

Abstract
The endothelium produces many substances that can regulate vascular tone. Acetylcholine is a widely used pharmacological tool to assess endothelial function. In general, acetylcholine binds to G-protein coupled muscarinic receptors that mediate a transient elevation in intracellular, free calcium. This intracellular rise in calcium is responsible for triggering several cellular responses, including the synthesis of nitric oxide, endothelium-derived hyperpolarizing factor, and eicosanoids derived from arachidonic acid. Endothelial arachidonic acid metabolism is also an important signaling pathway for mediating inflammation. Therefore, in conditions with sustained and excessive inflammation such as hypertension, arachidonic acid serves as a substrate for the synthesis of several vasoconstrictive metabolites, predominantly via the cyclooxygenase and lipoxygenase enzymes. Cyclooxygenase and lipoxygenase products can then activate G-protein coupled receptors expressed on vascular smooth muscle cells to causes contractile responses. As a result, acetylcholine-induced contraction due to arachidonic acid is a commonly observed feature of endothelial dysfunction and vascular inflammation in hypertension. In this review, we will critically analyze the literature supporting this concept, as well as address the potential underlying mechanisms, including the possibility that arachidonic acid signaling is diverted away from the synthesis of pro-resolving metabolites in conditions such as hypertension.
Keywords: Endothelium, arachidonic acid metabolites, vascular function, acetylcholine, hypertension
1. INTRODUCTION
Endothelial health has been a major topic of research since the 20th century. The endothelium plays an intricate role in vascular homeostasis by synthesizing and secreting several vasoactive factors, such as pro- and anti-inflammatory lipid mediators. In a healthy state, acute vascular inflammation is necessary for immune surveillance to eliminate pathogens and host-derived threats and to repair the associated tissue injury. It is a self-limiting process, leading to complete resolution that allows a return to homeostasis after threat eradication and subsequent damage repair [1]. The concern arises when there is prolonged or excessive inflammation, which can exacerbate the vascular damage and contribute to the genesis and/or progression of diseases. Accordingly, low-grade, chronic vascular inflammation is broadly accepted as a common factor in many cardiovascular diseases, including hypertension. Among various inflammatory mediators implicated in hypertension, metabolites and enzymes involved in arachidonic acid (AA) metabolism are of greater significance. Accordingly, arteries from hypertensive patients and animals present with a late phase acetylcholine-induced endothelium-dependent contraction (Figs. 1–3) due to AA-derived biologically active products. In contrast with other reviews about decreased acetylcholine-induced relaxation in hypertension, this mini-review will focus on the involvement of AA-derived lipids mediators in acetylcholine-induced endothelium-dependent contraction. Furthermore, given that AA-derived lipids also are crucial for “switching-off” inflammation, this mini-review will briefly address the possible mechanisms underlying the resolution of inflammation in vascular tissue.
Fig. (1).
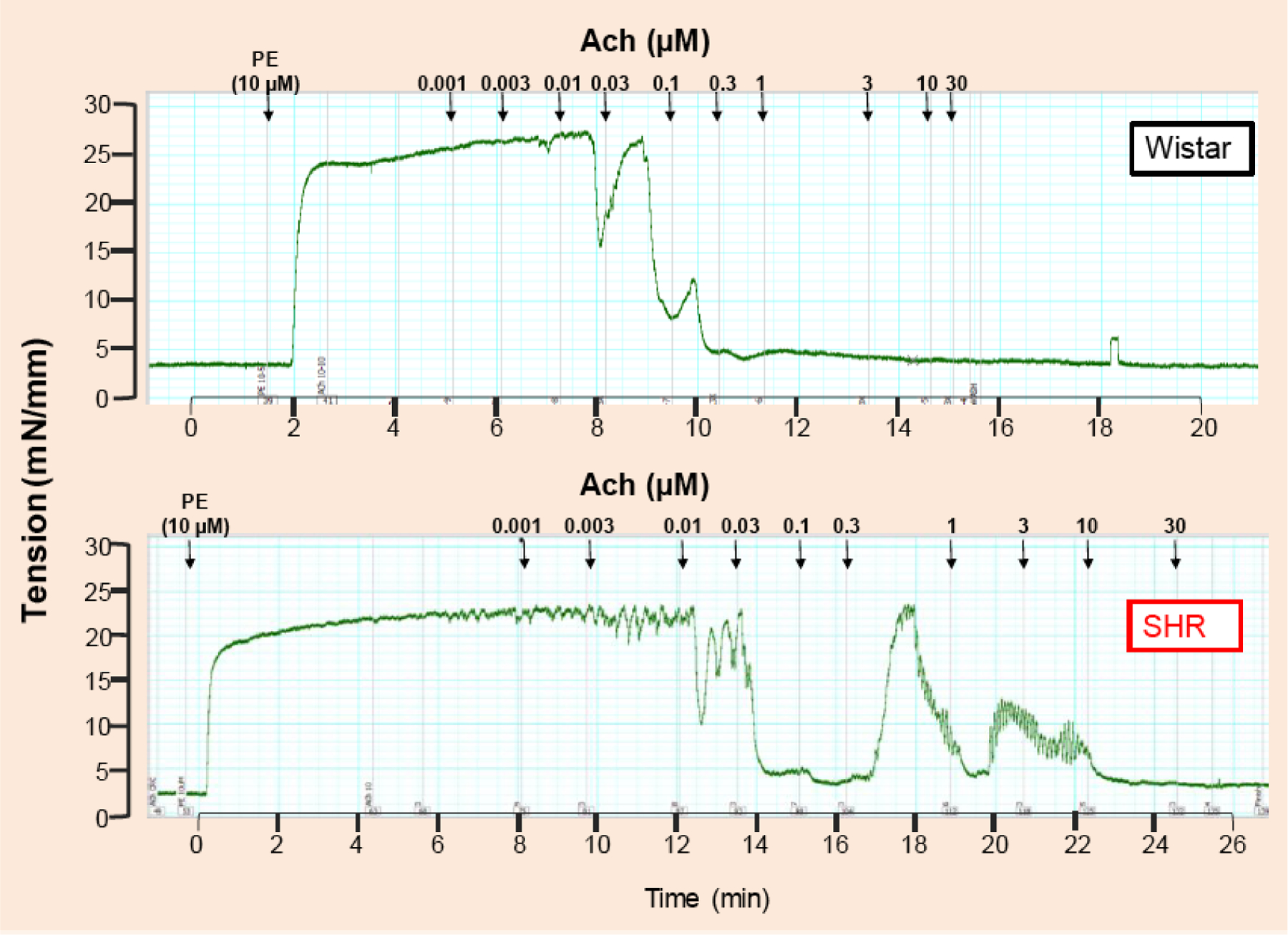
Typical traces represent concentration response curve to acetylcholine (1nM −30 μM) in mesenteric resistance arteries from 12-weeks old spontaneously hypertensive rats (SHR) and normotensive Wistar rats. The arteries were initially contracted with phenylephrine (10 μM). Please note that at the concentration of 0.03 μM acetylcholine, arteries from SHR are completely relaxed when compared to arteries from Wistar rats. This is a representative illustration of traces observed in our laboratory. However, similar findings are quantified in multiple studies [13–15, 35–37].
Fig. (3).
