, such as particular chromatin complexes or RNA, which are widely covered in the literature (Bracken et al., 2019; Khosraviani et al., 2019). The recent insights are synthesized into a unifying model for higher-order genome architecture in which the major features of spatial and temporal genome organization and function are emergent properties in a self-organizing system.
Architectural features of the genome
Eukaryotic genomes are organized via several ubiquitous architectural features. The basic organizational elements of the genome are the fibers, loops, domains, and compartments that chromatin forms, as well as chromosomes. These features organize genomes at multiple levels and over several length-scales (Fig. 1).
Figure 1. The organization of the eukaryotic genome.
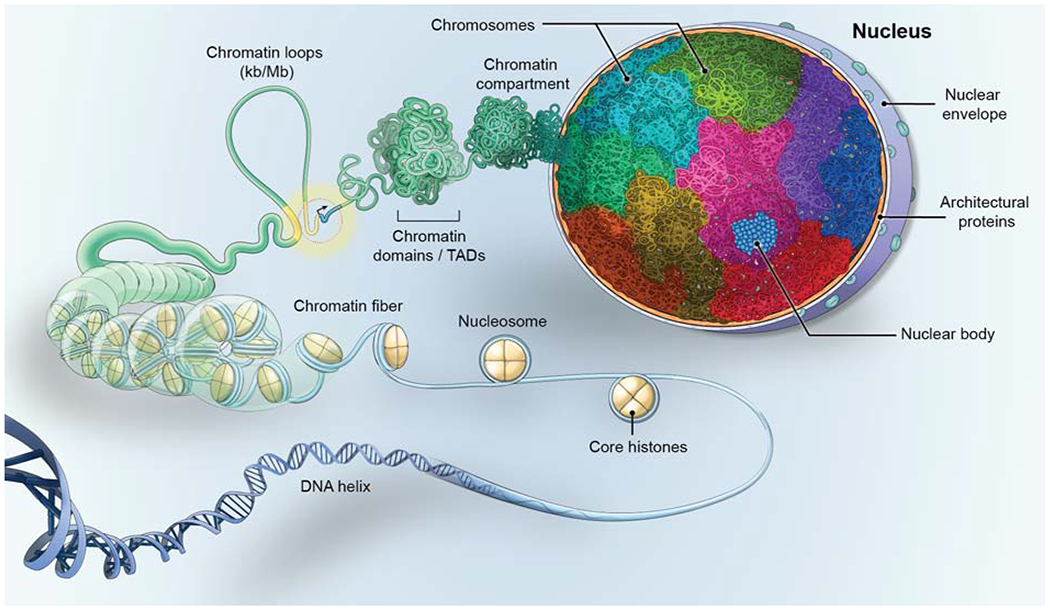
Genomes are organized at multiple levels. DNA is wrapped around the nucleosome, which is made up of an octamer of core-histones, forming the chromatin fiber which folds into loops, often bringing upstream gene regulatory elements (yellow), such as enhancers, into proximity to genes (blue) to control their transcription (black arrow). The fiber then folds into chromatin domains, referred to as topologically associating domains (TADs), which associate with each other to create chromatin compartments. The DNA of each chromosome occupies a distinct volume, or chromosome territory (multiple colors), within the cell nucleus, generating non-random patterns of chromosome and gene locations. In the DNA-free space, the nucleus also contains RNA and proteinaceous protein aggregates which form nuclear bodies (blue).
The chromatin fiber is made up of units of 146 bps of DNA wrapped around nucleosomes that consist of octamers of core histone proteins (Fig. 1, for in depth review see (Felsenfeld and Groudine, 2003). While the precise nature of the chromatin fiber has been extensively debated, recent observations from several orthogonal methods, including visualization in intact cells using EM tomography, support the view that the chromatin fiber is typically 5-24 nm in diameter throughout the nucleus and is irregularly folded into higher-order features, such as loops and domains (Maeshima et al., 2019; Ou et al., 2017). It is tempting to speculate that the observed small diameter and flexible nature of the chromatin fiber favors a dynamic state that allows for facile access of regulatory proteins and RNA to chromatin (Kim and Shendure, 2019; Maeshima et al., 2019; Ou et al., 2017).
The chromatin fiber then self-interacts to form loops (Fig. 1, for an in-depth review see (Dekker and Misteli, 2015; Vermunt et al., 2019). Chromatin loops are a universal feature of genomes in most organisms and they exist in various sizes, ranging from kbs to Mbs, and have multiple functions (Dekker and Misteli, 2015). At the smallest scale, loops mediate the interaction of regulatory elements, often enhancers with gene promoters, over distances of typically 10 to several hundred kbs (Dekker and Misteli, 2015; Halfon, 2020; Vermunt et al., 2019). Multiple enhancers may loop to form superenhancer clusters thought to integrate signaling events or to act redundantly on target genes (Halfon, 2020; Hnisz et al., 2017). Larger loops, of up to Mbs in length, contribute to the 3D compaction of the genome and are frequently used to regulate gene clusters in a precise temporal and spatial fashion via the sequential association of upstream elements with individual target genes. This type of regulation is particularly common in developmentally regulated genes, which require coordinated and accurate temporal and spatial control (Darbellay and Duboule, 2016; Vermunt et al., 2019). While it has traditionally been assumed that looping interactions, including promoter-enhancer pairing, require the direct physical interaction of two distantly located sites, an emerging view is that aggregates of phase-separated transcription factors may bring genome regions into spatial proximity without the need for direct chromatin-chromatin interaction (Khanna et al., 2019; Sabari et al., 2018) (see below).
The next level of genome organization are chromatin domains (Fig. 1; for in depth review see (Dekker and Mirny, 2016; Pombo and Dillon, 2015; Rowley and Corces, 2018). They were first morphologically described by in situ hybridization experiments (Cremer and Cremer, 2010; Shopland et al., 2006) and their universality subsequently confirmed by genome-wide, biochemical crosslinking approaches (Dixon et al., 2012; Lieberman-Aiden et al., 2009; Sexton et al., 2012). Based on their defining feature of representing genome regions that preferentially interact with each other rather than with their surrounding sequences, they are referred to as topologically associating domains (TADs) (Dixon et al., 2012; Lieberman-Aiden et al., 2009; Sexton et al., 2012). They are typically several hundred kbs in size and form by a loop extrusion mechanisms, in which the cohesin complex drives the formation of chromatin loops via its ATP-dependent molecular motor activity (Fudenberg et al., 2016; Mirny et al., 2019). The extruded loop then folds onto itself to form a domain whose boundaries are defined by the architectural chromatin protein CTCF (Dekker and Mirny, 2016). This model is supported by the recent demonstration of loop extrusion activity of yeast cohesin in vitro (Davidson et al., 2019). With regards to function, it has been proposed that the domain boundaries serve to restrict the interactions of regulatory elements to genes within the domain, thus generating regulatory units (Beagrie et al., 2017; Dekker and Mirny, 2016). The recent fine-mapping of TADs has revealed that extensive internal loop interactions occur and supports the notion that locally co-regulated genes are frequently found within the same TAD (Hsieh et al., 2020; Krietenstein et al., 2020). In agreement with this view, the disruption of TAD boundaries can lead to dysregulation of genes within the TAD (Lupianez et al., 2015; Narendra et al., 2015). However, a strong regulatory role of TADs has been challenged. Systematic analysis of the effect of enhancers on their target genes in Drosophila revealed that the presence or absence of chromatin domains has no strong effect on gene regulation (Ghavi-Helm et al., 2019). In addition, in-depth mapping of chromatin structure during Drosophila embryo development demonstrates that regulatory chromatin loops form prior to TADs (Espinola et al., 2020) and that developmentally distinct cell types have remarkably similar TAD structures (Ing-Simmons et al., 2020). Furthermore, single-cell analysis has shown that TAD structure is highly variable between individual cells (Cattoni et al., 2017; Finn and Misteli, 2019; Nagano et al., 2013). These findings suggest that chromatin domains are not strong determinants of gene function but primarily provide a structural framework for the accurate regulation of genes. Understanding the relevance of chromatin structure for genome function is arguable one of the major priorities of current work in the field.
Individual chromatin domains have a propensity to assemble into higher-order chromatin compartments (Fig. 1; for in depth review see (Dekker and Mirny, 2016; Rowley and Corces, 2018). This level of organization is morphologically evident in the spatial segregation of heterochromatin and euchromatin (Jagannathan et al., 2019; Shopland et al., 2006). In line with cytological observations, genome-wide biochemical analyses have assigned each genomic locus, based on the frequency of long-range interactions, to one of two nuclear compartments, A or B, which roughly correspond to eu- and heterochromatin, respectively (Dekker and Mirny, 2016; Lieberman-Aiden et al., 2009; Rowley and Corces, 2018). The higher-order organization of domains is also indicated by the detection of interactions between adjacent TADs by biochemical mapping (Fraser et al., 2015) and imaging studies have documented the coalescence of multiple domains into higher-order structures (Shopland et al., 2006; Wang et al., 2016).
Chromosomes are the major unit of genome organization and they exist in the nucleus in the form of chromosome territories. Throughout interphase, the DNA that makes up a single chromosome occupies a relatively compact, spatially restricted, territory of the nucleus rather than being dispersed through the nuclear space (Fig. 1; for in depth review see (Cremer and Cremer, 2010). In mammalian cells, a chromosome territory is typically 2-3μm in diameter, amorphously spherical, and characterized by a high surface-to-volume ratio since its interior is permeated by a complex network of anastomosed channels that are thought to ensure the access of regulatory factors to chromatin. The chromatin fibers of neighboring chromosomes often intermingle, but do not entangle, at their periphery, thus creating a continuum of chromatin throughout the nuclear space, although a lower density of chromatin at chromosome interfaces has been reported (Branco and Pombo, 2006; Cremer and Cremer, 2010).
The ultimate level of genome organization is the location of chromosomes and gene loci in the 3D space of the cell nucleus (Fig. 1; for in depth review see (Cremer and Cremer, 2010; Crosetto and Bienko, 2020; Takizawa et al., 2008). Simple measures of the non-random location of chromosomes and genes include their position relative to the nuclear center or periphery, their location relative to other loci or chromosomes, or to nuclear bodies (Croft et al., 1999; Takizawa et al., 2008). For example, inactive genes often locate near the nuclear edge and associate with peripheral heterochromatin, whereas active genes are often in the nuclear interior (Croft et al., 1999). While most loci and chromosomes show non-randomness in their position, and most genome organization patterns are tissue- and cell-type specific (Parada et al., 2004), the precise functional role of 3D positioning of genes remains unclear (Crosetto and Bienko, 2020; Shachar and Misteli, 2017). The molecular mechanisms which determine the location of a chromosome or gene are largely unknown, although several proteins and chromatin modifications have been identified that correlate with the 3D location of some genes (Crosetto and Bienko, 2020; Shachar et al., 2015).
Principles of genome organization
The architectural features of the genome, as described above, are now well characterized. But how do they come about? There are several general principles, whose interplay leads to the emergence of the major organizational features of the genome (Fig. 2).
Figure 2. Principles of genome organization.
