

SUMMARY
The lipidic cubic phase (LCP) technique has proved to facilitate the growth of high-quality crystals that are otherwise difficult to grow by other methods. However, crystal size optimization process could be time- and resource-consuming, if it ever happens. Therefore, improved techniques for structure determination using these small crystals is an important strategy in diffraction technology development. Microcrystal electron diffraction (MicroED) is a technique that uses a cryo-TEM to collect electron diffraction data and determine high-resolution structures from very thin micro- and nanocrystals. In this work, we have used modified LCP and MicroED protocols to analyze crystals embedded in LCP converted by 2-Methyl-2,4-pentanediol (MPD) or lipase, including proteinase K crystals grown in solution, cholesterol crystals and human adenosine A2A receptor crystals grown in LCP. These results set the stage for the use of MicroED to analyze microcrystalline samples grown in LCP, especially for those highly challenging membrane protein targets.
Keywords: Lipidic cubic phase (LCP), Microcrystal electron diffraction (MicroED), Microcrystallography, Additive phase conversion, Lipase hydrolysis, Proteinase K, Cholesterol, GPCR, Membrane protein
Graphical Abstract
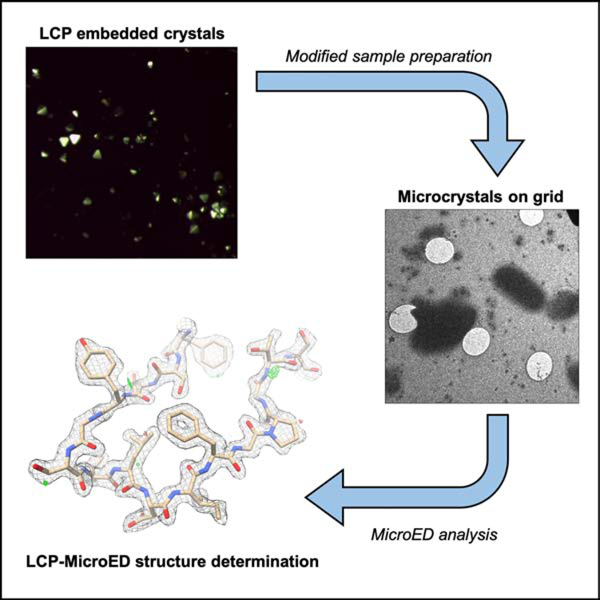
eTOC blurb
Lan et al. demonstrate modified microcrystal electron diffraction (MicroED) and lipidic cubic phase (LCP) sample preparation procedures, which can be used facilitate MicroED analysis of LCP embedded microcrystals. These results set the stage for the use of MicroED to analyze microcrystals that have been crystallized within LCP.
INTRODUCTION
Structural determination of membrane proteins has been difficult primarily due to their low expression and low stability once isolated from their native membrane environment. Despite these difficulties, the number of membrane protein crystal structures has increased in recent years due to multiple technical breakthroughs, including the lipidic cubic phase (LCP) technique, which provides a lipid environment close to that of the native membrane protein environment. Since the first high-resolution bacteriorhodopsin structure from LCP was determined in 1997 (Landau and Rosenbusch, 1996; Pebay-Peyroula et al., 1997), there are now over 120 unique membrane protein structures, covering a wide range of molecular sizes even to the bigger and bulkier proteins, resolved at atomic resolution from crystals formed in meso (Aherne et al., 2012; Al-Sahouri et al., 2018; Caffrey, 2008; Caffrey et al., 2012; Cherezov et al., 2006; El Ghachi et al., 2018; Huang et al., 2018; Huang et al., 2015; Ishchenko et al., 2017a; Ishchenko et al., 2017b; Jaeger et al., 2019; Johansson et al., 2017; Lan et al., 2018; Li and Caffrey, 2011; Li et al., 2015; Ma et al., 2017; Vogeley et al., 2016; Weinert et al., 2017; Weinert et al., 2019; Winkler et al., 2019; Xiang et al., 2016; Zabara et al., 2018). However, one challenge with this technique is that when membrane proteins crystallize in LCP, the crystals are often very small microcrystals in the initial condition that cannot withstand the radiation damage during synchrotron X-ray diffraction data collection, and the crystal size optimization may take months to years without assurances of improved diffraction quality. Serial femtosecond crystallography (SFX), which utilizes