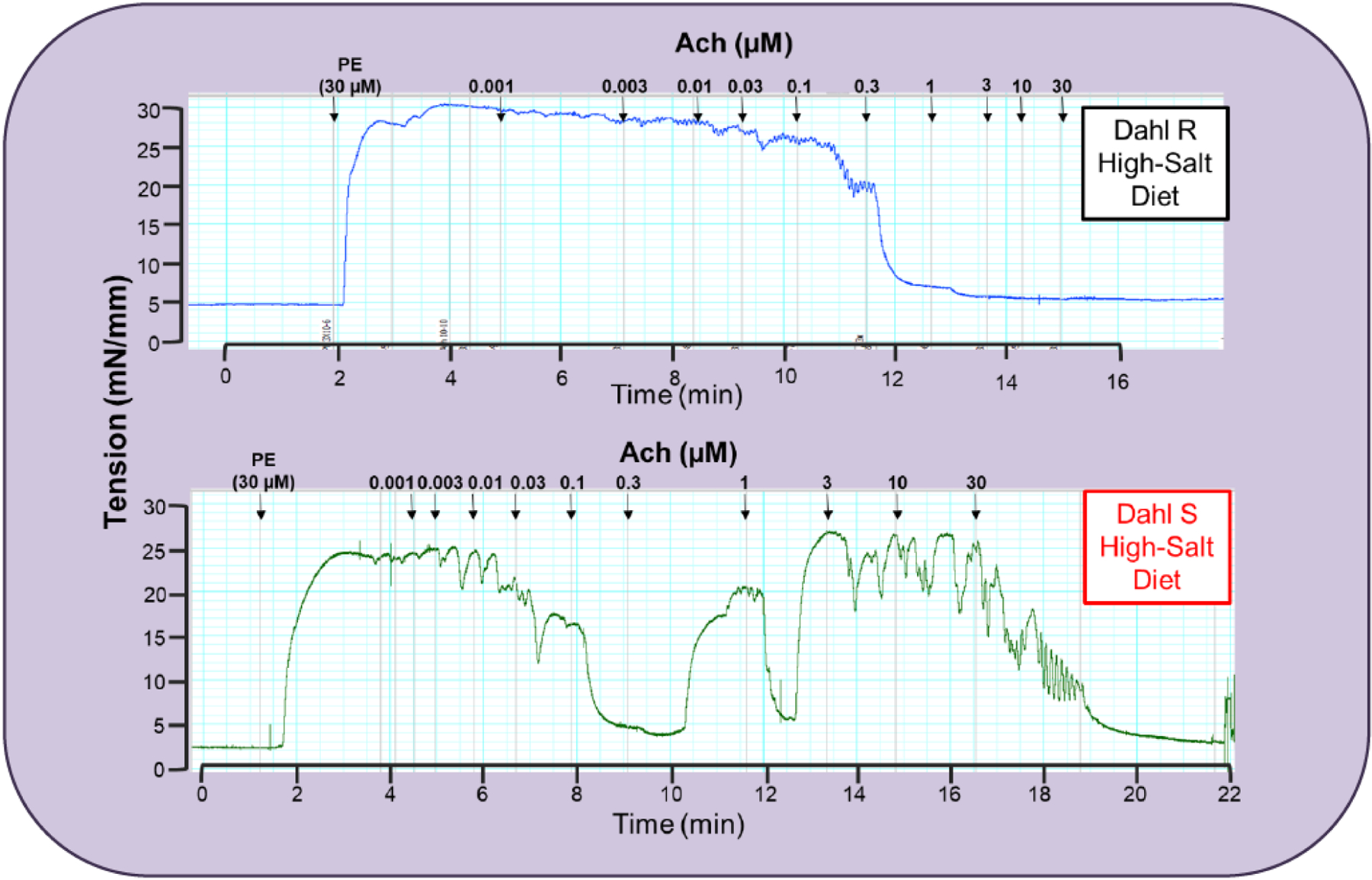
Typical traces represent concentration response curve to acetylcholine (1nM −30 μM) in mesenteric resistance arteries from 15-week old Dahl salt (S) sensitive and resistant (R) rats fed a 2% high-salt diet for 8 weeks. The arteries were initially contracted with phenylephrine (10 μM). Please note that at the concentration of 0.3 μM acetylcholine, arteries from Dahl S are completely relaxed when compared to arteries from Dahl R. This is a representative illustration of traces observed in our laboratory. However, similar findings are quantified in multiple studies [13–15, 35–37].
2. ACETYLCHOLINE-INDUCED ENDOTHELIUM-DEPENDENT CONTRACTION
In physiological conditions, vasculature oscillates between contraction and relaxation to maintain vascular tone and homeostasis. Over the last four decades, we have come to understand that endothelium plays a major role in regulation of vascular function. The endothelium produces many substances that can regulate vascular smooth muscle contraction, proliferation, migration, inflammation, and platelet function [2]. In general, vascular biology research laboratories use acetylcholine as a pharmacological tool to assess endothelial function (and dysfunction). Acetylcholine binds to G-protein coupled muscarinic receptors (mAChRs) on the endothelial cell membrane. These receptors are comprised of five subtypes (M1-M5). M1, M3, and M5 are coupled with the Gq protein and induce cytosolic calcium transients via phospholipase C signaling pathway. M2 and M4 are coupled with the Gi protein, which inhibits adenylyl cyclase [3]. A transient elevation of the intracellular, free calcium concentration is considered a key mechanism in triggering several cellular responses, such as the synthesis of nitric oxide (NO), endothelium-derived hyperpolarizing factor (EDHF) and eicosanoids derived from arachidonic acid (AA) [4–9]. Overall, healthy, isolated, resistance and conductance arteries release both vasodilator and vasoconstrictor factors, but the relative amount of vasodilators is higher, leading to full relaxation for long periods. In diseased states, this homeostatic scenario changes, resulting in an exacerbated production of vasoconstrictors molecules. For further details on how the endothelium regulates vascular function, please see references [10–11].
One of the major pathophysiological signatures in cardiovascular diseases is the presence of endothelial dysfunction, regardless of artery diameter (i.e., conductance and resistance arteries). Diseased conductance arteries are stiffer and larger than their normal controls, which increases systolic pressure. Diseased resistance arteries have several types of alterations that contribute to cardiovascular disease (i.e., reduced lumen diameter and hypertrophy of the vascular wall). Endothelium dysfunction is a result of an imbalance between endothelium-derived relaxing factors and endothelium-derived contracting factors. Endothelial dysfunction may lead to several abnormalities in the vasculature, such as an increase in maximum response and/or sensitivity to contractile factors, the decrease in maximum response and/or sensitivity to endothelium-dependent vasodilators, vascular remodeling, etc. In case of hypertension, especially in early stages or “mild” hypertension, a phenomenon known as acetylcholine-induced endothelium contraction occurs (Figs. 1 and 2). Endothelium-dependent contractions were first observed in isolated dog veins [12], although most studies today are performed on arteries. In 1981, Dr. Paul Vanhoutte was the first to report endothelium-derived vasoconstriction, showing that an unknown factor was being produced by the endothelium to cause an increased vascular tone. In physiological conditions, it has been seen that stretch applied to isolated canine basilar arteries caused the development of active tension in rings with endothelium but not in those in which the endothelium had been removed [13]. The authors suggested that the endothelium may contribute to the autoregulation of cerebral blood flow during increases in transmural pressure by the increased production and/or release of prostaglandins, which causes activation of the underlying vascular smooth muscle [13]. However, more probable is that the incidence of endothelium-dependent contractions is pathological, as they are prominent in arteries from animals that present vascular diseases, such as hypertension. In 1986, Dr. Vanhoutte, along with Dr. Thomas Lüscher, set out to investigate the decreased relaxation to acetylcholine in spontaneously hypertensive rats (SHR). Unexpectedly, they observed that acetylcholine caused endothelium-dependent contractions in arteries from SHR but not in those from Wistar Kyoto (WKY) rats [14]. In this study, aortas from hypertensive animals possessed a contraction to acetylcholine in concentrations ranged from ~100 nM - 10 μM. It was uncovered that the contraction to acetylcholine was also a characteristic of endothelium dysfunction [15–16].
Fig. (2).
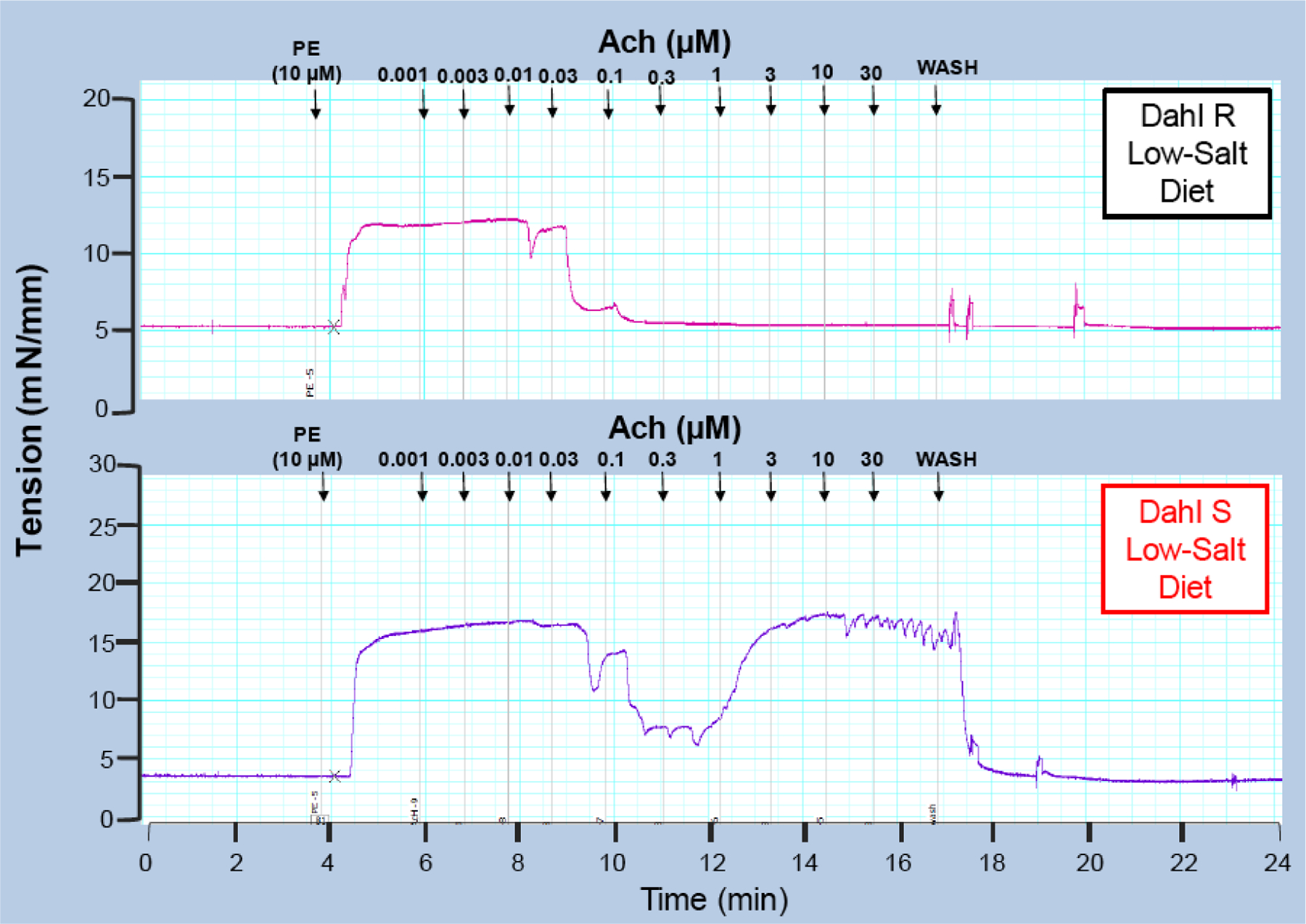
Typical traces represent concentration response curve to acetycholine (1nM −30 μM) in mesenteric resistance arteries from 8-weeks old Dahl salt (S) sensitive and resistant (R) rats fed a 0.3% low-salt diet. The arteries were initially contracted with phenylephrine (10 μM). This is a representative illustration of traces observed in our laboratory. However, similar findings are quantified in multiple studies [13–15, 35–37].