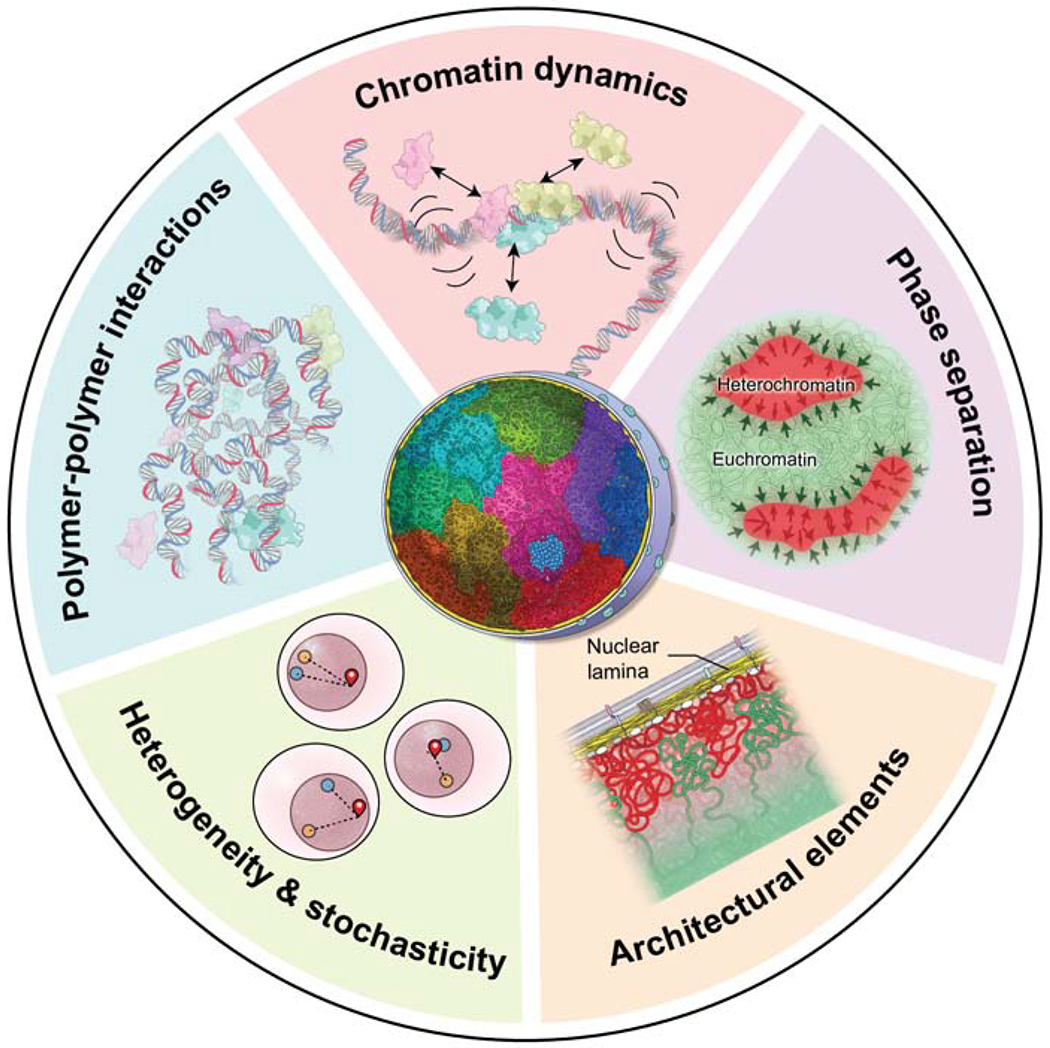
The interplay of several fundamental principles governs genome organization. (Light blue sector) Polymer-polymer interactions, mediated by chromatin binding proteins (light blue, pink, yellow), promote the formation of chromatin loops and shape the overall conformation of the chromatin fiber. (Pink sector) Chromatin undergoes local motion and chromatin-chromatin interactions are transient. Chromatin proteins (light blue, pink, yellow) undergo rapid cycles of association and dissociation with short residence times. (Purple sector) The process of phase separation involves the homotypic aggregation of proteins (arrows) and contributes to the formation and stabilization of chromatin-chromatin interactions and domains, including eu- and heterochromatin. (Orange sector) The physical interaction of chromatin with stable architectural elements of the nucleus such as the nuclear envelope limits the degree of freedom of a genome region and contributes to its non-random localization. (Green sector) the behavior of individual chromatin regions and genes (red, blue, yellow) varies stochastically in individual cells giving rise to extensive heterogeneity in genome organization and function amongst single cells in a population.
Polymer-polymer interactions
The genome is a polymer fiber and its folding is guided by principles of polymer physics (Bianco et al., 2020) (Fig. 2). Various computational chromatin polymer models have been developed and they quite accurately recapitulate many features of genome organization, pointing to polymer-polymer self-interactions as a major organizer of the genome (Parmar et al., 2019).
A driving role for intra-polymer interactions in genome organization is indicated by the finding that major chromatin features can be recreated in computational models by merely assuming that the chromatin fiber is a self-avoiding polymer that can undergo local diffusive motion and is constrained by its polymer nature (Barbieri et al., 2012; Jost et al., 2014; Parmar et al., 2019). In these models, key features of genome organization, including loops of various length scales, chromosome domains, and compartments spontaneously emerge as thermodynamically stable states (Barbieri et al., 2012; Jost et al., 2014). Simple polymer-interaction models are also sufficient to predict the local organization of gene loci, for example of the developmentally relevant Sox9 locus, the Bmp7 locus, or the Xist locus on the inactive X chromosome (Bianco et al., 2018). Polymer models can also account for several features of Hi-C datasets, which map chromatin-chromatin interactions genome-wide using chemical crosslinking methods, and these polymer models suggest that TADs and compartments are emergent properties of intra-polymer self-interactions (Falk et al., 2019; Jost et al., 2014; Nuebler et al., 2018). Domains of the size of TADs also emerge in simulations in which blocks of genome regions, such as euchromatin or heterochromatin, are assumed to undergo homotypic interactions along the chromatin fiber (Barbieri et al., 2012; Bianco et al., 2018). A striking example is the organization of chromatin in retinal rod cells of nocturnal animals, in which heterochromatin is located in the interior of the nucleus, which is the opposite of its typical enrichment at the periphery in most cell types (Solovei et al., 2009). Computational simulations of genome-wide interaction data recapitulate this inverted chromatin configuration merely by assuming that self-interactions occur among the heterochromatic regions of multiple chromosomes (Falk et al., 2019). Modeling of polymer-polymer interactions also reproduces changes in heterochromatin-lamina and heterochromatin re-organization in senescent cells (Sati et al., 2020). It is likely that similar mechanisms of polymer self-interactions are responsible for the formation of other homotypic chromatin compartments such as chromocenters, which contain multiple centromeres, or the nucleolus, in which multiple ribosomal genes cluster.
Chromatin interactions obviously do not occur on naked DNA; chromatin fibers are densely decorated with architectural proteins, including the core histones and accessory structural proteins. Chromatin is also acted upon by chromatin remodeling factors and by the transcriptional machinery (Brackley et al., 2016; Buckle et al., 2018). These chromatin-binding proteins significantly contribute to determining the patterns of chromatin-chromatin self-interactions along the fiber. Tellingly, the inclusion of target sites for chromatin-binding proteins in polymer models increases their accuracy (Brackley et al., 2016; Buckle et al., 2018).
Two of the most prominent chromatin binders that determine the formation of loops and domains are the chromosome cohesion factor, cohesin, and the chromatin architectural protein, CTCF, which has traditionally been thought to demarcate insulator sequences that separate functionally distinct genome regions (Rowley and Corces, 2018). Recent findings suggest that cohesin and CTCF cooperate to drive chromatin interactions by promoting the formation of chromatin loops and TADs (Dekker and Mirny, 2016; Rowley and Corces, 2018). In line with this function, CTCF is enriched at TAD boundaries and the deletion of CTCF-binding sites, or altered cohesin binding, destroys TADs (Nuebler et al., 2018; Rao et al., 2017; Wutz et al., 2017). The importance of chromatin-associated, protein-binding events as drivers of higher-order genome architecture is demonstrated by the finding that polymer modeling does not accurately recapitulate Hi-C data if CTCF or cohesin binding sites are not taken into account (Nuebler et al., 2018). While cohesin and CTCF are major players in mediating chromatin polymer interactions, it is likely that additional chromatin binding proteins, as well as non-coding RNAs, also contribute to higher-order genome organization (Quinodoz et al., 2018).
Genome dynamics
Several aspects of genome organization are highly dynamic (Fig. 2; for in depth review see (Shaban and Seeber, 2020). As demonstrated by live-cell imaging, a chromatin locus undergoes locally confined, diffusional motion, typically within a radius of ~1μm (Marshall et al., 1997). The extent of motion is largely independent of genome- or nucleus size and varies little during the cell cycle (Shaban and Seeber, 2020). In addition, long-range, directed motion of chromatin loci has been observed and appears to depend on nuclear actin and myosin (Caridi et al., 2019; Chuang et al., 2006). However, these long-range events are relatively rare and likely restricted to particular situations, such as the extrusion of damaged DNA from heterochromatin or large-scale genome reorganization over long time periods during differentiation (Caridi et al., 2019; Wang et al., 2020).
The ability of the chromatin fiber to dynamically explore its immediate surroundings by constrained diffusion is functionally important. It enables a locus to probe for potential interaction partners, for example for an enhancer to find its cognate promoter or for multiple genes to cluster into transcription hubs (Chen et al., 2018a; Gu et al., 2018; Shaban and Seeber, 2020). The extent of dynamic motion of chromatin appears to be affected by its functional state, including its transcriptional status (Germier et al., 2017; Shaban et al., 2020) and, while the local motion of the chromatin polymer occurs by constrained passive diffusion, it is indirectly dependent on energy via the action of ATP-dependent chromatin remodeling complexes, which periodically act on the chromatin fiber to alter its state and, in this way, promote the dynamic motion of the fiber (Shaban and Seeber, 2020).
In addition to the fluctuations and rearrangements of the chromatin fiber, the proteins that act on it are also highly dynamic (Hager et al., 2009; Kim and Shendure, 2019; Misteli, 2001). Proteins diffuse rapidly through the cell nucleus, enabling them to effectively sample the genome for appropriate binding sites (Hager et al., 2009; Kim and Shendure, 2019; Misteli, 2001). Photobleaching and single-molecule experiments show that protein-chromatin interactions are characterized by high off-rates, with most chromatin proteins binding to their target regions very transiently. Single-molecule tracking experiments demonstrate that a transcription factor typically spends >95% of its time in a diffusive state or is engaged in non-specific interactions, with dwell-times on the order of several 100ms; only a small fraction of a given transcription factor at any time is bound at specific sites with dwell-times of 10’s of seconds (Chong et al., 2018; Hager et al., 2009; Liu and Tjian, 2018). The dynamic nature of protein-chromatin interactions not only facilitates the targeting of transcription factors to their specific binding sites, but is also key to generating regulatory plasticity, because the short dwell time of transcription factors ensures that functional complexes on chromatin are not locked into a permanent state that would need to be reversed by the action of dedicated regulatory machinery in response to changes in cellular environment. Instead, short-lived, dynamic complexes offer the opportunity for rapid functional change by altering the composition of the regulatory complex as part of the dynamic exchange of complex components via continuous competitive binding (Hager et al., 2009). This principle is exemplified by the observation of the stable, yet highly dynamic, association of the RNA polymerase I machinery with rDNA promoters (Dundr et al., 2002) or the dynamic interplay of steroid receptors with their target genes in response to ligands (Paakinaho et al., 2017).
An important concept related to genome dynamics is the notion that overall stable steady-states can be generated from dynamic components (Misteli, 2001). This property is best illustrated by the dynamic behavior of nuclear bodies, such as the nucleolus or splicing speckles, which appear as overall stable structures but are made up of highly dynamic components (Phair and Misteli, 2000). Early photobleaching experiments demonstrated that proteins enriched in nuclear bodies, rapidly transition through these compartments with typical residence times of 10’s of seconds, yet the overall structure of the compartment remains stable over hours, representing a dynamic steady-state (Misteli, 2001; Phair and Misteli, 2000). The same principle applies to chromatin. While core histones have residence times on the order of hours, linker histones reside on chromatin only for minutes, and many architectural chromatin proteins, such as HMG proteins and the heterochromatin protein HP1, have dwell times on the order of seconds (Phair et al., 2004). Despite the transient nature of the individual binding events, the resulting chromatin states are stable due to the continuous replacement of dissociating proteins. The dynamic nature of steady-states generates functional plasticity by enabling rapid responses to alterations in the cellular milieu, but it also appears to be essential for the formation of chromatin domains via phase separation (Larson et al., 2017; Strom et al., 2017).