high-brilliance and ultra-fast X-ray pulses to capture single crystal diffraction patterns from LCP embedded membrane protein microcrystals before they are destructed, has recently been employed with great success (Al-Sahouri et al., 2018; Caffrey et al., 2014; Johansson et al., 2017; Liu et al., 2013; Nogly et al., 2018; Stauch and Cherezov, 2018; Weierstall et al., 2014). Nevertheless, the LCP-SFX technique is very time and resource intensive with limited experimental time as there are only six operational X-ray free electron laser facilities world-wide, currently. LCP serial crystallography technology has been successfully adapted for use with synchrotron radiation, with both monochromatic and polychromatic beamlines (Martin-Garcia et al., 2017; Martin-Garcia et al., 2019). Though this evolution provides exciting opportunities to structure determination from microcrystals, further developments are required to overcome existing hurdles. Moreover, serial crystallography methods require a very large number (in our experience, typically greater than 100,000) of microcrystal diffraction patterns to constitute a complete dataset for structure determination. Therefore, to improve the structural studies of important integral membrane proteins to resolve high-resolution details in a more high-throughput fashion, new methods for structure determination need to be developed in using the small crystals grown in LCP.
The advent of a new technique, microcrystal electron diffraction (MicroED) in 2013, offers an alternative for the structure determination of proteins from microcrystal samples (Shi et al., 2013). MicroED is a method that is used to collect electron diffraction patterns from sub-micrometer (mm) sized three-dimensional crystals in the electron microscope (Nannenga and Gonen, 2018). Microcrystals are deposited on electron microscope (EM) sample grids followed by sample blotting, as electrons cannot penetrate thick samples. To ensure the sample remains hydrated in the vacuum of the electron microscope and for reduced radiation damage, the samples are vitrified and kept at cryogenic temperatures. The continuous rotation data collection strategy for MicroED allows multiple diffraction patterns to be recorded from one single crystal with extremely low electron dose, resulting in a series of diffraction patterns that can be indexed, integrated, and processed with crystallographic data processing software without any prior knowledge of unit cell parameters or geometry (Nannenga et al., 2014). Since the initial implementation of MicroED, there have been further efforts in improving this method to determine structures of proteins, peptides, and small organic molecules (Gemmi et al., 2019; Gruene et al., 2018; Jones et al., 2018; Levine et al., 2020; Nannenga and Gonen, 2016; Nannenga and Gonen, 2019).
In this work, our goal is to combine MicroED with LCP microcrystallography methods (LCP-MicroED) to determine structures from microcrystals within the LCP matrix. Here, we report the first MicroED structures of a model soluble protein, proteinase K, that has been embedded in LCP. Additionally, we have demonstrated the approach is suitable for generating MicroED samples for crystals grown within LCP by collecting cholesterol MicroED data sets and diffraction patterns of a model G protein-coupled receptor (GPCR), human adenosine A2A receptor (A2AAR).