Significant to the pathogenesis of hypertension, acetylcholine-induced contraction is not unique to SHR (Fig. 1). In our laboratory, we routinely observe acetylcholine-induced contraction in resistance and conductance arteries from Dahl salt-sensitive (S) rats (Figs. 2 and 3). Salt-sensitivity is considered a hallmark of hypertension in blacks, including African Americans, given that it is found in 73% of hypertensive blacks [17]. Using inbred Dahl S rats, Dr. John Rapp [18] observed that the spontaneous elevation of blood pressure occurs in Dahl S rats fed a low salt diet, but not in Dahl salt-resistant (R) rats, due to unknown genetic influences and independent of environmental factors. According to Dr. Rapp’s measurements, spontaneous elevations in blood pressure from Dahl S rats begin at about ~4 weeks of age [18], and a high-salt diet accelerates this process leading to malignant hypertension, similar to humans. Corroborating, Dr. Rapp’s findings, we observed that arteries from young Dahl S rats (~8 weeks old) fed a low-salt diet also present acetylcholine-induced contractions (Fig. 2), but arteries from Dahl S fed a high salt diet (~15 weeks old) did not worsen this phenotype (Fig. 3). These data suggest that Dahl S already has endothelial dysfunction prior to malignant hypertension. Therefore, the genesis of acetylcholine-induced contraction may be, at least in part, due to genetic influences. Another characteristic that we frequently observe is resistance arteries from SHR or Dahl S high-salt start relaxing to acetylcholine at lower concentrations (100 nM-1 μM), which means that they are more sensitive to acetylcholine when compared to the arteries from normotensive animals (Fig. 3). However, at higher concentrations (0.3 μM), these arteries start contracting as previously described [13–14]. It is important to note that 0.3 μM acetylcholine, arteries should be relaxing back to baseline; however, hypertensive arteries have relaxed and start to contract again. Although acetylcholine-induced endothelium-dependent contraction was also observed in other conditions, such as aging and diabetes [19], it seems that this phenomenon is predominant in hypertension.
3. CULPRITS OF ACETYLCHOLINE-INDUCED CONTRACTION IN HYPERTENSION
Multiple studies have demonstrated contractions to endothelium-derived vasoactive factors in isolated arteries from animals with spontaneous hypertension and salt-sensitive hypertension [14, 20–24]. After that initial observation and many years of research later, mechanistic studies began uncovering possible “culprits” to this phenomenon. In line, it has been shown that endothelium pathways for reactive oxygen species (ROS), cyclooxygenase (COX) and lipoxygenase (LOX) products are the instigators and/or contributors to acetylcholine-induced endothelium-dependent contractions in hypertension [25–26].
In hypertension, there is no doubt that ROS, such as superoxide anions, hydroxyl radicals, and hydrogen peroxide are increased [27], and contribute to amplified contractile responses. ROS can increase contraction by quenching the bioavailability of NO [28] and also by uncoupling endothelial nitric oxide synthase (eNOS) (which itself can be a self-sustained source of ROS) [27]. ROS also leads to increased smooth muscle contraction and proliferation, platelet aggregation, and expression of adhesion molecules [29]. Moreover, ROS can depolarize vascular smooth muscle cells by inhibiting potassium channels and induce calcium sensitization, via activation of RhoA and Rho kinase activity [30]. However, while it is clear that ROS participates in acetylcholine-induced endothelium-dependent contractions, whether ROS are the primary instigators of this phenomenon is debatable. For example, it has been established that acetylcholine-induced endothelium-dependent contractions are decreased by superoxide dismutase, which breaks down superoxide anions to hydrogen peroxide, but not by catalase or deferoxamine, which scavenge hydrogen peroxide and hydroxyl radicals, respectively [31]. These observations suggest an important role for superoxide anions in acetylcholine-induced contraction [31–33].
On the other hand, it is also known that COX, a major downstream enzyme of AA signaling, is a significant source for ROS [34]. Specifically, we have observed that the incubation with the specific inhibitor of COX-2 decreased ROS generation in isolated arteries from ouabain-treated hypertensive rats [34]. While these rats present with malignant hypertension and their arteries were hyperreactive to noradrenaline and had oxidative stress, acetylcholine-induced contraction was not observed. These findings suggest that ROS generation is not the primary instigator of acetylcholine-induced endothelium-dependent contraction and that ROS only indirectly contribute to this phenomenon by reducing the bioavailability of NO and/or activating COX in the vascular smooth muscle cells [35]. Therefore, it is still unresolved whether ROS are a “cause” or an “effect” of endothelium-dependent, acetylcholine-induced contractions in hypertension. On the other hand, as it will be discussed below, it is established that AA-derived biologically active products are the cause of endothelium-dependent contractions elicited by acetylcholine.
3.1. Arachidonic Acid-Derived Bioactive Lipids
AA is the most common precursor of biologically active lipids metabolites [26], and it plays a vital role in the initiation of inflammation and the resolution of inflammation in physiological states. AA is the preferred substrate for the COX, LOX, and cytochrome P450 (CYP450) pathways to produce prostaglandins, leukotrienes, and epoxyeicosatrienoic acids (EETs), respectively [26, 36]. These lipid mediators are collectively referred to as eicosanoids, and these pathways are the target of approved drugs for the treatment of pain, allergies, inflammation, asthma, cardiovascular disorders, and, most recently, to inhibit tumor-associated inflammation [37]. A proper balance of substrate availability, enzyme expression and activity, and non-enzymatic oxidation is important for maintaining homeostasis. However, in hypertension, an unbalance in eicosanoids occurs that has a causative-effect in the genesis of acetylcholine-induced endothelium-dependent contractions predominantly through the COX, LOX, and CYP450 pathways that will be discussed below (Fig. 4).
Fig. (4).