Phase separation in genome organization
Phase separation is a driver of genome organization (Fig. 2; for in depth review see (Banani et al., 2017; Shin and Brangwynne, 2017). The basis of phase separation is the demixing of distinct protein populations when present above a saturation threshold due to their propensity to preferentially undergo homotypic rather than heterotypic interactions with other biomolecules, leading to the formation of segregated, dynamic phases (Banani et al., 2017; Shin and Brangwynne, 2017). Phase separation mediates the biogenesis of membraneless nuclear and cytoplasmic compartments, such as the nucleolus or stress granules, by promoting the aggregation of proteins to form distinct bodies (Banani et al., 2017; Shin and Brangwynne, 2017).
Phase separation also contributes significantly to chromatin organization (Kim and Shendure, 2019). The protein-protein and protein-RNA interactions required for phase separation is mediated by proteins that contain low complexity intrinsically disordered regions (IDRs), which are common in chromatin proteins and transcription factors (Banani et al., 2017; Shin and Brangwynne, 2017). The major heterochromatin protein HP1α forms phase-separated droplets both in vitro and in vivo and as such the formation of heterochromatin domains has been attributed to phase separation (Larson et al., 2017; Strom et al., 2017). Consistent with this view, HP1α is highly dynamic in heterochromatin and spreads along the chromatin fiber (Cheutin et al., 2003). The involvement of phase separation in heterochromatin formation has been suggested to account for the typically spherical shape of heterochromatin blocks, which may arise due to the demixing of HP1α-bound regions from HP1α-free regions and the coalescence of multiple heterochromatin blocks in 3D space (Larson et al., 2017; Sanulli et al., 2019; Strom et al., 2017). Furthermore, other major architectural proteins, including the linker histone H1, also phase separate and intrinsic features of chromatin, such as inter-nucleosome linker length, affect the phase separation properties of chromatin in vitro (Gibson et al., 2019; Kim and Shendure, 2019). In addition, histone modifications and chromatin binding proteins, including the histone acetylation reader BRD4, modulate the phase separation behavior of chromatin in vitro and in vivo (Gibson et al., 2019).
Phase separation events also act locally. Many eukaryotic transcription factors contain IDRs and can recruit RNA polymerase II into dynamic condensates which serve as hubs of transcription (Guo et al., 2019; Hnisz et al., 2017; Shaban et al., 2020). Furthermore, RNA polymerase II and the transcriptional Mediator co-activator complex form small, dynamic condensates (Boija et al., 2018; Cho et al., 2018; Chong et al., 2018). Similarly, Mediator and the transcription factor BRD4 accumulate on superenhancers in protein-rich aggregates and recruit the transcription machinery through their activator domains (Boija et al., 2018; Cho et al., 2018; Sabari et al., 2018). These observations point to phase separation as a driver of global and local genome organization (Hnisz et al., 2017; Yamamoto and Schiessel, 2016).
Architectural elements as genome constraints
The overall organization of genomes is strongly affected by their physical interaction with architectural elements of the cell nucleus, particularly the nuclear periphery and proteinaceous intranuclear bodies (Fig. 2; for in depth review see (Chen and Belmont, 2019; van Steensel and Belmont, 2017). The physical association of chromatin with these immobile features restricts its location in 3D space and limits the ability of a genome region to freely explore the nuclear space.
The nucleus of most metazoans contains a nuclear lamina structure, composed of an anastomosed meshwork of lamin intermediate filament proteins that lines the inner nuclear membrane and interacts with chromatin (van Steensel and Belmont, 2017). Reports over decades have described enrichment of condensed heterochromatin at the nuclear edge in many cell types, indicating that transcriptionally repressed chromatin is segregated to, and interacts with, the nuclear periphery (Fawcett, 1966). Genomic mapping has confirmed this notion and has demonstrated that these peripheral chromatin blocks, referred to as lamina associated domains (LADs), are defined by low gene density, the presence of LINE transposable elements, high A/T content, and the enrichment of histone modifications H3K9m2/3 and H2K27me3, which are all hallmarks of repressed genome regions (van Steensel and Belmont, 2017) (Fig. 2). Interspersed between LADs are inter-LADs, which contain predominantly active genome regions (van Steensel and Belmont, 2017). The molecular mechanisms by which chromatin interacts with the nuclear periphery are only poorly understood but, based on observations in C. elegans, they may involve histone modification readers that link the chromatin fiber either to lamins or are themselves anchored in the inner nuclear membrane (Gonzalez-Sandoval et al., 2015).
The nuclear envelope acts as a major constraint in the 3D organization of genomes because specific genome regions are tethered to the periphery (Fig. 2). When lamins are absent, spatial organization patterns are altered (Zheng et al., 2018) and LADs expand and become detached; loss of lamins also affects 3D organization at a genome-wide scale and results in repositioning of peripheral genome regions into the nuclear interior (Zheng et al., 2018). The constraining function of lamins is also demonstrated by the simultaneous imaging of all LADs of a single chromosome, which reveals that multiple LADs located on the same chromosome, but separated along the linear sequence, self-interact and are clustered (Luperchio et al., 2020). The LADs are segregated away from inter-LADs and, as expected, are in proximity to the nuclear envelope; the non-LAD regions of the same chromosome also congregate but protrude into the interior of the nucleus. This spatial segregation event depends on the presence of lamin A, demonstrating the constraining function of the lamina (Luperchio et al., 2020). Importantly, the peripheralization of genome regions also appears to have functional consequences as shown by experiments in which internal genome regions were artificially tethered to the periphery, which led to their repression or inability to be activated (Finlan et al., 2008; Kumaran and Spector, 2008; Reddy et al., 2008). These observations demonstrate the constraining role of the nuclear periphery in determining the location and function of genome regions.
A second major constraint in organizing the genome in 3D space are the various nuclear bodies present in the nuclear interior (Chen and Belmont, 2019) (Fig. 3B). While some nuclear bodies contain DNA and emerge as a consequence of the transcriptional activity of specific genomic sequences, such as the histone locus body which forms around clusters of histone genes, several prominent sub-nuclear bodies serve as physical constraints (Chen and Belmont, 2019; Chen et al., 2018b; Quinodoz et al., 2018). Prototypical examples for nuclear bodies as spatial genome organizers are the nucleolus and splicing factor speckles (Fig. 3B). Genome-wide mapping of simultaneously occurring chromatin interactions amongst multiple chromosomes shows that while transcriptionally active genome regions associate with splicing factors speckles, heterochromatin frequently associates with the edge of the nucleolus (Chen et al., 2018b; Quinodoz et al., 2018). PML bodies, which are enriched for the promyelocytic leukemia protein, have been suggested to have a similar role as they are themselves devoid of DNA but are abutted on all sides by chromatin (Ching et al., 2013). These observations highlight a constraining role for architectural elements of the cell nucleus in genome organization.
Figure 3. Drivers and constraints in genome organization.
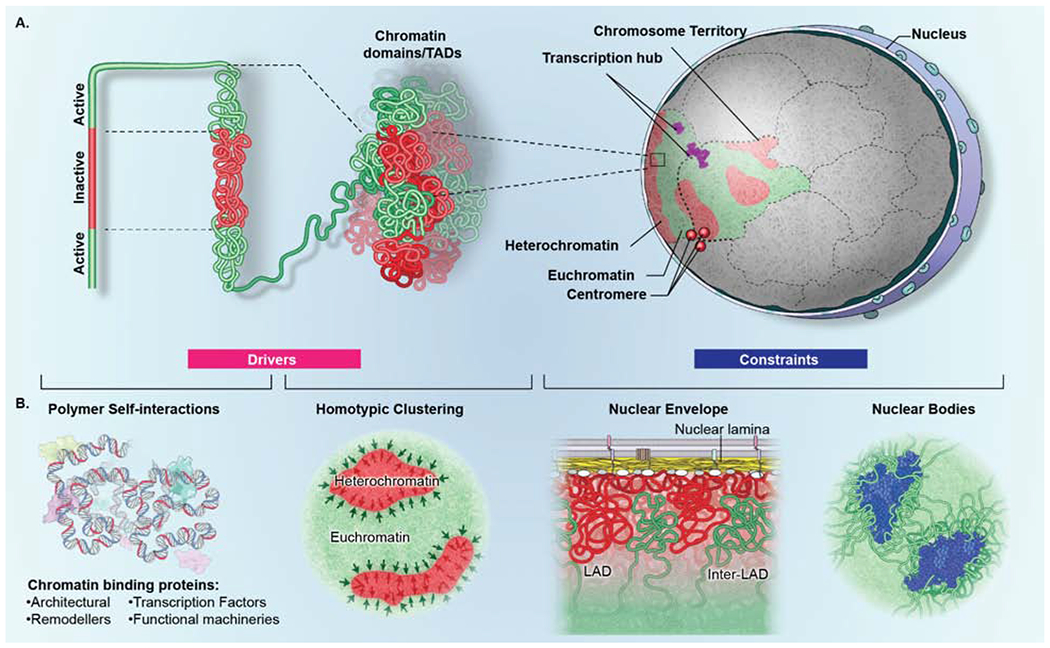
(A) Genome organization is determined by the interplay of drivers (pink box) and constraints (blue box). Higher-order organization is generated by self-interaction of transcriptionally active (green) and inactive (red) genome regions which fold onto themselves to form domains. Multiple homotypic (active or inactive) domains in turn aggregate both within a single chromosome and between domains on distinct chromosomes to form compartments. Homotypic interactions amongst multiple chromosomes generate transcription hubs (purple) and centromere aggregates (red spheres) which contain genes and centromeres located on distinct chromosomes, respectively. (B) (left) Drivers of genome organization include polymer-polymer interactions and phase separation. Homotypic polymer-polymer interactions among chromatin fibers form domains that are stabilized by phase separation (black arrows), including segregation of eu- and heterochromatin. (right) Constraints of genome organization include architectural features that physically interact with genome regions to influence their mobility and location, such as the nuclear lamina, which interacts with transcriptionally inactive lamina-associated domains (LADs) (red), interrupted by inter-LADs (green) or nuclear bodies, such as splicing speckles (blue), which preferentially associate with transcriptionally active chromatin.
Stochasticity and heterogeneity in genome organization and function
A further general principle of genome organization is its stochastic nature. Stochasticity in various forms is pervasively observed in gene expression and genome organization (Fig. 2; for in depth review see Symmons and Raj, 2016; Finn and Misteli, 2019).
Single-cell transcription analysis has revealed that the expression profiles of individual, genetically identical cells in a population vary considerably (Shah et al., 2018). A major contributor to transcriptional single-cell heterogeneity is the fact that most genes do not continuously engage the transcription machinery, but oscillate between dynamic on- and off-states and undergo “transcriptional bursting” (Rodriguez and Larson, 2020; Symmons and Raj, 2016). Imaging of transcriptional dynamics demonstrates that different genes have different on/off-cycle lengths with some genes exhibiting short bursts of activity with long intervening periods of inactivity, whereas others fire frequently with short periods of transcriptional silence (Rodriguez et al., 2019; Suter et al., 2011). Stochastic gene bursting is a universal phenomenon in organisms ranging from bacteria to mammals, is not limited to low-expressing genes, and has been implicated in many biological processes, including differentiation and response to immune signaling (Rodriguez and Larson, 2020; Symmons and Raj, 2016).
The molecular basis for transcriptional bursting is the inherently stochastic nature of the transcription process generated by the dynamic properties of the transcription machinery, which diffuses through the nuclear space and randomly collides with potential target sites where it undergoes short-lived interactions (Hager et al., 2009; Misteli, 2001). Given that there are typically only two copies of each gene in a nucleus, the likelihood of generating a productive transcription complex on a single allele is relatively low, particularly for genes that depend on low-abundance transcription factors. Furthermore, the expression of a gene is therefore an inherently stochastic event and coordinated firing of both alleles is unlikely. Indeed, single-cell imaging studies indicate that the two gene alleles in the same nucleus often show distinct transcriptional states and distinct chromatin organization (Finn et al., 2019; Rodriguez et al.,2019).