Proteinase K had been used extensively as a model protein to generate samples for both MicroED and new LCP-based serial crystallography method development (de la Cruz et al., 2017; Hattne et al., 2018; Martin-Garcia et al., 2017; Martin-Garcia et al., 2019; Martynowycz et al., 2019a). Cholesterol crystals grown in LCP were used to elucidate the structure determination of small molecules by MicroED method. A2AAR was chosen as a model GPCR crystallized in LCP using previously published condition that resulted in a high-resolution SFX structure as well as the structures determined by serial crystallography studies at synchrotrons (Batyuk et al., 2016; Martin-Garcia et al., 2017; Martin-Garcia et al., 2019). By treating the LCP samples with different reagents to lower the viscosity of the LCP, we further optimized two strategies – dilution using MPD and treatment of the sample with lipase - that led to high-quality MicroED samples. Both strategies were used on the LCP-proteinase K samples to successfully determine the structure of proteinase K at 2.0 Å resolution using MicroED. Diffraction datasets collected from MPD-treated cholesterol crystals and A2AAR crystals diffracted to ~1 Å and 4.5 Å, respectively. Cholesterol diffraction data were processed, with unit cell parameters matching previously known cholesterol crystals (Varsano et al., 2018), and maps from the low-completeness data sets were generated. We also observed that the A2AAR diffraction pattern is consistent with previously published results (Batyuk et al., 2016; Martin-Garcia et al., 2017; Martin-Garcia et al., 2019).
RESULTS
Though LCP crystallization has proved significant success in structure determination of membrane proteins, due to the intrinsic high viscosity of LCP matrix (Caffrey and Cherezov, 2009), this technique is not well-suited to standard MicroED sample preparation protocols. In order to identify sample preparation conditions and protocols that would allow MicroED to be used on samples embedded in LCP, we chose the well-studied model proteins, Proteinase K and A2AAR, as well as a small molecule cholesterol, that have previously been used to benchmark both new LCP-based X-ray diffraction (Batyuk et al., 2016; Martin-Garcia et al., 2019) and MicroED methods (Hattne et al., 2016). Proteinase K microcrystals were grown in batch and reconstituted into the LCP to be used for further studies on LCP sample preparation for MicroED. As an initial test for crystals that were grown within the LCP matrix rather than embedded after crystal growth, cholesterol was crystallized in LCP and MicroED data were collected on these crystals. Finally, A2AAR microcrystals were grown in LCP and directly treated for LCP-MicroED sample preparation, and single diffraction patterns were collected.
LCP sample conversions for EM grid deposition
We initially focused on the identification and optimization of sample preparation conditions that would allow the collection of MicroED data from crystals embedded or grown in LCP. Because the viscosity of LCP matrix is too high to be directly deposited on EM grids and effectively blotted thin enough to be penetrated by the electron beam, we adapted two different strategies to reduce the viscosity: 1) by mixing with certain additives to convert into a less viscous liquid analogue of cubic phase, which could be a sponge phase (Caffrey, 2015; Qiu and Caffrey, 2000); 2) treatment with lipase to hydrolyze matrix lipids and convert LCP into a two-liquid phase system of water/glycerol solution and oleic acid (Nollert and Landau, 1998; Nollert et al., 2002).
First, we screened the following additives at various concentrations: 2-Methyl-2,4-pentanediol (MPD), PEG200, PEG400, Jeffamine M600, t-butanol, ethylene glycol, and 1,4-butanediol (Caffrey and Cherezov, 2009; Wadsten et al., 2006). These seven agents are commonly used in traditional protein crystallization screening and cryoprotection solutions. Though the previous studies have proposed a less viscous lipid mesophase might form with the addition of these additives (Liu et al., 2012; Wadsten et al., 2006), the condition of utilizing them to liberate crystals from LCP matrix was not well defined. Initial tests were conducted with blank LCP by mixing of the host lipid monoolein (MO) and proteinase K precipitant buffer without protein. All seven additives could convert the blank LCP to a less viscous lipid mesophase in syringe mixing, in the range of 6–18% with MPD, 24–40% with PEG200, 32–48% with PEG400, 9–20% with Jeffamine M600, 11–20% with t-butanol, 15–33% with ethylene glycol, and 28–41% with 1,4-butanediol, respectively, which are consistent with previous sponge phase transition studies (Wadsten et al., 2006).