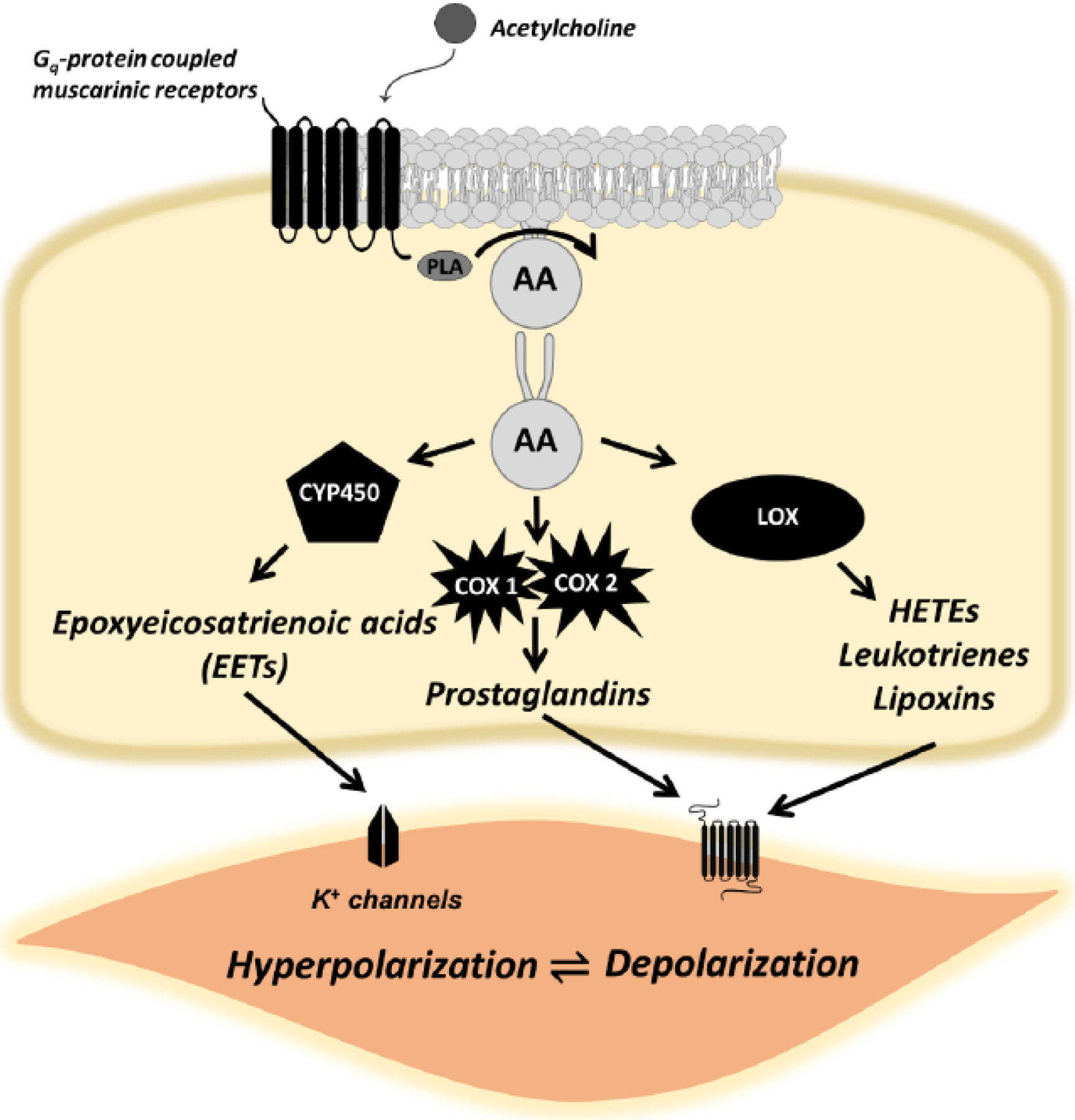
The Janus face of acetylcholine and endothelium-dependent, arachidonic acid signaling. In physiological conditions, acetylcholine promotes the synthesis of vasodilating eicosanoids and other endothelium-derived hyperpolarizing factors (e.g., nitric oxide). However, in pathophysiological conditions such as hypertension, there is aberrant arachidonic acid signaling due to excessive inflammation. As a result, acetylcholine promotes the synthesis of vasoconstrictor metabolites, predominantly from cyclooxygenase and lipoxygenase that can act on vascular smooth muscle cells. As a result, acetylcholine causes endothelium-dependent contractile responses, as opposed to vasodilation.
3.1.1. Cyclooxygenases
The COX pathways produce pro-inflammatory or pro-resolving lipid mediators from the COX-1 or COX-2 isoforms. Although COX-1 and COX-2 are 65% homologous, they are regulated differently and can function independently within the same cell [38]. COX-1 uses fatty acids like AA preferentially, while COX-2 uses fatty acids and glycerol, because of this COX-2 is capable of making mediators that COX-1 cannot [39]. COX-1 is constitutively expressed in tissues but can also be overexpressed [40]. COX-2 is also constitutive, but its activity is induced mainly in inflammatory states. However, it has also been shown that COX-2 participates in physiological states. Specifically, it has been shown that cultured bovine aortic endothelial cells (BAECs) and human umbilical vein endothelial cells (HUVECs) exposed to steady fluid shear stress of 10 dyn/cm2 for five hours upregulates COX-2 and prostacyclin (a potent vasodilator) [41]. The authors observed the relationship between the mechanosensor platelet endothelial cell adhesion molecule-1 (PECAM-1) and the intracellular mechanoresponsive molecules phosphatidylinositol 3-kinase (PI3K), focal adhesion kinase (FAK), and mitogen-activated protein kinase p38 in the fluid shear stress induction of COX-2 expression and PGI2 release. Knockdown of PECAM-1 expression inhibited fluid shear stress-induced activation of α5β1-integrin, upregulation of COX-2, and release of PGI2 [41]. In addition, there is a third distinct COX isozyme, called COX-3, whose discovery explained, in part, the pharmacology of acetaminophen [42]. Less is known about COX-3, which seems to be found in the cerebral cortex and cardiac tissue and appears to be involved in centrally mediated pain. COX-3 is a splice variant for COX-1. Therefore, drugs that favorably block COX-1 also appear to act at COX-3.
The COX pathway converts AA to endoperoxides that are then converted by their respective synthases into five main prostaglandins (PGs): PGD2, PGE2, PGF2α, PGI2 (prostacyclin), and thromboxane (Tx)A2. Because prostanoids are a family of lipid mediators produced from fatty acids, and hence are generally regarded as hydrophobic compounds, it was thought in earlier times that they were incorporated into the cell membrane and exerted their action by perturbing lipid fluidity [43]. However, each prostanoid has a unique activity profile, not exactly overlapping with others, which indicates that each prostanoid has a specific site of action. Hence, the concept of prostanoid action via prostanoid receptors gradually appeared [43]. Supporting the concept that prostanoids indeed act via receptor, it was shown that some prostanoid actions induce changes in intracellular cAMP levels, phosphatidylinositol (PI) turnover, and free Ca2+ concentrations [43]. Currently, we know that each prostanoid (TxA2, PGI2, PGE2, PGF2, and PGD2) has a specific receptor (TP, IP, EP, FP, and DP, respectively) [43]. These receptors are G protein-coupled receptors, and there are eight types and subtypes that are encoded by different genes, but as a whole constitute a subfamily in the superfamily of the rhodopsin-type receptors [43]. Prostanoid receptors have been classically grouped into three categories, depending on the type of G-protein they bind to the contractile receptors EP1, FP and TP are coupled to Gq and activate phospholipase C, leading to intracellular calcium increases; the relaxant type, which comprises IP, DP1, EP2 and EP4, signal through Gs to induce adenylate cyclase activity; and the inhibitory receptor EP3 couples to Gi, leading to a decrease in intracellular cAMP content [44]. Promiscuously, prostanoids can bind to multiple receptors or receptor isoforms and are therefore able to activate diverse G-proteins and signal transduction pathways. Accordingly, it has been shown that in aorta from SHR and WKY, different prostaglandins studied, TXA2, PGH2, PGF2α, PGE2, PGD2, PGI2 as well as 8-isoprostane, were able to induce vascular contraction via TP receptors, given that the contractions were abolished by S18886, a specific antagonist for the TP receptor [45].
In hypertension, COX-1 and 2 expression and activity are enhanced in conductance and resistance arteries [34, 46], consequently leading to an increase in prostanoids production. Incubation with non-selective (indomethacin) or selective inhibitors of COX-1 and −2, such as N[−2-(cyclohexyloxy)-4-nitrophenyl]-methanesulfonamide (NS398) or 2-[(1-oxopenytyl)oxy]-benzoic acid (valeryl salicylate) prevented acetylcholine-induced endothelium-dependent contractions [48]. As mentioned above, TXA2 is a potent vasoconstrictor, and its synthesis is increased in endothelial cells from hypertensive animals [36]. However, the use of a selective inhibitor of TXA2 synthase only partially prevented the endothelium-dependent contractions indicating that TXA2 is only one factor contributing to contraction [47–52]. On the other hand, acetylcholine-induced endothelium-dependent contractions were completely inhibited by treatment with a TXA2/PGH2 antagonist (O NO-3708), but not with a TXA2 synthase inhibitor (OKY-046) [53]. The authors suggested that since these contractile responses were completely suppressed by TXA2/PGH2 antagonist, but were not affected by a TXA2 synthase inhibitor, they are most likely mediated by endothelium-derived PGH2 [53]. There was also a statistically significant correlation between the acetylcholine-induced contractions and blood pressure. However, in vivo administration of another TXA2/PGH2 antagonist (ONO-8809) (10 or 30 micrograms per body per day) for 3 weeks (5–8 weeks of age) did not affect blood pressure in SHR or WKY. These observations suggest that the increase in endothelium-derived contractile factors in SHR is more likely to be a result than a cause of hypertension in SHR [53].
Another prostaglandin associated with endothelium-dependent contractions in hypertension is PGI2. Notably, PGI2 is the most abundant prostaglandin in hypertensive endothelial cells [49–52]. In physiological and acute conditions, the induction of COX-2 promotes the production of PGI2, which is a vasodilator, however in chronic inflammatory conditions, as seen in hypertensive animals, COX-2 upregulation induces endothelium-dependent contractions via PGI2 [36]. In hypertension, PGI2 synthase activity and expression are increased in endothelial cells, which leads to an exacerbated production of PGI2 via activation of TP receptors to induce contraction [20, 49–50]. Exogenous prostacyclin induced a concentration-dependent relaxation in mesenteric resistance arteries from WKY rats. However, in arteries from non-treated SHR and both WKY and SHR treated with aldosterone (0.05 mg/kg/day) for 3 weeks, prostacyclin concentrations below 0.1 μM induced concentration-dependent relaxation whereas concentrations equal or above 0.1 μM have a biphasic effect, characterized by an initial contractile response followed by a relaxing phase [54]. These authors also observed that the treatment with aldosterone-induced an exacerbated COX-2 expression in both strains. However, chronic aldosterone administration did not significantly modify blood pressure levels. These suggest that another mechanism, different from high blood pressure, maybe the initiator of endothelial-induced contraction in hypertension.