The functional heterogeneity observed for gene expression is mirrored by a high degree of heterogeneity in the organization of genomes in individual cells (Finn and Misteli, 2019; Rowley and Corces, 2018). In addition to the heterogeneity of gross morphological features, such as the variable appearance and number of nuclear bodies or the location of alleles in individual nuclei, most chromatin features are highly variable between cells. Imaging experiments show that chromatin loops only occur with relatively low frequencies in individual cells in the population, typically on the order of 5-30% (Bintu et al., 2018; Cattoni et al., 2017; Finn et al., 2019; Mateo et al., 2019). Super-resolution imaging and single-cell biochemical experiments extended this notion to chromatin domains by demonstrating that the structure and boundaries of a given TAD vary considerably between individual cells in the population (Bintu et al., 2018; Boettiger et al., 2016; Finn et al., 2019; Mateo et al., 2019; Nagano et al., 2013; Stevens et al., 2017; Wang et al., 2016). Similarly, while the configuration of chromatin domains along the length of the chromosome follows a preferred arrangement, the pattern is variable between individual cells (Cardozo Gizzi et al., 2019; Mateo et al., 2019; Nir et al., 2018; Wang et al., 2016). In agreement with the finding of unsynchronized firing patterns of the two alleles in a nucleus, single-cell imaging also reveals a lack of coordination in higher-order genome organization between alleles in the same nucleus (Finn et al., 2019). Furthermore, while a specific promoter-enhancer loop may be present at one allele, it may be absent at the same time on the other allele (Chen et al., 2018a; Finn et al., 2019).
These observations point to pervasive stochasticity and heterogeneity as being fundamental features of genomes. Their functional relevance is indicated by the fact that computational models that include stochastic aspects of gene expression and organization appear to better reflect experimental observations. For example, the use of a cohort of genome configurations, rather than a single conformation, results in more precise models of chromatin interaction data (Kalhor et al., 2011; Shi and Thirumalai, 2019; Tjong et al., 2016). Similarly, constraining computational models of genome organization with experimentally determined frequencies of interactions yields higher accuracy (Shi and Thirumalai, 2019). These observations suggest that the stochastic and heterogeneous nature of genomes is an intrinsic property of their structure and function.
The self-organizing genome
As outlined above, genomes are characterized by a high degree of order represented by ubiquitously conserved architectural features, such as chromatin loops and domains, nuclear bodies, and by non-random patterns, such as the location of genes and chromosomes in 3D space. In addition, the transcriptional program of a given cell is stable and defines its overall state. At the same time, genome organization and gene expression are also highly dynamic, variable and stochastic. How can these two apparently conflicting aspects of genome organization – steady-state stability and intrinsic variability - be reconciled? One hint comes from the realization that the major characteristics of genome organization, including a dynamic, stable steady-state, and a high degree of heterogeneity and variability, are hallmarks of a self-organizing system. The principle of self-organization is ubiquitous in nature and, when applied to the genome, provides a unifying mechanism to account for many of its structural and functional features.
The genome as a self-organizing system
Self-organizing systems are defined by their ability to spontaneously adjust their features by integrating the input of all their components without the need of external organizing agents. Prominent examples of self-organization in biology range from the flocking behavior of birds and insects, to the generation of cell and tissue patterns in morphogenesis and development, and the 3D folding of proteins (Camazine et al., 2003).
Genomes exhibit all defining properties of a self-organizing system. These include high dynamics, overall steady-state stability, and a high degree of heterogeneity. In addition, a key feature of self-organizing systems is the presence of regulatory feedback loops, which enable the system to adjust its overall state in response to a changing environment, while maintaining its internal steady-state stability (To and Maheshri, 2010). Positive and negative feedback loops are widely used in genome function (Milo et al., 2002). For example, developmental transcription networks often involve positive feedback loops to enhance their effects and to ensure regulation at long timescales. Feedback loops are also used to amplify low frequency events generated by variability in the population, thus creating a means for individual cells to sample a wide range of stochastic inputs and to adjust the system accordingly (Milo et al., 2002; Symmons and Raj, 2016). While most feedback loops operate at the level of gene expression, biochemical and mechanical feedback mechanisms mediated by protein-protein interactions also exist, such as the sensing of mechanical forces (Hannezo and Heisenberg, 2019).
In a self-organizing system, the properties that arise from the collaborative functioning of the system’s components, such as its architectural features, are referred to as emergent properties. A prominent example of an emergent property in cell biology are the filaments of the cytoskeleton, which exhibit vastly different mechanical properties than those of their monomeric components (Camazine et al., 2003). As outlined below, most organizational features of the genome, such as chromatin loops and domains, compartments, and the 3D location of genes and chromosomes, can be considered emergent properties of a self-organizing genome.
A further property of self-organizing system is the presence of an attractor, which is defined as the overall state at which the system is at equilibrium. In the case of the genome, the overall attractor is the gene expression profile that defines a cell type. The concept of attractors is well established in the field of cellular differentiation, where each cell state is defined as an attractor and the process of differentiation represents the transition between attractors (Macarthur et al., 2009). Transitions between attractor states are mediated by changes in gene expression programs that define a cell type, and they are typically accompanied by alterations in large scale genome topology (Bertero et al., 2019; Bonev et al., 2017; Paulsen et al., 2019).
The organization of genomes by self-organization is distinct from a deterministic model in which genomes function in a static fashion with limited feedback between structure and function and in which the organization and location of genome regions is determined by specific mechanisms, for example involving genetic zip-codes, adaptor proteins that read them and site-specific tethering mechanisms that define the location of gene loci (for in depth discussion of these models see (Misteli, 2007)). Taken together, the dynamic and stochastic nature of the genome, combined with the presence of genetic feedback loops and a strong attractor in the form of cell-type defining gene expression programs, confers to the genome all major hallmarks of self-organization.
Architectural genome features as emergent properties
Applied to the genome, self-organization can account for the formation of many of the architectural features of the genome, including loops, domains, compartments, and the 3D position of genes and chromosomes. These features emerge as the result of the integrated action of organization drivers (particularly polymer-polymer interactions and phase separation) and of constraints (particularly nuclear architectural elements), in the context of self-organization (Fig. 3, 4).
Figure 4. Formation of chromatin features by self-organization.
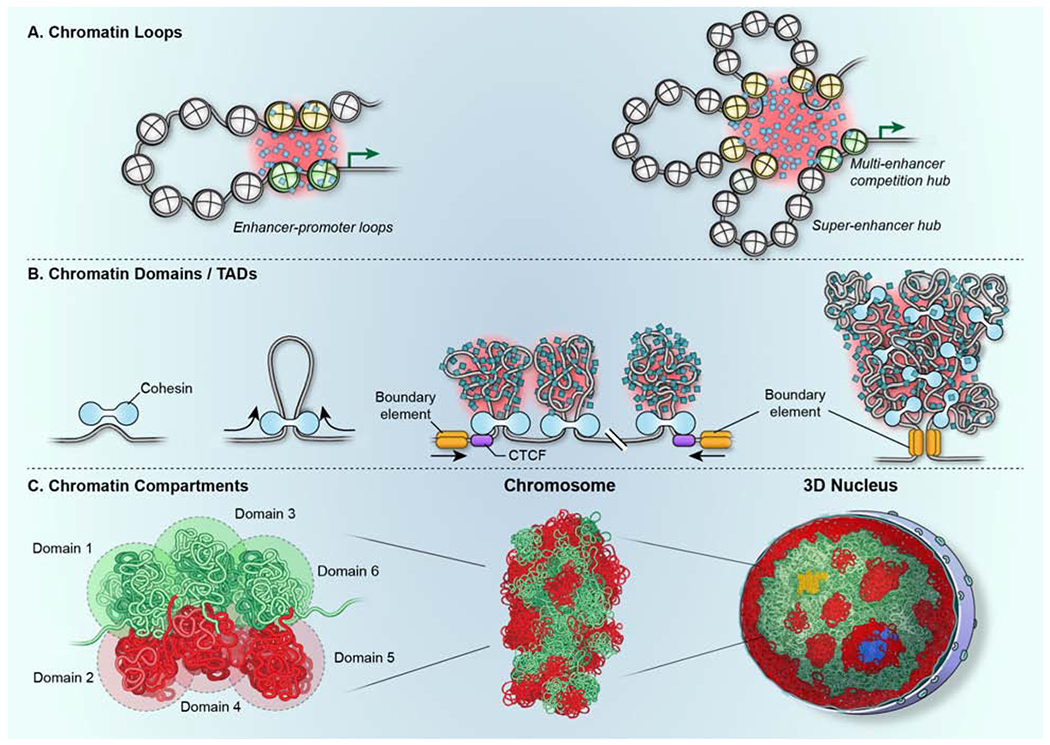
(A) Chromatin loops, including promoter-enhancer loops form through the close physical proximity of genome regions. Their interactions are stabilized by the formation of phase-separated aggregates (shaded in red) of relevant chromatin proteins and transcription factors (blue squares). Multiple enhancers (yellow nucleosomes) may associate with the same gene promoter (green nucleosomes) to activate the same gene target (green arrow) in multi-enhancer hubs or in superenhancers. (B) TADs form via loop-extrusion generated by the cohesin motor (light blue). The CTCF protein (purple) defines the boundary of the domain (gold) by determining the location of cohesin. The internal structure of TADs is determined by heterogenous polymer chromatin-chromatin interactions, which are likely stabilized by phase separation (shaded in red). Black arrows denote the direction of movement of DNA through the cohesin complex. (C) (left, middle) Chromatin compartments form via the association of multiple homotypic domains (1-6; green and red) to ultimately form chromosomes. (middle, right) Multiple chromosomes associate via interactions between homotypic domains across the 3D structure of the nucleus to form large-scale blocks of heterochromatin (red) and euchromatin (green). Nuclear bodies (blue) serve as anchoring points. Transcription hubs (yellow) form due to the coalescence of multiple active genome regions located on multiple chromosomes.
Formation of chromatin loops
Chromatin loops form via intra-polymer self-associations (Furlong and Levine, 2018; Vermunt et al., 2019) (Fig. 3A, 4A). The spatial proximity of two interacting regions in 3D space is an obvious requirement for the formation of a functionally active loop. During loop extrusion, cohesin constrains the degree of freedom of the chromatin fiber and promotes chromatin-chromatin interactions (Hansen, 2020). Alternatively, loops may form via stochastic chromatin-chromatin interactions as part of their diffusive motion (Chen et al., 2018a; Khanna et al., 2019). Once the two interacting regions are spatially juxtaposed, their interaction is stabilized by associated chromatin proteins (Vermunt et al., 2019; Khanna et al., 2019) (Fig. 4A). Visualization of promoter-enhancer interactions in living cells documents the rapid motion of the chromatin fiber followed by sustained physical interaction upon transcriptional activation of the target gene (Chen et al., 2018a). Similarly, in VDJ recombination which relies on long-range interactions of regions in the immunoglobulin heavy chain locus, the interacting segments are limited to local motion (Khanna et al., 2019). Observations of the motion dynamics of the VDJ locus is consistent with the association of interacting regions with a gel-like phase formed by self-interacting chromatin proteins. This phase-separated condensate limits chromatin motion and thus increases the probability of persistent chromatin-chromatin interactions (Khanna et al., 2019) (Fig. 4A). Phase separation mediated by self-interacting chromatin proteins has also been implicated in the coalescence of multiple enhancers (Hnisz et al., 2017; Sabari et al., 2018). Of note is that in this hub model, the phase separated condensate bridges the gap between the interacting chromatin fibers and loops form without direct physical chromatin-chromatin interaction (Fig. 4A).
Formation of chromatin domains
The formation of TADs involves loop extrusion (Fudenberg et al., 2016; Mirny et al., 2019; Nuebler et al., 2018) (Fig 3A, 4B). The extruded chromatin then folds onto itself to form a domain via polymer-polymer self-interactions (Fig. 4B). While the process of loop extrusion per se is a directed process, the organization of the internal TAD structure relies on self-organization via a network of intra-TAD chromatin-chromatin interactions, likely stabilized by phase-separation. In support, high-resolution mapping has demonstrated that the internal structure of TADs consists of a heterogenous collection of transient, highly heterogenous loops, which interact extensively (Hsieh et al., 2020; Krietenstein et al., 2020). These intra-TAD polymer interactions might be facilitated by phase-separated supramolecular condensates of chromatin-associated proteins that engulf the chromatin domain (Erdel and Rippe, 2018; Gibson et al., 2019) (Fig. 4B). Indeed, in vitro and in vivo evidence supports the phase separation interactions of linker histones and architectural chromatin proteins (Gibson et al., 2019; Kim and Shendure, 2019). The observed heterogeneity in chromatin-chromatin interactions within a TAD (Finn et al., 2019; Hsieh et al., 2020; Krietenstein et al., 2020) is in line with their formation by self-organization since the network of intra-fiber interactions differs stochastically amongst individual cells. In this scenario, self-organization via phase separation stabilizes, but also buffers, the variable internal structure of TADs, while delineating boundaries between adjacent domains (Fig. 3, 4B).