We then tested the absorption of the resulting less viscous lipid mesophase on blotting paper, in order to empirically determine how to blot these converted LCP samples in order to generate a thin layer sample suitable for vitrification for MicroED. In this step, samples were expelled out of the mixing syringe and deposited on the blotting paper, without external blotting force applied. Compared with the LCP droplet (Figure 1a), which retained its shape and did not blot on the filter paper, the seven additive-converted less viscous lipid mesophase samples showed absorption on the filter paper (Figure 1b–h). MPD-converted (Figure 1b) shows the most significant blotting relative to the other additive-converted samples. When similar blotting tests were conducted on EM grids, the MPD-converted lipid mesophase samples produced the most consistently thin samples for successful TEM visualization. Therefore, MPD was selected for further experiments with proteinase K, cholesterol, and A2AAR crystals embedded in LCP.
Figure 1. LCP phase converting by the addition of additives or the lipase treatment to generate low-viscous liquid-like sample suitable for MicroED grid preparation.
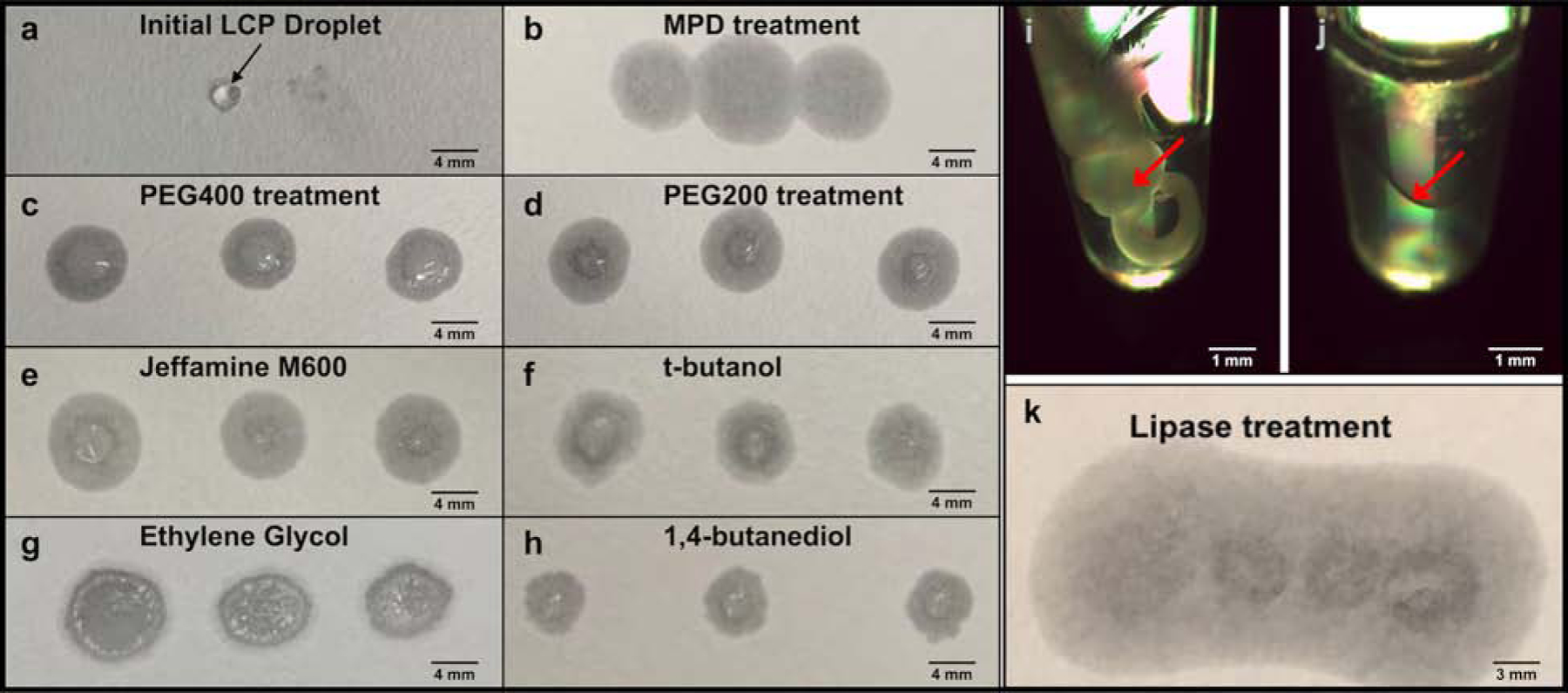
a) high-viscous LCP sample could not penetrate the blotting paper, rather to stay on the paper surface as a solid droplet. b-h) the treatment with the phase converting buffer supplemented with seven additives, MPD (b), PEG400 (c), PEG200 (d), Jeffamine M600 (e), t-butanol (f), ethylene glycol (g), and 1,4-butanediol (h), respectively, converted the LCP sample to a less viscous liquid-like phase, which penetrated the blotting paper. i-k) the lipase hydrolysis treatment of LCP sample to form two liquid phases, i) the LCP stream (white solid stream in the tube highlighted in red arrow) with freshly prepared lipase solution mixed in a ratio of 1:1 (v/v) before treatment, j) the LCP sample was separated into two liquid phases (phase separation surface denoted in red arrow) after 14-hour treatment; k) the lipase hydrolyzed sample penetrated the blotting paper without LCP residue on the paper surface.