3.1.2. Lipoxygenases
The LOX pathway produces hydroperoxides from AA [55]. LOX enzymes are expressed in several cells, such as immune, epithelial, and tumor cells that display a variety of physiological functions, including inflammation, skin disorder, and tumorigenesis. In humans, there are five LOXs: the 5S-(arachidonate: oxygen 5-oxidoreductase), 12R-(arachidonate 12-lipoxygenase, 12R-type), 12S-(arachidonate: oxygen 12-oxidoreductase), and two distinct 15S-(arachidonate: oxygen 15-oxidoreductase) LOXs that oxygenate arachidonic acid in different positions along the carbon chain and form the corresponding 5S-, 12S-, 12R-, or 15S-hydroperoxides, respectively [56]. In general, 15-LOX synthesizes anti-inflammatory 15-hydroxyeicosatetraenoic acid (HETE), 5-LOX and 12-LOX induce pro-inflammatory mediators, and 5-LOX produces 5-HETE and leukotrienes, which are potent chemoattractants.
Lipoxygenase inhibitors have been reported to reduce the angiotensin II-induced contraction on isolated arteries. Specifically, contractions induced by angiotensin II in isolated human internal mammary artery were inhibited by phenidone 100 μM (cyclooxygenase and lipoxygenase inhibitor), baicalein 100 μM (5-, 12- and 15-lipoxygenases inhibitor), AA861 10 μM (5-lipoxygenase inhibitor) and MK571 1 μM (CysLT1 receptor antagonist) [57]. Interestingly, increases in the systolic pressor response to angiotensin II were reduced by two different LOX inhibitors, baicalein (30 mg/kg) and esculetin (60 mg/kg) in rats, but not by the cyclooxygenase inhibitor indomethacin, suggesting that LOX pathway may have an important role in mediating the pressor effect of angiotensin II [58]. DelliPizzi et al. [59] demonstrated that in cytosolic fractions of aortae taken from aortic coarctation-induced hypertensive rats, 12-lipoxygenase protein was increased as compared to normotensive controls. Aortic rings from hypertensive, but not from normotensive rats, exhibited a basal tone that was reduced by the lipoxygenase inhibitors cinnamyl-3,4-dihydroxy-α-cyanocinnamate (CDC,10 μM) and 5,8,11-eicosatriynoic acid (ETI, 10 uM) [59]. Corroborating previous studies, these authors observed that CDC (8mg/kg administered subcutaneously) did not affect the blood pressure of normotensive rats but decreased that of hypertensive rats. Only a few studies evaluated the possible mechanisms of LOX-derived products and endothelium-dependent contraction in hypertension [59]. Accordingly, it has been shown that SHR and WKY orally treated for 3 weeks with cysteinyl leukotrienes biosynthesis inhibitor, MK-886 (0.1 mg/ml), did not present changes in blood pressure. However, in arteries incubated with NOS inhibitor N-nitro-L-arginine (L-NNA; 100 μM) and contracted to prostaglandin F2α (PGF2α)-induced tone, acetylcholine was able to evoke concentration-dependent contractions in SHR aorta [60]. Further, incubation with MK-886 (10 μM), the 5-lipoxygenase (5-LO) inhibitor AA861 (10 μmol/l), or the cysteinyl leukotriene receptor antagonist MK571 (1 μmol/l) was able to decrease these acetylcholine-induced contractions. Overall this suggests that LOX-derived products may be associated in the endothelium-dependent contraction to acetylcholine, but not in the regulation of blood pressure in SHR [60].
As previously described, AA is also a substrate for LOX to produce leukotrienes and lipoxins [26, 55]. Leukotrienes and lipoxins are two functionally different classes of LOX-derived eicosanoids. Leukotrienes are potent pro-vasoconstrictors mediators and directly and indirectly stimulate fibroblast chemotaxis, proliferation, and collagen synthesis. On the other hand, lipoxins counter-regulate the pro-inflammatory actions of leukotrienes and activate the resolution of the inflammatory response [61]. Lipoxin A4 (LXA4) was discovered in 1984 through interaction(s) between the 5- and 15-lipoxygenase pathways in human leukocytes [62]. The generation of LXA4 is a very rapid process that aspirin does not inhibit. In fact, aspirin has been shown to trigger the production of LXA4 through acetylation of COX-2 that metabolizes arachidonic acid to 15(R)-hydroxyeicosatetraenoic acid. This metabolite is then converted via lipoxygenase to LXA4, also known as ‘aspirin-triggered lipoxin.’ This process is augmented during inflammation, atherosclerosis, and thrombosis [63]. Interestingly, LXA4 binds the formyl peptide receptor-2 (FPR2), a subfamily of G protein-coupled receptors. FPR activation in the cardiovascular system has been shown to have functional implications, such as modulation of vascular tone and blood pressure [64]. Specifically, we observed that LXA4 induced concentration-dependent contractions via FPR-2 activation, and both RhoA/Rho kinase inhibitor (Y-27632, 1 μm) and ROS scavenger (tempol, 1mM) decreased this contraction. Also, endothelium removal, COX and NAD(P)H oxidase inhibitors (indomethacin and apocynin, respectively, both 10μM) attenuate the LXA4-induced contraction. LXA4 potentiated phenylephrine-induced contraction and inhibited acetylcholine-induced relaxation. In another study, we observed that FPR-1 activation induces actin polymerization in vascular smooth muscle cells. The absence of FPR-1 in the vasculature significantly decreased vascular contraction and induced loss of myogenic tone to elevated intraluminal pressures via disruption of actin polymerization [65]. Therefore, it is possible to infer that LXA4 may contribute to further acetylcholine-induced contraction in conditions where its production is exacerbated.
3.1.3. Cytochrome P450
Endothelial and vascular smooth cells also generate CYP450 metabolites from AA that modulates endothelial cell function and vascular homeostasis. CYP450 produces two main eicosanoid products, EETs, formed by CYP epoxygenases, and HETEs, formed by CYP hydroxylases [66]. Similar to PGI2, EETs are recognized as endothelium-derived relaxing factors [26]. EETs have been proposed to be the unidentified endothelium-derived hyperpolarizing factor because they hyperpolarize and relax vascular smooth muscle cells by activating calcium-sensitive potassium (KCa2+) channels [66]. On the other hand, 20-HETE is the predominant CYP hydroxylase synthesized by vascular smooth muscle cells [67]. 20-HETE generation is directly correlated with myogenic tone in renal and cerebral arteries [68]. Once formed, 20-HETE increases smooth muscle contraction by inhibiting large-conductance potassium channels (BKCa2+), inducing depolarization, and further increasing intracellular calcium [68]. The elevation of 20-HETE production in malignant hypertension was first reported in 1989 [69]. 20-HETE inhibition decreases blood pressure in angiotensin II and endothelin-induced hypertension [70–71]. Interestingly, it has been shown that Dahl S rats present a decrease in 20-HETE production, leading to salt-sensitive hypertension development [72]. As described above, it is well known that the hyperpolarization in response to acetylcholine in small arteries is closely regulated by CYPP450–dependent enzymes [73]. However, it is currently unknown whether CYP450 products may be associated with acetylcholine-induced endothelium-dependent contractions in hypertension.
4. CAN WE RESOLVE ACETYLCHOLINE-INDUCED ENDOTHELIUM-DEPENDENT CONTRACTIONS?
Excessive production of inflammatory mediators, such as leukotrienes and prostaglandins, in damaged tissue, triggers the transition from acute inflammation to chronic inflammation [74]. Failure to resolve inflammation or chronic activation of the inflammatory response accelerates tissue injury and can eventually develop into disease. As complex as inflammation is, resolution of inflammation is a physiological response to “switch off” the inflammatory cascade. Cessation of acute inflammation is an active process that involves the biosynthesis of specialized pro-resolving mediators [1]. Pro-resolving mediators are endogenously derived from the AA, omega-6 fatty acid, and omega-3 fatty acids, including eicosapentaenoic acid and docosahexaenoic acid. As described above, AA-bioactive lipids play a fundamental role in acetylcholine-induced endothelium-dependent contraction in hypertension; therefore, it is possible that unresolved vascular inflammation may also play a role in this process. Unfortunately, there is little or no evidence beyond expert opinion to support this concept, so, as a result, we will briefly discuss pro-resolving mediators that were shown to be involved in vascular function, such as LXA4 and resolvins, and only infer that acetylcholine-induced endothelium-dependent contraction may also occur because the mechanisms behind the resolution of inflammation failed to resolve this process in hypertension.
4.1. Resolution of Inflammation
Specialized pro-resolving mediators can be anti-inflammatory at transcriptional and translation levels and are capable of activating NO and PGI2 production, phagocytosis, and efferocytosis [75]. Although there are four identified specialized pro-resolving lipid mediators: resolvins, protectins, LXA4, and maresins, this review will focus on lipoxins and resolvins.