Formation of chromatin compartments and chromosome territories
While loop extrusion is a major driver of TAD formation, the genesis of higher-order chromatin compartments appears to be primarily mediated by phase separation (Mirny et al., 2019) (Fig. 3, 4C). In line with self-organization, the formation of chromatin compartments involves homotypic interactions among genome regions that are located distantly on individual chromosomes or on separate chromosomes. Through these interactions, genome regions coalesce into distinct domains to generate phase-separated structures (Nuebler et al., 2018) (Fig. 3, 4C). This self-organization mechanism predicts that chromatin of a similar type, such as heterochromatin or euchromatin, coalesces while de-mixing from heterotypic chromatin. In line with this prediction, the spatial separation of active and inactive chromosome regions, congregation of centromeres, and segregation of transcriptionally inactive LADs from transcriptionally active inter-LADs have been reported (Jagannathan et al., 2019; Luperchio et al., 2020; Shopland et al., 2006). Furthermore, in computational simulations, changes in genome compartmentalization, including the effects of loss of cohesin and/or CTFC on compartment formation (Nuebler et al., 2018) and the observed inversion of heterochromatin towards the nuclear interior in retinal rod cells (Falk et al., 2019), can only be recreated by including phase separation in polymer-based modeling approaches.
Despite distinct mechanisms involved in the formation of TADs and compartments, there is interplay between them as is illustrated by the finding, based on computational simulations, that the loop extrusion events that generate TADs interfere with compartment formation, while the loss of TAD boundaries leads to a reduction in TAD formation, but has no effect on compartments (Mirny et al., 2019; Nuebler et al., 2018). Conversely, the stimulation of loop extrusion via the depletion of the cohesin-unloading factor WAPL results in increased occupancy of cohesin and leads to more sharply define TADs but weakened compartment patterns (Mirny et al., 2019; Nuebler et al., 2018). These observations demonstrate that the active loop extrusion process counteracts compartmental segregation, which occurs via phase separation and may be thought of as a default mechanism that brings multiple TADs into proximity to form higher order compartments (Fig. 3, 4C).
The same principles that generate compartments apply to the formation of chromosome territories (Fig. 3, 4C). Just as phase-separation mediates associations between local homotypic genome regions to form compartments, it also promotes the self-interaction of homotypic domains separated over larger distances along the length of a single chromosome. As a consequence, the overall topology of a chromosome is determined by the network of homotypic interactions amongst chromatin domains, which lead to the clustered arrangement of chromatin domains and to the formation of a near-spherical chromosome territory (Cremer and Cremer, 2010; Wang et al., 2016).
Determining the 3D location of chromosomes and genes
The mapping of the 3D locations of chromosomes and genes has shown that gene loci assume non-random, yet probabilistic, positions in the cell nucleus (Crosetto and Bienko, 2020; Shachar and Misteli, 2017). Some cellular factors and histone modifications that contribute to determining the location of genes have been identified but they do not reflect a dedicated localization machinery; rather these factors are part of general nuclear processes, such as replication or transcription (Crosetto and Bienko, 2020; Shachar et al., 2015). It thus seems likely, in line with self-organization, that the 3D position of a genome region is determined by its overall functional status, as defined by the proteins that associate with it, combined with the sum of its homotypic interactions with other genome regions and its association with constraining architectural elements of the nucleus (Fig. 3). For example, given the propensity of heterochromatin to associate with the nuclear periphery, transcriptionally silent portions of chromosomes are more likely to orient towards the nuclear periphery whereas the transcriptionally active regions are more likely in the interior (Luperchio et al., 2020). Transcriptionally inactive regions also tend to interact with internal heterochromatin domains and with heterochromatin around the nucleolus, whereas active regions are attracted to nuclear splicing speckles (Chen et al., 2018b; Quinodoz et al., 2018) (Fig. 3). These interactions generate the observed overall gradient of transcriptional activity from the interior to the periphery. Due to the presence of multiple influences acting on a genome region, multi-stable states of the same region co-exist in a population and individual genes assume distinct locations in single cells, which is reflected in the well-documented probabilistic and heterogenous distribution of gene locations in a population (Crosetto and Bienko, 2020; Shachar and Misteli, 2017).
The probabilistic genome: structure-function relationship in the genome
A central question in genome biology is how structural features relate to the function of individual genes and the genome as a whole. Correlations between genome architecture and function abound. Most prominently, the presence of heterochromatin often correlates with transcriptional repression, and the formation of promoter-enhancer loops is linked to the transcriptional activation of target genes. However, most of these correlations are incomplete, and several observations point to two important, and still underappreciated, concepts in understanding the interplay of structure and function: First, genome function is a driver of genome structure; while the architectural features of the genome influence its function, the local and global activity of the genome also shapes its structure via feedback mechanisms (Fig. 5, 6A). Second, it is becoming evident that structural features do not act as binary on-off switches, but as modulators of function (Fig. 6B). The combination of bi-directional feedback between structure and function and the modulatory role of architectural genome features renders genome function an overall probabilistic process (Fig. 6C).
Figure 5. Genome function drives structure.
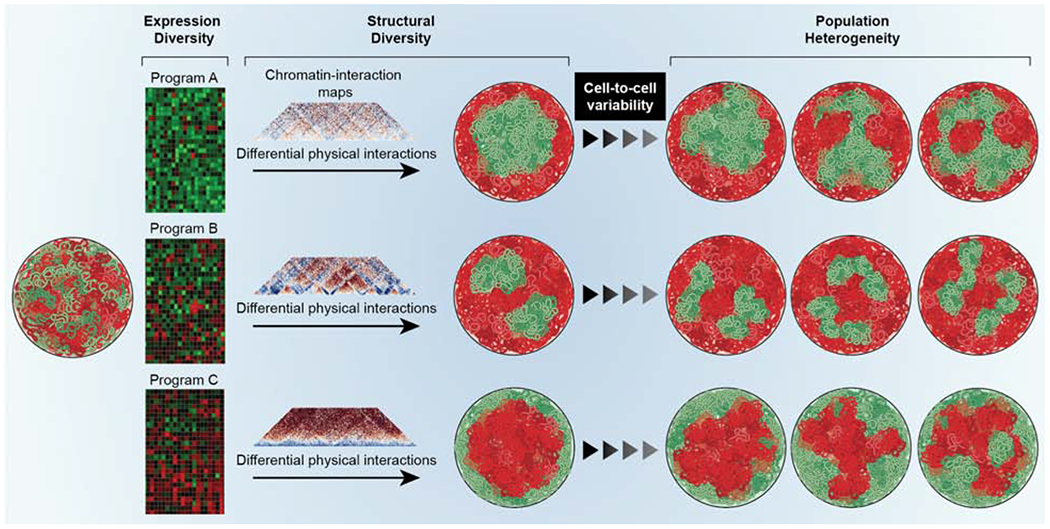
All cells of an organism contain genomes of identical sequence (left single nucleus), but different cell-types express distinct gene expression programs (heatmaps) and consequently have different genome topologies as detected by chromatin interaction maps. The cell-type specific homotypic chromatin-chromatin interactions drive higher-order genome organization and generate distinct overall genome topologies in different cell types, resulting in cell-type specific patterns of euchromatin (green) and heterochromatin (red). (right) Within a cell type, the heterogeneity of chromatin-chromatin interactions generates cell-to-cell variability in the population.
Figure 6. The probabilistic genome.
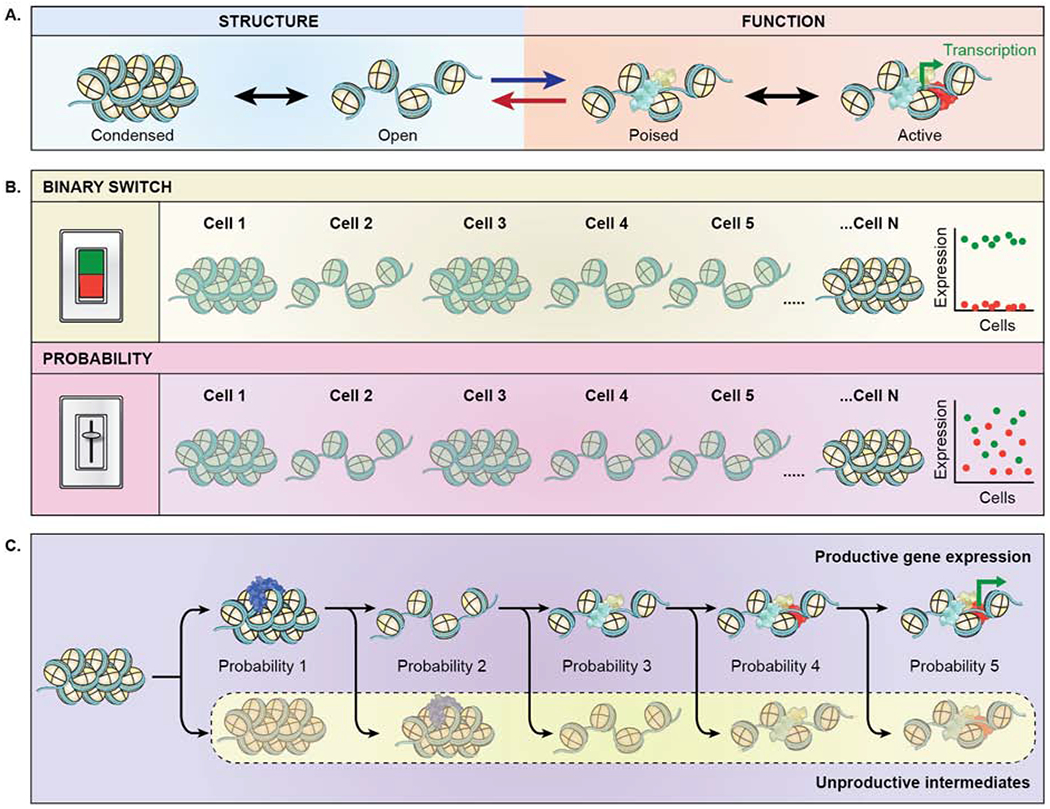
(A) Bi-directional interplay of chromatin structure (blue shaded area) and function (orange shaded area). Chromatin structure affects gene function (blue forward arrow). Chromatin structure reversibly oscillates between a condensed and open state, which facilitates association of transcription factors (blue, yellow), leading to a poised stated and upon association of RNA polymerase (red) enables transcription (green bent arrow). Conversely, transcription affects structure (red reverse arrow), by maintaining an open chromatin structure. (B) Chromatin structure does not act as a deterministic, binary switch, but rather as a probabilistic modulator of function. (green panel) In a binary switch model, the expression level of a given gene in individual cells is either fully on (green points) or fully off (red points) reflecting the open and closed configuration of the gene locus in single cells. (pink panel) In a modulatory model, gene expression levels are heterogeneous (red, green points) in individual cells reflecting the probabilistic nature of gene activity in open and closed chromatin. Variable expression levels are typically observed experimentally. (C) Gene expression is a multi-step processes and is inherently probabilistic. Each step required to activate a gene represents an equilibrium of a productive event towards gene activation versus an unproductive event with a certain probability. Probability 1: stable association of a chromatin remodeling factors (blue) vs. transient interaction as they diffuse through the nucleus. Probability 2: maintenance of decondensed chromatin vs. reversion to condensed state. Probability 3: association of early transcription factors (light blue, yellow) which promotes the likelihood of association of additional transcription factors vs. disassociation of early transcription factor. Probability 4: association of RNA polymerase (red) vs. loss of activating transcription factors. Probability 5: transcriptional activation (bent green arrow) vs. unproductive dissociation of RNA polymerase. As a result of the probabilistic nature of each step, the activation process as a whole is relatively inefficient, thus generating the stochastic patterns of activation observed for most genes.