In addition to the additive treatment to convert LCP to less viscous lipid mesophases, we investigated an alternative strategy by treating LCP with lipase to hydrolyze the host lipid molecules and transition the cubic phase to a two-liquid phase system (Nollert et al., 2002). Blank LCP was again used to find an optimal hydrolysis ratio of LCP with lipase, as well as the minimum treatment time to completely separate LCP into two liquid phases. LCP sample was expelled from the syringe mixer into a 0.2 mL microfuge tube, and freshly prepared lipase solution at 50 mg/mL was directly added on top of LCP sample without additional pipetting. It was found that after a 14-hour treatment in a 1:1 ratio of LCP and freshly prepared lipase solution incubated at 20 °C, the solid cubic phase (Figure 1i) was completely separated into two liquid layers (Figure 1j). This lipase hydrolyzed sample also penetrated the blotting paper without any visible LCP residua on the surface (Figure 1k). For those protein targets that do not require lipid molecules involved in crystal packing, this enzymic release method can be used to clean the LCP matrix for crystal liberation. As with the MPD treated samples described above, this strategy was then tested with LCP-proteinase K crystal samples to study crystal survival and grid preparation for data collection.
Microcrystal survival during LCP conversion
We then followed the batch crystallization method to grow proteinase K microcrystals in solution (crystal size may range between 5–100 μm as shown in Figure 2a, but only less than 5 μm sized with less than ~0.5 μm thick crystals would be targeted for MicroED data collection as in Figure 3a and 3e) and reconstituted them into LCP by dual-syringe mixing with the host lipid monoolein in a lipid:crystal solution ratio of 3:2 (v/v). Proteinase K microcrystals survived reconstitution into LCP (Figure 2b) and were used for phase conversion test with MPD or lipase treatment.
Figure 2. Proteinase K microcrystals were imaged before and after LCP phase converting.