LXA4 is the most studied and characterized member of the lipoxin family. As described above, LXA4 is biosynthesized from AA, and it has potent anti-inflammatory and resolution capabilities [76]. Interestingly, aspirin triggers the production of LXA4 through acetylation of COX-2 that metabolizes arachidonic acid to 15(R)-hydroxyeicosatetraenoic acid. This acid is quickly converted to LXA4 by LOX [64]. In arteries, LOX-5 biosynthesizes LXA4, and in platelets, LOX-12 biosynthesize LXB4 [74, 77]. The formation of LXA4 within the vascular lumen and wall during inflammation places this lipid in a strategically advantageous site for modulation of vascular function. Accordingly, von der Weid et al. [78] demonstrated that LXA4 induces endothelium-dependent relaxation in mesenteric arteries and aortic segments. This study showed that LXA4 (1 μmol/l) in rat aortic rings contracted with phenylephrine resulted in relaxation [78]. In opposition, Feuerstein and Siren [79] showed that intravenous LXA4 concentration-dependently constricted mesenteric arteries, but did not alter blood pressure or heart rate. Supporting these data, we showed that LXA4 induces contraction in aortic rings via FPR-2 and RhoA activation in the vascular smooth muscle cells [64]. Nascimento-Silva et al. [80] demonstrated that LXA4 suppresses NADPH oxidase-mediated ROS generation in endothelial cells. Also, another study demonstrated that LXA4 attenuates lipopolysaccharide-induced intracellular ROS in microglia cells by inhibiting cytoplasmic NADPH oxidase subunit p47(phox) translocation to the cell membrane and NADPH oxidase activity [81]. Recently it was observed that plasma LXA4 levels were higher in preeclamptic women compared to in non-pregnant and normotensive pregnant women. Nonetheless, endogenous LXA4 concentration seems to be insufficient to attenuate inflammation in preeclampsia because these women still showed features of systemic inflammatory response syndrome despite the increased levels of LXA4 [82].
Resolvins, previously referred to as bioactive lipid signals [83], are also lipid mediators that inhibit neutrophil transmigration in between endothelial cells, among other functions. Resolvins are derived from omega-3 polyunsaturated fatty acids. Humans obtain eicosapentaenoic acid and docosahexaenoic acid from marine oils that are eaten or supplemented [83–84]. Resolvins were originally isolated from murine dorsal air pouches treated with aspirin. They were also isolated from co-cultured endothelial cells and neutrophils [83]. Transcellular formation of resolvins can occur with the formation of 18R-hydroxyeicosapentaenoic acid by endothelial cells expressing COX-2 and treated with aspirin [85]. Interestingly, selective COX-2 inhibitors block resolvins formation, but neither indomethacin nor acetaminophen can.
Similar to LXA4, resolvins also attenuate nuclear factor kappa B activation and ROS generation [86–87], stimulate macrophages to phagocytose apoptotic neutrophils [88], and inhibit cytokine release and cell migration [89–90]. These lipids also modulate pro-inflammatory leukocyte expression and disrupt TXA2-mediated platelet aggregation [91]. Interestingly, a study conducted by Rathod et al. found that young women are protected against systemic inflammation-induced endothelial dysfunction due to accelerated resolution of inflammation, which is mediated by the D-series resolvin pathway [92].
To summarize these data, it is clear that LXA4 and resolvins are important pro- and anti-inflammatory mediators and play a role in vascular health. However, there is still a gap in the literature in understanding the mechanism associated with the resolution of inflammation, vascular dysfunction, and hypertension. Specifically, it is unclear if disturbances in LXA4 and resolvins pathways could be associated and, perhaps, a cause of vascular inflammation and, subsequently, acetylcholine-induced- endothelium-dependent contractions in hypertension.
CONCLUSION AND PERSPECTIVES
Acetylcholine-induced endothelium-dependent contractions are considered a hallmark for the most studied models of hypertension, such as SHR and Dahl S rats. However, this phenomenon’s precise mechanism is still debated, which makes it unclear if this process is fundamental for the genesis of hypertension or if it is exacerbating vascular tissue injury leading to malignant hypertension. Conflicting arguments are suggesting that elevations of blood pressure may be or may not be the cause of this phenomenon. However mechanistically, it is acceptable that AA and its metabolites play critical roles in this process. With this thought, we question whether a decrease in resolution of inflammation would participate in the genesis and/or maintenance of endothelium-dependent contraction to acetylcholine.
ACKNOWLEDGEMENTS
This mini-review is dedicated to the memory of Paul Michel Vanhoutte (1940-2019).
FUNDING
This work was supported by National Institutes of Health (NIGMS: R00GM11888 - C.F.W.), National Science Foundation (NOA-AGEP: 1432878 - J.M.E), and American Heart Association (18POST34060003 - C.G.M.)
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
CONSENT FOR PUBLICATION
Not applicable.
REFERENCES
- [1].Serhan CN. Novel Pro-Resolving Lipid Mediators in Inflammation Are Leads for Resolution Physiology. Nature 2014; 510(7503): 92–101. 10.1038/nature13479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Barton M The discovery of endothelium-dependent contraction: the legacy of Paul M. Vanhoutte. Pharmacol Res 2011; 63(6): 455–62. 10.1016/j.phrs.2011.02.013 [DOI] [PubMed] [Google Scholar]
- [3].Radu BM, Osculati AMM, Suku E, et al. All muscarinic acetylcholine receptors (M1-M5) are expressed in murine brain microvascular endothelium. Sci Rep 2017; 7(1): 5083 10.1038/s41598-017-05384-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lückhoff A, Busse R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflugers Arch 1990; 416(3): 305–11. 10.1007/BF00392067 [DOI] [PubMed] [Google Scholar]
- [5].Randriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature 1993; 364(6440): 809–14. 10.1038/364809a0 [DOI] [PubMed] [Google Scholar]
- [6].Tran QK, Ohashi K, Watanabe H. Calcium signalling in endothelial cells. Cardiovasc Res 2000; 48(1): 13–22. doi: 10.1016/s0008-6363(00)00172-3 [DOI] [PubMed] [Google Scholar]
- [7].Lückhoff A, Pohl U, Mülsch A, Busse R. Differential role of extra- and intracellular calcium in the release of EDRF and prostacyclin from cultured endothelial cells. Br J Pharmacol 1988; 95(1): 189–196. doi: 10.1111/j.1476-5381.1988.tb16564.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Trepakova ES, Csutora P, Hunton DL, Marchase RB, Cohen RA, Bolotina VM. Calcium influx factor directly activates store-operated cation channels in vascular smooth muscle cells. J Biol Chem 2000; 275(34): 26158–63. 10.1074/jbc.M004666200 [DOI] [PubMed] [Google Scholar]
- [9].Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol 2004; 6(2): 113–20. 10.1038/ncb1089 [DOI] [PubMed] [Google Scholar]
- [10].Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J 2010; 4: 302–12. 10.2174/1874192401004010302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Félétou M The Endothelium: Part 1: Multiple Functions of the Endothelial Cells—Focus on Endothelium-Derived Vasoactive Mediators. San Rafael (CA): Morgan & Claypool Life Sciences; 2011. Chapter 4, Endothelium-Dependent Regulation of Vascular Tone. Available from: https://www.ncbi.nlm.nih.gov/books/NBK57147/ [PubMed]
- [12].De Mey JG, Vanhoutte PM. Heterogeneous behavior of the canine arterial and venous wall. Importance of the endothelium. Circ Res 1982; 51(4): 439–47. 10.1161/01.RES.51.4.439 [DOI] [PubMed] [Google Scholar]
- [13].Katusic ZS, Shepherd JT, Vanhoutte PM. Endothelium-dependent contraction to stretch in canine basilar arteries. Am J Physiol 1987; 252(3 Pt 2): H671–3. [DOI] [PubMed] [Google Scholar]
- [14].Lüscher TF, Vanhoutte PM. Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension 1986; 8(4): 344–8. 10.1161/01.HYP.8.4.344 [DOI] [PubMed] [Google Scholar]
- [15].Vanhoutte PM, Feletou M, Taddei S. Endothelium-dependent contractions in hypertension. Br J Pharmacol 2005; 144(4): 449–58. 10.1038/sj.bjp.0706042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhou Y, Varadharaj S, Zhao X, Parinandi N, Flavahan NA, Zweier JL. Acetylcholine causes endothelium-dependent contraction of mouse arteries. Am J Physiol Heart Circ Physiol 2005; 289(3): H1027–H1032. doi: 10.1152/ajpheart.00226.2005 [DOI] [PubMed] [Google Scholar]