Genome function drives genome structure
Individual cell types are defined by particular gene expression programs. Cell types are also characterized by distinct patterns of global genome organization, for example, the extent and location of heterochromatin domains (Fig. 5). Several considerations suggest that the nature of the active gene expression program in a cell type shapes genome organization both locally and globally (van Steensel and Furlong, 2019).
Patterns of active and inactive genome regions differ considerably between cell types (Fig. 5). The differential usage of gene sets amongst cell types generates distinct networks of cell-type specific, chromatin-chromatin interactions. Since these interactions drive higher-order genome organization, cell-type specific configurations of chromatin domains and compartments emerge (Fig. 5). One prediction from genome function as a driver of genome organization is that the overall morphological appearance of genomes should differ between cell types and that genome morphologies should diverge with the increasing separation of lineages. In line with these predictions, embryonic stem (ES) cells, which are totipotent and express most parts of the genome at low levels, are characterized by mostly homogeneous chromatin and by the near-complete absence of repressive heterochromatin, whereas differentiated cells that express distinct sets of genes exhibit a wide range of genome morphologies depending on cell type (Schlesinger and Meshorer, 2019). Distinct local and global chromatin topologies have been documented based on morphological features and by interaction mapping in numerous differentiation and development processes, including hematopoiesis, cardiogenesis, ES cell differentiation, mouse development and senescence (Bertero et al., 2019; Bonev et al., 2017; Paulsen et al., 2019; Rajapakse et al., 2009; Sati et al., 2020).
Gene activity also influences local chromatin structure. The stochastic pulsatile cycles of activity and inactivity of a gene generates cyclical alterations in chromatin organization. During the transcriptionally active periods, chromatin remains open but condenses upon cessation of a gene’s transcription suggesting that active transcription maintains chromatin in an open state (van Steensel and Furlong, 2019). In support, steroid receptor target genes undergoing rapid condensation upon treatment with a transcriptional inhibitor (Muller et al., 2001) and ongoing transcription is required to maintain open chromatin structure upon influenza A infection (Heinz et al., 2018). Furthermore, acute inhibition of transcription results in a genome-wide loss of local promoter-enhancer interactions without affecting higher-order organization at the level of domains or compartments (Heinz et al., 2018; Hsieh et al., 2020). Perhaps most tellingly, in Drosophila, global transcriptional status is a strong predictor of chromatin domains, and transcriptional inhibition results in the loss of domain structure (Rowley et al., 2017). Strikingly, transcription data alone is sufficient to computationally model genome structure in flies (Rowley et al., 2017). These results support the notion that rather than merely reflecting transcriptional activity, the process of transcription shapes chromatin structure both locally and globally (Fig. 5).
Genome structure as a modulator of genome function
Genome structure is clearly related to genome function as indicated by extensive correlations between chromatin condensation state and gene activity (Fig. 6A). However, it is equally clear that structure alone does not determine the functional status of a gene. At the simplest level, not all genes in condensed chromatin are repressed, and many promoter-enhancer interactions occur dynamically without necessarily producing a gene expression event (Chen et al., 2018a). The question then arises: what role does structure play in genome function? The most likely scenario is one in which structural chromatin features act as modulators of genome function rather than as binary on/off switches (Fig. 6B).
The modulatory effect of chromatin structure is evident in measurements of the accessibility of target sequences to transcription factors in compact versus open chromatin. The association properties and effects of transcription factors are strongly influenced by the local chromatin environment, including nucleosome position, DNA supercoiling, and chromatin modifications. Importantly, these effects are not binary and typically show a gradual change rather than a complete loss or gain of function upon disruption (Donovan et al., 2019; Kim and Shendure, 2019). Similarly, in large-scale mapping of promoter-enhancer loops in Drosophila only a weak correlation was found between chromatin structure and gene activity and loss of chromatin interactions or domain structure only had minor effects on the activity of target genes, pointing to, at best, a modulatory role of these interactions (Ghavi-Helm et al., 2019). Along the same lines, developmentally relevant enhancer-promoter interactions form prior to TADs in early fly development and developmentally distinct cell types exhibit similar TAD structures, suggesting that the gene expression program in these cells is not determined by higher-order structure (Espinola et al., 2020; Ing-Simmons et al., 2020). Furthermore, comparative mapping in fly embryos of the Ubx and abd-A TADs, which contain important development genes, revealed that a moderate ~2-fold change in contact frequencies is accompanied by a ~7-fold change in the expression of genes in the TADs (Mateo et al., 2019), demonstrating non-linearity of the structure-function relationship and a modulatory role of structural features and. Taken together, these observations suggest that structural features of chromatin modulate but do not determine function in a binary fashion (Fig. 6B).
The modulatory role of chromatin features, combined with the dynamic nature of the molecular events that act on genomes, particularly transient transcription-factor binding, suggests that, in line with self-organization, gene expression should be thought of as a probabilistic, rather than a deterministic process, in which each step occurs with a certain probability, but not with certainty (Fig. 6C). While the completion of each step enhances the probability of the subsequent step to occur, the efficiency of the overall process is determined by the cumulative probabilities of all steps (Fig. 6C). Other genome functions, such as replication and repair, are similarly based on dynamic cellular machinery and depend on chromatin structure, and likely also occur in a probabilistic fashion.
Creating genome plasticity and robustness
Biological systems, including genomes, are characterized by two apparently conflicting properties. On the one hand, they must be plastic in order to adapt to varying environments or respond to external cues. On the other, they must be robust to be able to maintain their overall state in the face of environmental fluctuations. Self-organizing systems are inherently robust and plastic (Camazine et al., 2003) and the self-organizing nature of genomes can account for the plasticity and robustness of genome function and reconciles the co-existence of these two disparate properties.
Plasticity refers to the ability of a cell to change its state in response to external stimuli. Transcriptional plasticity is critical for cells to adjust to their environment and occurs via rapid alteration of their transcriptional program (Symmons and Raj, 2016). The dynamic and probabilistic nature of protein-protein, protein-chromatin and chromatin-chromatin interactions in the context of self-organization contributes to creating plastic transcriptional networks as it enables gene expression networks to responsively adjust their activity by rapidly modulating key genes (Symmons and Raj, 2016).
The ultimate reflection of genome plasticity is the process of differentiation, during which gene expression programs are altered to generate new, stable cellular states. These transitions occur gradually in the population, but stochastically amongst individual cells, as indicated by the observation that differentiation-relevant genes are inactive in ES cells, but are expressed at low levels, but with high variance, in precursor cells, and are then expressed at high levels with low variance in differentiated cells (Wada et al., 2018). In line with probabilistic and combinatorial self-organization, differentiation processes in vitro are typically inefficient and occur asynchronously in the population with only some cells responding to a given differentiation signal (Symmons and Raj, 2016). The differential response of individual cells reflects the stochastic heterogeneity of the gene expression programs represented in single cells in a population; those cells that have a gene expression program that is prone to respond to a given stimulus will enter the differentiation pathway more rapidly than those that do not. A testable prediction to link genome structure to functional genome plasticity is that the degree of stochastic variability in gene expression during differentiation should decrease and should be matched by the degree of structural variability observed amongst individual cells.
At the same time, as cellular systems are highly plastic, they must also be robust. Robustness describes the ability of a system to maintain its overall state despite external and internal perturbations (Felix and Barkoulas, 2015). When applied to the genome, this means that while the expression of individual genes occurs stochastically, the overall gene expression program that defines a cell type remains stable in a single cell. The presence of extensive feedback loops and the inherently dynamic nature of chromatin-chromatin and protein-chromatin interactions in self-organizing systems ensures the stability and robustness of gene expression programs.
The degree of robustness of a gene is determined by the extent of variability of its firing rate (Rodriguez and Larson, 2020). Transcriptional regulatory mechanisms ensure robustness of gene expression at multiple levels. Many regulatory regions, particularly enhancers, often contain several binding sites of the same transcription factor, ensuring persistent baseline occupancy (Felix and Barkoulas, 2015; Macneil and Walhout, 2011; Small et al., 1992). Furthermore, multiple enhancers, typically located in the same chromatin domain, are often able to act on a target gene, and the presence of several enhancers has been demonstrated to increase robustness (Frankel et al., 2010; Macneil and Walhout, 2011). In addition, high cellular concentration of regulatory transcription factors, as is frequent for developmentally relevant transcription factors, increases the likelihood of firing and thus enhances the persistent expression of a gene (Kang et al., 2013; Papadopoulos et al., 2019). Transcription factors also often act as groups of paralogs with closely related functional properties, which generate redundancy in ensuring the occupancy of regulatory binding sites (Felix and Barkoulas, 2015; Macneil and Walhout, 2011). Finally, the formation of persistent chromatin structures, such as stable domains and loops, enhances robustness in developmental contexts (Paliou et al., 2019).
While some genes show limited robustness and their expression is variable, others are highly robust in their spatial and temporal expression amongst individual cells (Bar-Even et al., 2006). A prominent group of genes with high robustness are developmentally regulated genes that require accurate expression during narrow windows of time in particular locations of an embryo to ensure the precise execution of a developmental program. Developmentally regulated genes generally exhibit lower levels of variability in their expression, are typically controlled by abundant transcription factors, and have relatively frequent chromatin-chromatin interactions, as would be expected for regulatory events that need to be tightly controlled (Felix and Barkoulas, 2015; Nijhout, 2002). In addition, tightly regulated developmental genes are typically components of feedback-loops to ensure their robust expression (Nijhout, 2002; Symmons and Raj, 2016). The high degree of robustness of developmentally relevant genes makes sense in the light of evolution, which has likely tilted the balance between robustness and plasticity for these genes to ensure precise execution of a tightly controlled developmental program rather than the ability to respond to a broad range of environmental cues as is desirable for other cell types.
Taken together, these observations suggest that the presence of feedback loops and the probabilistic nature of dynamic interactions of proteins with chromatin and of chromatin-chromatin interactions shape genome architecture and contribute significantly to enabling plasticity and robustness in gene expression programs in the context of self-organization.
Concluding remarks
Decades of experimental observations have converged to generate a unifying model that explains the prominent architectural features of genomes and that accounts for the variability in genome organization observed in individual cells. The available data support the notion that the major features of higher-order genome architecture are emergent properties in a self-organizing system that is driven by the functional status of the genome. Self-organization accounts for the formation of major hallmarks of spatial genome organization, from local loops and chromatin domains to the 3D location of genes and chromosomes. It also explains the considerable variability of architectural features of genomes at the single cell level, yet ensures plasticity and robustness of gene expression programs. In this view, the non-random architectural features of genome organization are the result of the superimposition of multiple driver events, particularly polymer-polymer interactions and phase separation, as well as of constraints, such as physical interactions with architectural elements of the nucleus. Importantly, the overall morphological appearance of the genome in a cell is thus ultimately determined by the bi-directional, dynamic interplay of the functional status of the genome, defined by its transcriptional program, and its architectural features (Fig. 5).
While we have gained a better understanding of the non-random features of genome organization, challenges remain. To start with, the molecular basis of higher-order genome organization remains poorly characterized. The identification of select chromatin proteins, such as CTCF and cohesin and of RNA, as determinants of chromatin structure is an important step, but additional cellular factors will need to be identified to fully delineate the mechanistic foundation of genome organization. Arguably, the next frontier in genome biology is the full elucidation of how structure relates to function. This will require the development of new tools to more precisely probe how structure affects function which do not rely on correlating structure with function, as is currently common done in mutation and knock-down experiments. These new approaches may include nanomanipulation tools, such as optogenetic methods, non-invasive nanobead-based force generators, or synthetic biology approaches to create and manipulate chromatin structures in living cells. Together, these methods will enable the targeted exploration of the functional relevance of architectural features while uncovering the molecular and biophysical mechanisms that drive them.