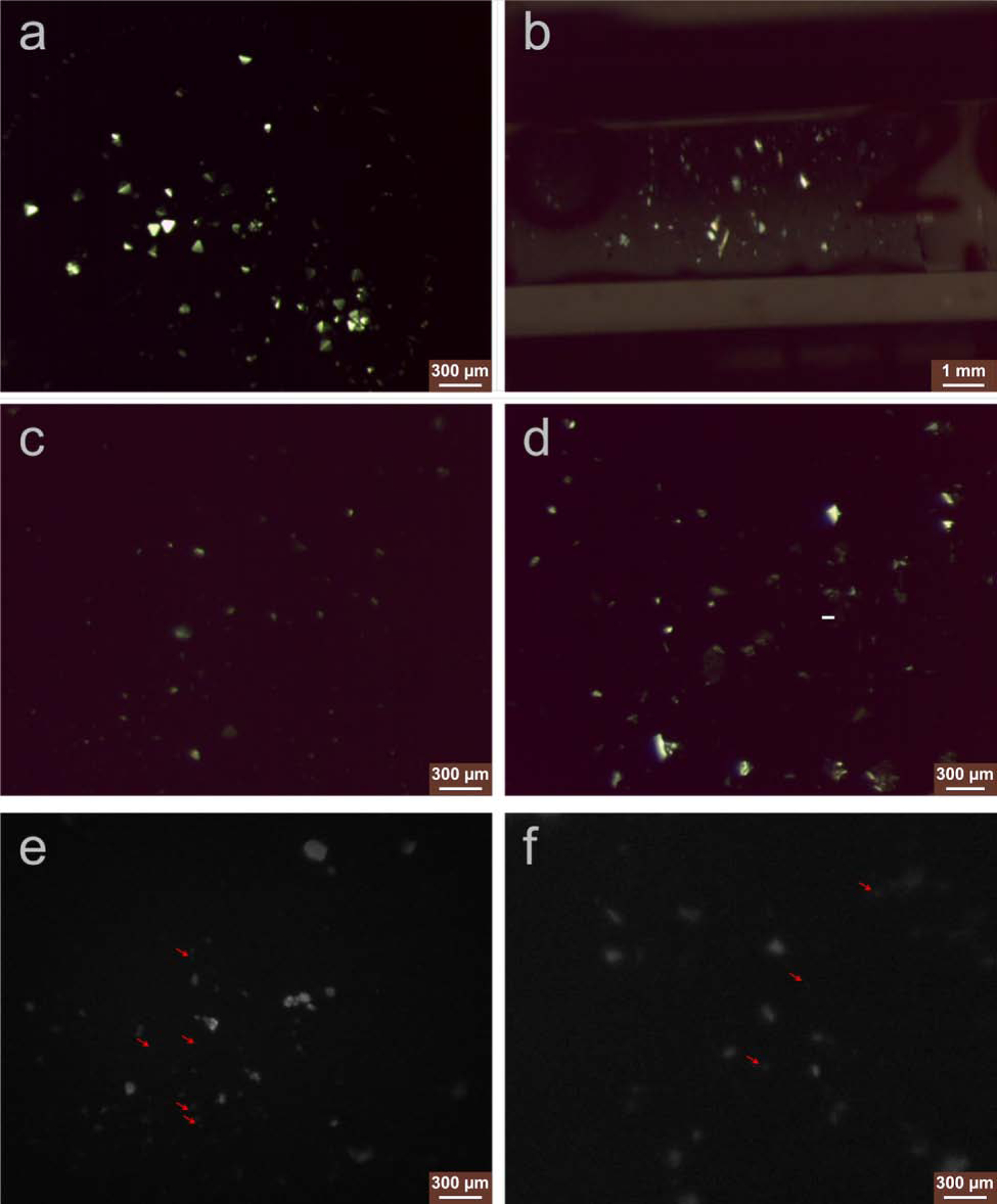
Microcrystals of proteinase K grown in batch method (a) and reconstituted into the LCP matrix by syringe mixing (b), viewed with cross polarizer light. Proteinase K microcrystals embedded in LCP survived after the LCP phase conversion by the converting buffer supplemented with 12.5% MPD (c) or LCP hydrolyzed by the lipase treatment (d), viewed with cross polarizer light. LCP-proteinase K samples converted by MPD (e) and lipase treatment (f) were successfully blotted on the glow-discharged EM grids used for MicroED data collection, and when grids were viewed with UV, the presence of crystals could still be seen on the blotted grids. Red arrows pointed proteinase K microcrystals with size less than 5 μm and thinner than ~0.5 μm that were used for MicroED data collection.
Figure 3. LCP-MicroED structure of proteinase K.