- [17].Svetkey LP, McKeown SP, Wilson AF. Heritability of salt sensitivity in black Americans. Hypertension 1996; 28(5): 854–8. 10.1161/01.HYP.28.5.854 [DOI] [PubMed] [Google Scholar]
- [18].Rapp JP. Dahl salt-susceptible and salt-resistant rats. A review. Hypertension 1982; 4(6): 753–63. 10.1161/01.HYP.4.6.753 [DOI] [PubMed] [Google Scholar]
- [19].Wong SL, Leung FP, Lau CW, et al. Cyclooxygenase-2-derived prostaglandin F2alpha mediates endothelium-dependent contractions in the aortae of hamsters with increased impact during aging. Circ Res 2009; 104(2): 228–35. 10.1161/CIRCRESAHA.108.179770 [DOI] [PubMed] [Google Scholar]
- [20].Rapoport RM, Williams SP. Role of prostaglandins in acetylcholine-induced contraction of aorta from spontaneously hypertensive and Wistar-Kyoto rats. Hypertension 1996; 28(1): 64–75. 10.1161/01.HYP.28.1.64 [DOI] [PubMed] [Google Scholar]
- [21].Zhou MS, Nishida Y, Chen QH, Kosaka H. Endothelium-derived contracting factor in carotid artery of hypertensive Dahl rats. Hypertension 1999; 34(1): 39–43. 10.1161/01.HYP.34.1.39 [DOI] [PubMed] [Google Scholar]
- [22].Zhou MS, Kosaka H, Tian RX, et al. L-Arginine improves endothelial function in renal artery of hypertensive Dahl rats. J Hypertens 2001; 19(3): 421–9. 10.1097/00004872-200103000-00010 [DOI] [PubMed] [Google Scholar]
- [23].Tesfamariam B, Brown ML, Deykin D, Cohen RA. Elevated glucose promotes generation of endothelium-derived vasoconstrictor prostanoids in rabbit aorta. J Clin Invest 1990; 85(3): 929–32. 10.1172/JCI114521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dantas AP, Scivoletto R, Fortes ZB, Nigro D, Carvalho MH. Influence of female sex hormones on endothelium-derived vasoconstrictor prostanoid generation in microvessels of spontaneously hypertensive rats. Hypertension 1999; 34(4 Pt 2): 914–9. 10.1161/01.HYP.34.4.914 [DOI] [PubMed] [Google Scholar]
- [25].Tang EH, Vanhoutte PM. Prostanoids and reactive oxygen species: team players in endothelium-dependent contractions. Pharmacol Ther 2009; 122(2): 140–9. 10.1016/j.pharmthera.2009.02.006 [DOI] [PubMed] [Google Scholar]
- [26].Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch 2010; 459(6): 881–95. 10.1007/s00424-010-0804-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lassègue B, Griendling KK. Reactive oxygen species in hypertension; An update. Am J Hypertens 2004; 17(9): 852–60. 10.1016/j.amjhyper.2004.02.004 [DOI] [PubMed] [Google Scholar]
- [28].Wong MS, Vanhoutte PM. COX-mediated endothelium-dependent contractions: from the past to recent discoveries. Acta Pharmacol Sin 2010; 31(9): 1095–102. 10.1038/aps.2010.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Vanhoutte PM. Nitric oxide: from good to bad. Ann Vasc Dis 2018; 11(1): 41–51. 10.3400/avd.ra.17-00134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jin L, Ying Z, Webb RC. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am J Physiol Heart Circ Physiol 2004; 287(4): H1495–500. 10.1152/ajpheart.01006.2003 [DOI] [PubMed] [Google Scholar]
- [31].Katusic ZS, Vanhoutte PM. Superoxide anion is an endothelium-derived contracting factor. Am J Physiol 1989; 257(1 Pt 2): H33–7. [DOI] [PubMed] [Google Scholar]
- [32].Katusic ZS, Shepherd JT, Vanhoutte PM. Endothelium-dependent contractions to calcium ionophore A23187, arachidonic acid, and acetylcholine in canine basilar arteries. Stroke 1988; 19(4): 476–9. 10.1161/01.STR.19.4.476 [DOI] [PubMed] [Google Scholar]
- [33].Cosentino F, Sill JC, Katusić ZS. Role of superoxide anions in the mediation of endothelium-dependent contractions. Hypertension 1994; 23(2): 229–35. 10.1161/01.HYP.23.2.229 [DOI] [PubMed] [Google Scholar]
- [34].Wenceslau CF, Davel AP, Xavier FE, Rossoni LV. Long-term ouabain treatment impairs vascular function in resistance arteries. J Vasc Res 2011; 48(4): 316–26. 10.1159/000322576 [DOI] [PubMed] [Google Scholar]
- [35].Yang D, Félétou M, Boulanger CM, et al. Oxygen-derived free radicals mediate endothelium-dependent contractions to acetylcholine in aortas from spontaneously hypertensive rats. Br J Pharmacol 2002; 136(1): 104–10. 10.1038/sj.bjp.0704669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Félétou M, Huang Y, Vanhoutte PM. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol 2011; 164(3): 894–912. 10.1111/j.1476-5381.2011.01276.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Greene ER, Huang S, Serhan CN, Panigrahy D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat 2011; 96(1–4): 27–36. 10.1016/j.prostaglandins.2011.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Davidge ST. Prostaglandin H synthase and vascular function. Circ Res 2001; 89(8): 650–60. 10.1161/hh2001.098351 [DOI] [PubMed] [Google Scholar]
- [39].Smith WL, Song I. The enzymology of prostaglandin endoperoxide H synthases-1 and −2. Prostaglandins Other Lipid Mediat 2002; 68–69: 115–28. 10.1016/S0090-6980(02)00025-4 [DOI] [PubMed] [Google Scholar]
- [40].Doroudi R, Gan LM, Selin Sjögren L, Jern S. Effects of shear stress on eicosanoid gene expression and metabolite production in vascular endothelium as studied in a novel biomechanical perfusion model. Biochem Biophys Res Commun 2000; 269(1): 257–64. 10.1006/bbrc.2000.2279 [DOI] [PubMed] [Google Scholar]
- [41].Russell-Puleri S, Dela Paz NG, Adams D, et al. Fluid shear stress induces upregulation of COX-2 and PGI2 release in endothelial cells via a pathway involving PECAM-1, PI3K, FAK, and p38. Am J Physiol Heart Circ Physiol 2017; 312(3): H485–500. 10.1152/ajpheart.00035.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chandrasekharan NV, Dai H, Roos KL, et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci USA 2002; 99(21): 13926–31. 10.1073/pnas.162468699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev 1999; 79(4): 1193–226. 10.1152/physrev.1999.79.4.1193 [DOI] [PubMed] [Google Scholar]
- [44].Alfranca A, Iñiguez MA, Fresno M, Redondo JM. Prostanoid signal transduction and gene expression in the endothelium: role in cardiovascular diseases. Cardiovasc Res 2006; 70(3): 446–56. 10.1016/j.cardiores.2005.12.020 [DOI] [PubMed] [Google Scholar]
- [45].Simonet S, Descombes JJ, Vallez MO, Dubuffet T, Lavielle G, Verbeuren TJS. S 18886, a new thromboxane (TP)-receptor antagonist is the active isomer of S 18204 in all species, except in the guinea-pig. Adv Exp Med Biol 1997; 433: 173–6. 10.1007/978-1-4899-1810-9_35 [DOI] [PubMed] [Google Scholar]
- [46].Tang EH, Vanhoutte PM. Gene expression changes of prostanoid synthases in endothelial cells and prostanoid receptors in vascular smooth muscle cells caused by aging and hypertension. Physiol Genomics 2008; 32(3): 409–18. 10.1152/physiolgenomics.00136.2007 [DOI] [PubMed] [Google Scholar]
- [47].Koga T, Takata Y, Kobayashi K, Takishita S, Yamashita Y, Fujishima M. Age and hypertension promote endothelium-dependent contractions to acetylcholine in the aorta of the rat. Hypertension 1989; 14(5): 542–8. [DOI] [PubMed] [Google Scholar]
- [48].Gluais P, Lonchampt M, Morrow JD, Vanhoutte PM, Feletou M. Acetylcholine-induced endothelium-dependent contractions in the SHR aorta: the Janus face of prostacyclin. Br J Pharmacol 2005; 146(6): 834–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kato T, Iwama Y, Okumura K, Hashimoto H, Ito T, Satake T. Prostaglandin H2 may be the endothelium-derived contracting factor released by acetylcholine in the aorta of the rat. Hypertension 1990; 15(5): 475–81. 10.1161/01.HYP.15.5.475 [DOI] [PubMed] [Google Scholar]
- [50].Ge T, Hughes H, Junquero DC, Wu KK, Vanhoutte PM, Boulanger CM. Endothelium-dependent contractions are associated with both augmented expression of prostaglandin H synthase-1 and hypersensitivity to prostaglandin H2 in the SHR aorta. Circ Res 1995; 76(6): 1003–10. 10.1161/01.RES.76.6.1003 [DOI] [PubMed] [Google Scholar]
- [51].Gluais P, Paysant J, Badier-Commander C, Verbeuren T, Vanhoutte PM, Félétou M. In SHR aorta, calcium ionophore A-23187 releases prostacyclin and thromboxane A2 as endothelium-derived contracting factors. Am J Physiol Heart Circ Physiol 2006; 291(5): H2255–64. 10.1152/ajpheart.01115.2005 [DOI] [PubMed] [Google Scholar]
- [52].Gluais P, Vanhoutte PM, Félétou M. Mechanisms underlying ATP-induced endothelium-dependent contractions in the SHR aorta. Eur J Pharmacol 2007; 556(1–3): 107–14. 10.1016/j.ejphar.2006.10.050 [DOI] [PubMed] [Google Scholar]