Our understanding of genome organization has come a long way. The powerful combination of imaging and biochemical approaches has led to the characterization of the fundamental features and principles of genome organization. These efforts are the foundation to now embark on the elucidation of how structure relates to function in the genome in health and disease. With the realization that genome architecture is an emergent property of a self-organizing system, the next phase of studying the genome is now upon us.
Acknowledgement
I thank members of the Misteli lab and numerous colleagues in the field for critical and constructive input. I thank Erina He, NIH Medical Arts, for creating the figures. Work in the Misteli lab on genome organization is supported by the Intramural Research Program of the NIH, NCI, Center for Cancer Research and by NIH Common Fund 4D Nucleosome Program (Grant U54 DK107890).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The author declares no competing interests.
References
- Banani SF, Lee HO, Hyman AA, and Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Even A, Paulsson J, Maheshri N, Carmi M, O’Shea E, Pilpel Y, and Barkai N (2006). Noise in protein expression scales with natural protein abundance. Nat Genet 38, 636–643. [DOI] [PubMed] [Google Scholar]
- Barbieri M, Chotalia M, Fraser J, Lavitas LM, Dostie J, Pombo A, and Nicodemi M (2012). Complexity of chromatin folding is captured by the strings and binders switch model. Proc Natl Acad Sci U S A 109, 16173–16178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagrie RA, Scialdone A, Schueler M, Kraemer DC, Chotalia M, Xie SQ, Barbieri M, de Santiago I, Lavitas LM, Branco MR, et al. (2017). Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero A, Fields PA, Ramani V, Bonora G, Yardimci GG, Reinecke H, Pabon L, Noble WS, Shendure J, and Murry CE (2019). Dynamics of genome reorganization during human cardiogenesis reveal an RBM20-dependent splicing factory. Nat Commun 10, 1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco S, Chiariello AM, Conte M, Esposito A, Fiorillo L, Musella F, and Nicodemi M (2020). Computational approaches from polymer physics to investigate chromatin folding. Curr Opin Cell Biol 64, 10–17. [DOI] [PubMed] [Google Scholar]
- Bianco S, Lupianez DG, Chiariello AM, Annunziatella C, Kraft K, Schopflin R, Wittler L, Andrey G, Vingron M, Pombo A, et al. (2018). Polymer physics predicts the effects of structural variants on chromatin architecture. Nat Genet 50, 662–667. [DOI] [PubMed] [Google Scholar]
- Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, and Zhuang X (2018). Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu CT, and Zhuang X (2016). Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529, 418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, et al. (2018). Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855 e1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, et al. (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572 e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Brien GL, and Verrijzer CP (2019). Dangerous liaisons: interplay between SWI/SNF, NuRD, and Polycomb in chromatin regulation and cancer. Genes Dev 33, 936–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackley CA, Johnson J, Kelly S, Cook PR, and Marenduzzo D (2016). Simulated binding of transcription factors to active and inactive regions folds human chromosomes into loops, rosettes and topological domains. Nucleic Acids Res 44, 3503–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco MR, and Pombo A (2006). Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations. PLoS Biol 4, e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckle A, Brackley CA, Boyle S, Marenduzzo D, and Gilbert N (2018). Polymer Simulations of Heteromorphic Chromatin Predict the 3D Folding of Complex Genomic Loci. Mol Cell 72, 786–797 e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camazine S, Deneubourg JL, Franks NR, Sneyd J, Theraulaz G, and Bonabeau E (2003). Self-Organization in Biological Systems (Princeton University Press; ). [Google Scholar]
- Cardozo Gizzi AM, Cattoni DI, Fiche JB, Espinola SM, Gurgo J, Messina O, Houbron C, Ogiyama Y, Papadopoulos GL, Cavalli Gv et al. (2019). Microscopy-Based Chromosome Conformation Capture Enables Simultaneous Visualization of Genome Organization and Transcription in Intact Organisms. Mol Cell 74, 212–222 e215. [DOI] [PubMed] [Google Scholar]
- Caridi CP, Plessner M, Grosse R, and Chiolo I (2019). Nuclear actin filaments in DNA repair dynamics. Nat Cell Biol 21, 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoni DI, Cardozo Gizzi AM, Georgieva M, Di Stefano M, Valeri A, Chamousset D, Houbron C, Dejardin S, Fiche JB, Gonzalez I, et al. (2017). Single-cell absolute contact probability detection reveals chromosomes are organized by multiple low-frequency yet specific interactions. Nat Commun 8, 1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Levo M, Barinov L, Fujioka M, Jaynes JB, and Gregor T (2018a). Dynamic interplay between enhancer-promoter topology and gene activity. Nat Genet 50, 1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, and Belmont AS (2019). Genome organization around nuclear speckles. Curr Opin Genet Dev 55, 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang Y, Wang Y, Zhang L, Brinkman EK, Adam SA, Goldman R, van Steensel B, Ma J, and Belmont AS (2018b). Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J Cell Biol 217, 4025–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheutin T, McNairn AJ, Jenuwein T, Gilbert DM, Singh PB, and Misteli T (2003). Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science 299, 721–725. [DOI] [PubMed] [Google Scholar]
- Ching RW, Ahmed K, Boutros PC, Penn LZ, and Bazett-Jones DP (2013). Identifying gene locus associations with promyelocytic leukemia nuclear bodies using immuno-TRAP. J Cell Biol 201, 325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, and Cisse II (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, Dugast-Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, Heckert A, Banala S, Lavis L, Darzacq X, et al. (2018). Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang CH, Carpenter AE, Fuchsova B, Johnson T, de Lanerolle P, and Belmont AS (2006). Long-range directional movement of an interphase chromosome site. Curr Biol 16, 825–831. [DOI] [PubMed] [Google Scholar]
- Cremer T, and Cremer M (2010). Chromosome territories. Cold Spring Harb Perspect Biol 2, a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft JA, Bridger JM, Boyle S, Perry P, Teague P, and Bickmore WA (1999). Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol 145, 1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosetto N, and Bienko M (2020). Radial Organization in the Mammalian Nucleus. Front Genet 11, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbellay F, and Duboule D (2016). Topological Domains, Metagenes, and the Emergence of Pleiotropic Regulations at Hox Loci. Curr Top Dev Biol 116, 299–314. [DOI] [PubMed] [Google Scholar]
- Davidson IF, Bauer B, Goetz D, Tang W, Wutz G, and Peters JM (2019). DNA loop extrusion by human cohesin. Science 366, 1338–1345. [DOI] [PubMed] [Google Scholar]
- Dekker J, and Mirny L (2016). The 3D Genome as Moderator of Chromosomal Communication. Cell 164, 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, and Misteli T (2015). Long-Range Chromatin Interactions. Cold Spring Harb Perspect Biol 7, a019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan BT, Huynh A, Ball DA, Patel HP, Poirier MG, Larson DR, Ferguson ML, and Lenstra TL (2019). Live-cell imaging reveals the interplay between transcription factors, nucleosomes, and bursting. EMBO J 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr M, Hoffmann-Rohrer U, Hu Q, Grummt I, Rothblum LI, Phair RD, and Misteli T (2002). A kinetic framework for a mammalian RNA polymerase in vivo. Science 298, 1623–1626. [DOI] [PubMed] [Google Scholar]
- Erdel F, and Rippe K (2018). Formation of Chromatin Subcompartments by Phase Separation. Biophys J 114, 2262–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinola SM, Götz M, Fiche J-B, Bellec M, Houbron C, Cardozo Gizzi AM, Lagha M, and Nollmann M (2020). Cis-regulatory chromatin loops arise before TADs and gene activation, and are independent of cell fate during development. Biorxiv 10.1101/2020.07.07.191015. [DOI] [PubMed] [Google Scholar]
- Falk M, Feodorova Y, Naumova N, Imakaev M, Lajoie BR, Leonhardt H, Joffe B, Dekker J, Fudenberg G, Solovei I, et al. (2019). Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett DW (1966). On the occurrence of a fibrous lamina on the inner aspect of the nuclear envelope in certain cells of vertebrates. Am J Anat 119, 129–145. [DOI] [PubMed] [Google Scholar]
- Felix MA, and Barkoulas M (2015). Pervasive robustness in biological systems. Nat Rev Genet 16, 483–496. [DOI] [PubMed] [Google Scholar]
- Felsenfeld G, and Groudine M (2003). Controlling the double helix. Nature 421, 448–453. [DOI] [PubMed] [Google Scholar]
- Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, and Bickmore WA (2008). Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet 4, e1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn EH, and Misteli T (2019). Molecular basis and biological function of variability in spatial genome organization. Science 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn EH, Pegoraro G, Brandao HB, Valton AL, Oomen ME, Dekker J, Mirny L, and Misteli T (2019). Extensive Heterogeneity and Intrinsic Variation in Spatial Genome Organization. Cell 176, 1502–1515 e1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel N, Davis GK, Vargas D, Wang S, Payre F, and Stern DL (2010). Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466, 490–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser P, and Bickmore W (2007). Nuclear organization of the genome and the potential for gene regulation. Nature 447, 413–417. [DOI] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, and Mirny LA (2016). Formation of Chromosomal Domains by Loop Extrusion. Cell Rep 15, 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong EEM, and Levine M (2018). Developmental enhancers and chromosome topology. Science 361, 1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germier T, Kocanova S, Walther N, Bancaud A, Shaban HA, Sellou H, Politi AZ, Ellenberg J, Gallardo F, and Bystricky K (2017). Real-Time Imaging of a Single Gene Reveals Transcription-Initiated Local Confinement. Biophys J 113, 1383–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavi-Helm Y, Jankowski A, Meiers S, Viales RR, Korbel JO, and Furlong EEM (2019). Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat Genet 51, 1272–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, and Rosen MK (2019). Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484 e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Sandoval A, Towbin BD, Kalck V, Cabianca DS, Gaidatzis D, Hauer MH, Geng L, Wang L, Yang T, Wang X, et al. (2015). Perinuclear Anchoring of H3K9-Methylated Chromatin Stabilizes Induced Cell Fate in C. elegans Embryos. Cell 163, 1333–1347. [DOI] [PubMed] [Google Scholar]
- Gu B, Swigut T, Spencley A, Bauer MR, Chung M, Meyer T, and Wysocka J (2018). Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science 359, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall’Agnese A, Hannett NM, Spille JH, Afeyan LK, Zamudio AV, Shrinivas K, et al. (2019). Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager GL, McNally JG, and Misteli T (2009). Transcription dynamics. Mol Cell 35, 741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfon MS (2020). Silencers, Enhancers, and the Multifunctional Regulatory Genome. Trends Genet 36, 149–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannezo E, and Heisenberg CP (2019). Mechanochemical Feedback Loops in Development and Disease. Cell 178, 12–25. [DOI] [PubMed] [Google Scholar]
- Hansen AS (2020). CTCF as a boundary factor for cohesin-mediated loop extrusion: evidence for a multi-step mechanism. Nucleus 11, 132–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Texari L, Hayes MGB, Urbanowski M, Chang MW, Givarkes N, Rialdi A, White KM, Albrecht RA, Pache L et al. (2018). Transcription Elongation Can Affect Genome 3D Structure. Cell 174, 1522–1536 e1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TS, Cattoglio C, Slobodyanyuk E, Hansen AS, Rando OJ, Tjian R, and Darzacq X (2020). Resolving the 3D Landscape of Transcription-Linked Mammalian Chromatin Folding. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ing-Simmons E, Vaid R, Mannervik M, and Vaquerizas JM (2020). Independence of 3D chromatin conformation and gene regulation during Drosophila dorsoventral patterning. Biorxiv 10.1101/2020.07.07.18679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan M, Cummings R, and Yamashita YM (2019). The modular mechanism of chromocenter formation in Drosophila. Elife 8, e43938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost D, Carrivain P, Cavalli G, and Vaillant C (2014). Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res 42, 9553–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalhor R, Tjong H, Jayathilaka N, Alber F, and Chen L (2011). Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat Biotechnol 30, 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Piliszek A, Artus J, and Hadjantonakis AK (2013). FGF4 is required for lineage restriction and salt-and-pepper distribution of primitive endoderm factors but not their initial expression in the mouse. Development 140, 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna N, Zhang Y, Lucas JS, Dudko OK, and Murre C (2019). Chromosome dynamics near the sol-gel phase transition dictate the timing of remote genomic interactions. Nat Commun 10, 2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosraviani N, Ostrowski LA, and Mekhail K (2019). Roles for Non-coding RNAs in Spatial Genome Organization. Front Cell Dev Biol 7, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, and Shendure J (2019). Mechanisms of Interplay between Transcription Factors and the 3D Genome. Mol Cell 76, 306–319. [DOI] [PubMed] [Google Scholar]