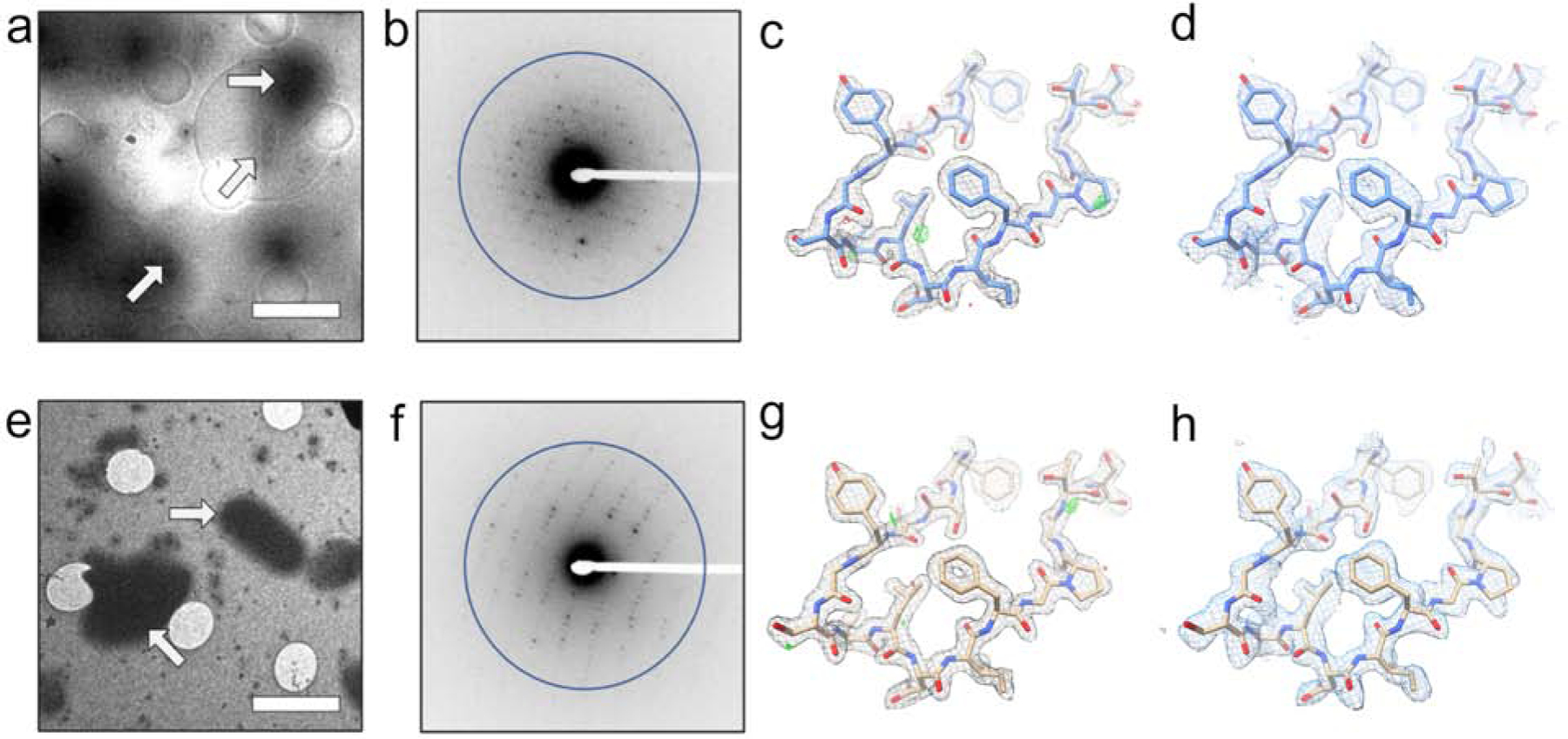
Both MPD treated samples (a-d) and lipase treated samples (e-h) produced grids where crystals could be identified in the cryo-TEM (a - MPD and e - lipase). MicroED data collection (representative diffraction patterns with resolution ring of 2.0 Å shown as b-MPD and f-lipase) on these crystals from both treatments produced structures at 2.0 Å. The 2Fo-Fc maps (c – MPD and g – lipase) and composite omit maps (d – MPD and h – lipase) show clear density surrounding the models. Scale bars in a and e represent 4 μm. The density maps in c and g and composite omit maps in d and h are contoured at 1.5σ and 1.0σ, respectively. The 2Fo-Fc map in c and g is contoured at 3.0σ (green) and −3.0σ (red).