- [53].Iwama Y, Kato T, Muramatsu M, et al. Correlation with blood pressure of the acetylcholine-induced endothelium-derived contracting factor in the rat aorta. Hypertension 1992; 19(4): 326–32. 10.1161/01.HYP.19.4.326 [DOI] [PubMed] [Google Scholar]
- [54].Xavier FE, Aras-López R, Arroyo-Villa I, et al. Aldosterone induces endothelial dysfunction in resistance arteries from normotensive and hypertensive rats by increasing thromboxane A2 and prostacyclin. Br J Pharmacol 2008; 154(6): 1225–35. 10.1038/bjp.2008.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mashima R, Okuyama T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol 2015; 6: 297–310. 10.1016/j.redox.2015.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Izzo AA, Mitchell JA. Eicosanoid turnover (version 2019.5) in the IUPHAR/BPS Guide to Pharmacology Database. GtoPdb CITE 2019; 2019(5) [Google Scholar]
- [57].Stanke-Labesque F, Devillier P, Bedouch P, Cracowski JL, Chavanon O, Bessard G. Angiotensin II-induced contractions in human internal mammary artery: effects of cyclooxygenase and lipoxygenase inhibition. Cardiovasc Res 2000; 47(2): 376–83. 10.1016/S0008-6363(00)00112-7 [DOI] [PubMed] [Google Scholar]
- [58].Stern N, Golub M, Nozawa K, et al. Selective inhibition of angiotensin II-mediated vasoconstriction by lipoxygenase blockade. Am J Physiol 1989; 257(2 Pt 2): H434–43. [DOI] [PubMed] [Google Scholar]
- [59].DelliPizzi A, Guan H, Tong X, Takizawa H, Nasjletti A. Lipoxygenase-dependent mechanisms in hypertension. Clin Exp Hypertens 2000; 22(2): 181–92. 10.1081/CEH-100100071 [DOI] [PubMed] [Google Scholar]
- [60].Lefebvre B, Caron F, Bessard G, Stanke-Labesque F. Effect of 5-lipoxygenase blockade on blood pressure and acetylcholine-evoked endothelium-dependent contraction in aorta from spontaneously hypertensive rats. J Hypertens 2006; 24(1): 85–93. 10.1097/01.hjh.0000198027.76729.b8 [DOI] [PubMed] [Google Scholar]
- [61].Kowal-Bielecka O, Kowal K, Distler O, Gay S. Mechanisms of Disease: leukotrienes and lipoxins in scleroderma lung disease--insights and potential therapeutic implications. Nat Clin Pract Rheumatol 2007; 3(1): 43–51. 10.1038/ncprheum0375 [DOI] [PubMed] [Google Scholar]
- [62].Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci USA 1984; 81(17): 5335–9. 10.1073/pnas.81.17.5335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kantarci A, Van Dyke TE. Lipoxins in chronic inflammation. Crit Rev Oral Biol Med 2003; 14(1): 4–12. 10.1177/154411130301400102 [DOI] [PubMed] [Google Scholar]
- [64].Wenceslau CF, McCarthy CG, Szasz T, Webb RC. Lipoxin A4 mediates aortic contraction via RHOA/RHO kinase, endothelial dysfunction and reactive oxygen species. J Vasc Res 2014; 51(6): 407–17. 10.1159/000371490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wenceslau CF, McCarthy CG, Szasz T, Calmasini FB, Mamenko M, Webb RC. Formyl peptide receptor-1 activation exerts a critical role for the dynamic plasticity of arteries via actin polymerization. Pharmacol Res 2019; 141: 276–90. 10.1016/j.phrs.2019.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Node K, Huo Y, Ruan X, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999; 285(5431): 1276–9. 10.1126/science.285.5431.1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Imig JD. Epoxyeicosatrienoic Acids and 20-Hydroxyeicosatetraenoic Acid on Endothelial and Vascular Function. Adv Pharmacol 2016; 77: 105–41. 10.1016/bs.apha.2016.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Fleming I Cytochrome p450 and vascular homeostasis. Circ Res 2001; 89(9): 753–62. 10.1161/hh2101.099268 [DOI] [PubMed] [Google Scholar]
- [69].Sacerdoti D, Escalante B, Abraham NG, McGiff JC, Levere RD, Schwartzman ML. Treatment with tin prevents the development of hypertension in spontaneously hypertensive rats. Science 1989; 243(4889): 388–90. 10.1126/science.2492116 [DOI] [PubMed] [Google Scholar]
- [70].Fan F, Muroya Y, Roman RJ. Cytochrome P450 eicosanoids in hypertension and renal disease. Curr Opin Nephrol Hypertens 2015; 24(1): 37–46. 10.1097/MNH.0000000000000088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wu CC, Gupta T, Garcia V, Ding Y, Schwartzman ML. 20-HETE and blood pressure regulation: clinical implications. Cardiol Rev 2014; 22(1): 1–12. 10.1097/CRD.0b013e3182961659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Fan F, Roman RJ. Effect of Cytochrome P450 Metabolites of Arachidonic Acid in Nephrology. J Am Soc Nephrol 2017; 28(10): 2845–55. 10.1681/ASN.2017030252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chen G, Cheung DW. Modulation of endothelium-dependent hyperpolarization and relaxation to acetylcholine in rat mesenteric artery by cytochrome P450 enzyme activity. Circ Res 1996; 79(4): 827–33. 10.1161/01.RES.79.4.827 [DOI] [PubMed] [Google Scholar]
- [74].Freire MO, Van Dyke TE. Natural resolution of inflammation. Periodontol 2000 2013; 63(1): 149–64. 10.1111/prd.12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Serhan CN, Chiang N. Lipid-derived mediators in endogenous anti-inflammation and resolution: lipoxins and aspirin-triggered 15-epilipoxins. ScientificWorldJournal 2002; 2: 169–204. 10.1100/tsw.2002.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Serhan CN, Chiang N, Dalli J, Levy BD. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol 2014; 7(2)a016311 10.1101/cshperspect.a016311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Chiang N, Serhan CN, Dahlén SE, et al. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev 2006; 58(3): 463–87. 10.1124/pr.58.3.4 [DOI] [PubMed] [Google Scholar]
- [78].von der Weid PY, Hollenberg MD, Fiorucci S, Wallace JL. Aspirin-triggered, cyclooxygenase-2-dependent lipoxin synthesis modulates vascular tone. Circulation 2004; 110(10): 1320–5. 10.1161/01.CIR.0000140985.89766.CB [DOI] [PubMed] [Google Scholar]
- [79].Feuerstein G, Siren AL. Mesenteric vascular responses to i.v. administration of lipoxin A4 and lipoxin B4 in the conscious rat. FEBS Lett 1988; 232(1): 51–5. 10.1016/0014-5793(88)80384-3 [DOI] [PubMed] [Google Scholar]
- [80].Nascimento-Silva V, Arruda MA, Barja-Fidalgo C, Fierro IM. Aspirin-triggered lipoxin A4 blocks reactive oxygen species generation in endothelial cells: a novel antioxidative mechanism. Thromb Haemost 2007; 97(1): 88–98. 10.1160/TH06-06-0315 [DOI] [PubMed] [Google Scholar]
- [81].Wu Y, Zhai H, Wang Y, et al. Aspirin-triggered lipoxin A4 attenuates lipopolysaccharide-induced intracellular ROS in BV2 microglia cells by inhibiting the function of NADPH oxidase. Neurochem Res 2012; 37(8): 1690–6. 10.1007/s11064-012-0776-3 [DOI] [PubMed] [Google Scholar]
- [82].Perucci LO, Santos PC, Ribeiro LS, et al. Lipoxin A4 Is Increased in the Plasma of Preeclamptic Women. Am J Hypertens 2016; 29(10): 1179–85. 10.1093/ajh/hpw053 [DOI] [PubMed] [Google Scholar]
- [83].Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med 2000; 192(8): 1197–204. 10.1084/jem.192.8.1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest 2018; 128(7): 2657–69. 10.1172/JCI97943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kohli P, Levy BD. Resolvins and protectins: mediating solutions to inflammation. Br J Pharmacol 2009; 158(4): 960–71. 10.1111/j.1476-5381.2009.00290.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Arita M, Yoshida M, Hong S, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci USA 2005; 102(21): 7671–6. 10.1073/pnas.0409271102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Gronert K, Kantarci A, Levy BD, et al. A molecular defect in intracellular lipid signaling in human neutrophils in localized aggressive periodontal tissue damage. J Immunol 2004; 172(3): 1856–61. 10.4049/jimmunol.172.3.1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007; 447(7146): 869–74. 10.1038/nature05877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Arita M, Bianchini F, Aliberti J, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med 2005; 201(5): 713–22. 10.1084/jem.20042031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol 2008; 9(8): 873–9. 10.1038/ni.1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Dona M, Fredman G, Schwab JM, et al. Resolvin E1, an EPA-derived mediator in whole blood, selectively counterregulates leukocytes and platelets. Blood 2008; 112(3): 848–55. 10.1182/blood-2007-11-122598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Rathod KS, Kapil V, Velmurugan S, et al. Accelerated resolution of inflammation underlies sex differences in inflammatory responses in humans. J Clin Invest 2017; 127(1): 169–82. 10.1172/JCI89429 [DOI] [PMC free article] [PubMed] [Google Scholar]