- Krietenstein N, Abraham S, Venev SV, Abdennur N, Gibcus J, Hsieh TS, Parsi KM, Yang L, Maehr R, Mirny LAV et al. (2020). Ultrastructural Details of Mammalian Chromosome Architecture. Mol Cell 78, 554–565 e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran RI, and Spector DL (2008). A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J Cell Biol 180, 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MOV et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, and Tjian R (2018). Visualizing transcription factor dynamics in living cells. J Cell Biol 217, 1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luperchio TR, Sauria MEG, Hoskins VE, Wong X, DeBoy E, Gaillard M-C, Tsang P, Pekrun K, Ach RA, Yamada NA, et al. (2020). The repressive genome compartment is established early in the cell cycle before forming the lamina associated domains. Biorxiv 10.1101/481598. [DOI] [Google Scholar]
- Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macarthur BD, Ma’ayan A, and Lemischka IR (2009). Systems biology of stem cell fate and cellular reprogramming. Nat Rev Mol Cell Biol 10, 672–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macneil LT, and Walhout AJ (2011). Gene regulatory networks and the role of robustness and stochasticity in the control of gene expression. Genome Res 21, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima K, Ide S, and Babokhov M (2019). Dynamic chromatin organization without the 30-nm fiber. Curr Opin Cell Biol 58, 95–104. [DOI] [PubMed] [Google Scholar]
- Marshall WF, Straight A, Marko JF, Swedlow J, Dernburg A, Belmont A, Murray AW, Agard DA, and Sedat JW (1997). Interphase chromosomes undergo constrained diffusional motion in living cells. Curr Biol 7, 930–939. [DOI] [PubMed] [Google Scholar]
- Mateo LJ, Murphy SE, Hafner A, Cinquini IS, Walker CA, and Boettiger AN (2019). Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, and Alon U (2002). Network motifs: simple building blocks of complex networks. Science 298, 824–827. [DOI] [PubMed] [Google Scholar]
- Mirny LA, Imakaev M, and Abdennur N (2019). Two major mechanisms of chromosome organization. Curr Opin Cell Biol 58, 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T (2001). Protein dynamics: implications for nuclear architecture and gene expression. Science 291, 843–847. [DOI] [PubMed] [Google Scholar]
- Misteli T (2007). Beyond the sequence: cellular organization of genome function. Cell 128, 787–800. [DOI] [PubMed] [Google Scholar]
- Muller WG, Walker D, Hager GL, and McNally JG (2001). Large-scale chromatin decondensation and recondensation regulated by transcription from a natural promoter. J Cell Biol 154, 33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, and Fraser P (2013). Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, and Reinberg D (2015). CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhout HF (2002). The nature of robustness in development. Bioessays 24, 553–563. [DOI] [PubMed] [Google Scholar]
- Nir G, Farabella I, Perez Estrada C, Ebeling CG, Beliveau BJ, Sasaki HM, Lee SD, Nguyen SC, McCole RB, Chattoraj Sv et al. (2018). Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet 14, e1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuebler J, Fudenberg G, Imakaev M, Abdennur N, and Mirny LA (2018). Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc Natl Acad Sci U S A 115, E6697–E6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, and O’Shea CC (2017). ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paakinaho V, Presman DM, Ball DA, Johnson TA, Schiltz RL, Levitt P, Mazza D, Morisaki T, Karpova TS, and Hager GL (2017). Single-molecule analysis of steroid receptor and cofactor action in living cells. Nat Commun 8, 15896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliou C, Guckelberger P, Schopflin R, Heinrich V, Esposito A, Chiariello AM, Bianco S, Annunziatella C, Helmuth J, Haas Sv et al. (2019). Preformed chromatin topology assists transcriptional robustness of Shh during limb development. Proc Natl Acad Sci U S A 116, 12390–12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos DK, Skouloudaki K, Engstrom Y, Terenius L, Rigler R, Zechner C, Vukojevic V, and Tomancak P (2019). Control of Hox transcription factor concentration and cell-to-cell variability by an auto-regulatory switch. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada LA, McQueen PG, and Misteli T (2004). Tissue-specific spatial organization of genomes. Genome Biol 5, R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar JJ, Woringer M, and Zimmer C (2019). How the Genome Folds: The Biophysics of Four-Dimensional Chromatin Organization. Annu Rev Biophys 48, 231–253. [DOI] [PubMed] [Google Scholar]
- Paulsen J, Liyakat Ali TM, Nekrasov M, Delbarre E, Baudement MO, Kurscheid S, Tremethick D, and Collas P (2019). Long-range interactions between topologically associating domains shape the four-dimensional genome during differentiation. Nat Genet 51, 835–843. [DOI] [PubMed] [Google Scholar]
- Phair RD, and Misteli T (2000). High mobility of proteins in the mammalian cell nucleus. Nature 404, 604–609. [DOI] [PubMed] [Google Scholar]
- Phair RD, Scaffidi P, Elbi C, Vecerova J, Dey A, Ozato K, Brown DT, Hager G, Bustin M, and Misteli T (2004). Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol Cell Biol 24, 6393–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo A, and Dillon N (2015). Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol 16, 245–257. [DOI] [PubMed] [Google Scholar]
- Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Yv et al. (2018). Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 174, 744–757 e724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse I, Perlman MD, Scalzo D, Kooperberg C, Groudine M, and Kosak ST (2009). The emergence of lineage-specific chromosomal topologies from coordinate gene regulation. Proc Natl Acad Sci U S A 106, 6679–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320 e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy KL, Zullo JM, Bertolino E, and Singh H (2008). Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452, 243–247. [DOI] [PubMed] [Google Scholar]
- Rodriguez J, and Larson DR (2020). Transcription in Living Cells: Molecular Mechanisms of Bursting. Annu Rev Biochem. [DOI] [PubMed] [Google Scholar]
- Rodriguez J, Ren G, Day CR, Zhao K, Chow CC, and Larson DR (2019). Intrinsic Dynamics of a Human Gene Reveal the Basis of Expression Heterogeneity. Cell 176, 213–226 e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, and Corces VG (2018). Organizational principles of 3D genome architecture. Nat Rev Genet 19, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, Nichols MH, Lyu X, Ando-Kuri M, Rivera ISM, Hermetz K, Wang P, Ruan Y, and Corces VG (2017). Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol Cell 67, 837–852 e837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at superenhancers links phase separation and gene control. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanulli S, Trnka MJ, Dharmarajan V, Tibble RW, Pascal BD, Burlingame AL, Griffin PR, Gross JD, and Narlikar GJ (2019). HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575, 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sati S, Bonev B, Szabo Q, Jost D, Bensadoun P, Serra F, Loubiere V, Papadopoulos GL, Rivera-Mulia JC, Fritsch Lv et al. (2020). 4D Genome Rewiring during Oncogene-Induced and Replicative Senescence. Mol Cell 78, 522–538 e529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger S, and Meshorer E (2019). Open Chromatin, Epigenetic Plasticity, and Nuclear Organization in Pluripotency. Dev Cell 48, 135–150. [DOI] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, and Cavalli G (2012). Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148, 458–472. [DOI] [PubMed] [Google Scholar]
- Shaban HA, Barth R, Recoules L, and Bystricky K (2020). Hi-D: nanoscale mapping of nuclear dynamics in single living cells. Genome Biol 21, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaban HA, and Seeber A (2020). Monitoring the spatio-temporal organization and dynamics of the genome. Nucleic Acids Res 48, 3423–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar S, and Misteli T (2017). Causes and consequences of nuclear gene positioning. J Cell Sci 130, 1501–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar S, Voss TC, Pegoraro G, Sciascia N, and Misteli T (2015). Identification of Gene Positioning Factors Using High-Throughput Imaging Mapping. Cell 162, 911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Takei Y, Zhou W, Lubeck E, Yun J, Eng CL, Koulena N, Cronin C, Karp C, Liaw EJV et al. (2018). Dynamics and Spatial Genomics of the Nascent Transcriptome by Intron seqFISH. Cell 174, 363–376 e316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, and Thirumalai D (2019). Conformational heterogeneity in human interphase chromosome organization reconciles the FISH and Hi-C paradox. Nat Commun 10, 3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, and Brangwynne CP (2017). Liquid phase condensation in cell physiology and disease. Science 357. [DOI] [PubMed] [Google Scholar]
- Shopland LS, Lynch CR, Peterson KA, Thornton K, Kepper N, Hase J, Stein S, Vincent S, Molloy KR, Kreth Gv et al. (2006). Folding and organization of a contiguous chromosome region according to the gene distribution pattern in primary genomic sequence. J Cell Biol 174, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small S, Blair A, and Levine M (1992). Regulation of even-skipped stripe 2 in the Drosophila embryo. EMBO J 11, 4047–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovei I, Kreysing M, Lanctot C, Kosem S, Peichl L, Cremer T, Guck J, and Joffe B (2009). Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 137, 356–368. [DOI] [PubMed] [Google Scholar]
- Spector DL, and Lamond AI (2011). Nuclear speckles. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanek D, and Fox AH (2017). Nuclear bodies: news insights into structure and function. Curr Opin Cell Biol 46, 94–101. [DOI] [PubMed] [Google Scholar]
- Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, Leeb M, Wohlfahrt KJ, Boucher W, O’Shaughnessy-Kirwan A, et al. (2017). 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter DM, Molina N, Gatfield D, Schneider K, Schibler U, and Naef F (2011). Mammalian genes are transcribed with widely different bursting kinetics. Science 332, 472–474. [DOI] [PubMed] [Google Scholar]
- Symmons O, and Raj A (2016). Wha’s Luck Got to Do with It: Single Cells, Multiple Fates, and Biological Nondeterminism. Mol Cell 62, 788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa T, Meaburn KJ, and Misteli T (2008). The meaning of gene positioning. Cell 135, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjong H, Li W, Kalhor R, Dai C, Hao S, Gong K, Zhou Y, Li H, Zhou XJ, Le Gros MA, et al. (2016). Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proc Natl Acad Sci U S A 113, E1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To TL, and Maheshri N (2010). Noise can induce bimodality in positive transcriptional feedback loops without bistability. Science 327, 1142–1145. [DOI] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Furlong EEM (2019). The role of transcription in shaping the spatial organization of the genome. Nat Rev Mol Cell Biol 20, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermunt MW, Zhang D, and Blobel GA (2019). The interdependence of gene-regulatory elements and the 3D genome. J Cell Biol 218, 12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Wallerich S, and Becskei A (2018). Stochastic Gene Choice during Cellular Differentiation. Cell Rep 24, 3503–3512. [DOI] [PubMed] [Google Scholar]
- Wang A, Kolhe JA, Gioacchini N, Baade I, Brieher WM, Peterson CL, and Freeman BC (2020). Mechanism of Long-Range Chromosome Motion Triggered by Gene Activation. Dev Cell 52, 309–320 e305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Su JH, Beliveau BJ, Bintu B, Moffitt JR, Wu CT, and Zhuang X (2016). Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz G, Varnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, Schoenfelder S, Jessberger G, Muhar M, Hossain MJV et al. (2017). Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J 36, 3573–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, and Schiessel H (2016). Transcription Driven Phase Separation in Chromatin Brush. Langmuir 32, 3036–3044. [DOI] [PubMed] [Google Scholar]
- Zheng X, Hu J, Yue S, Kristiani L, Kim M, Sauria M, Taylor J, Kim Y, and Zheng Y (2018). Lamins Organize the Global Three-Dimensional Genome from the Nuclear Periphery. Mol Cell 71, 802–815 e807. [DOI] [PMC free article] [PubMed] [Google Scholar]