For phase conversion, we tested MPD supplementation in the proteinase K precipitant solution with a range of 6–18% MPD in 0.5% increments as a phase converting buffer. This conversion buffer was then mixed with LCP-proteinase K in the syringe. We observed that the converting buffer supplemented with more than 12.5% MPD resulted in fewer/no proteinase K microcrystals or crystals with dissolved edges (crystal image not shown). Therefore, 12.5% MPD was chosen as the maximum concentration capable of to reducing the viscosity of the LCP sample (Figure 1b) without dissolving the embedded proteinase K crystals (Figure 2c).
LCP-proteinase K samples were treated with lipase (1:1 (v/v) ratio) along with additional crystallization precipitant solution added as a supplement to ensure the stability of the PK crystals after release from the LCP. Lipid hydrolysis was monitored every 1 hour for the first 14 hours, and every 20 minutes afterward. After 18-hours of lipase treatment, the sample composed of 1:1:2 (v/v/v) ratio of lipase:LCP-proteinase K:precipitant solution was completely hydrolyzed and separated into two liquid layers with the PK crystals being released from the LCP matrix into the glycerol rich phase. We observed that the liquid phase contained crystals of similar size and density to that of proteinase K microcrystals in the LCP-proteinase K sample before treatment (Figure 2d).
With these sample preparation methods, the low-viscosity LCP-microcrystal solution is applied to glow-discharged carbon coated EM grids, blotted with filter paper and vitrified in a method that in similar to preparing EM grids for non-LCP MicroED samples (Shi et al., 2016). MPD-induced less viscous lipid mesophase sample was further diluted by adding the converting buffer in a 1:1 (v/v) ratio, which generated a thin layer on EM grid where crystals could still be identified by UV microscopy (Figure 2e). Lipase treatment also produced grids with a similar level of microcrystals visible by UV (Figure 2f).
To further test the viability of the sample treatment strategy to crystals grown in LCP, cholesterol and the membrane protein A2AAR were crystallized in LCP and then examined in the MPD-treatment method. Cholesterol or A2AAR laden LCP sample was crystallized in the presence of 28% PEG400 in the initial crystallization precipitant solution, which transformed the cubic phase into a less viscous intermediate phase. Once crystals formed to the size of 2–3 μm needle shaped for cholesterol (Figure 4a) and ~ 5x5x2 μm3 for A2AAR (Figure 5a), excess precipitant solution was removed, and only 7% MPD was needed to convert the LCP by syringe mixing in a gentle manner. Further dilution by adding the converting buffer was applied before depositing crystals to the EM grid. A2AAR crystals on the grids were shown to have survived the addition of 7% MPD and the deposition process via UV microscopy (Figure 5b). The diffraction quality of A2AAR microcrystals following LCP conversion was also examined by X-ray diffraction using a microfocus beamline. These crystals retained their diffracting power to 2.4 Å resolution (Figure 5e), indicating the treatment with MPD does not significantly reduce the crystal quality.
Figure 4. Cholesterol microcrystals and MicroED diffraction.

After phase conversion with MPD, cholesterol microcrystals were located on the EM grids (a) and a still electron diffraction pattern was recorded with resolution ring of 0.9 Å (b) before the continuous-rotation MicroED data collection in cryo-TEM. (c) The model and density map derived from the LCP-MicroED cholesterol data viewed along the direction with high-completeness. Scale bar in a represents 3 μm.
Figure 5. A2AAR microcrystals monitoring and MicroED diffraction.
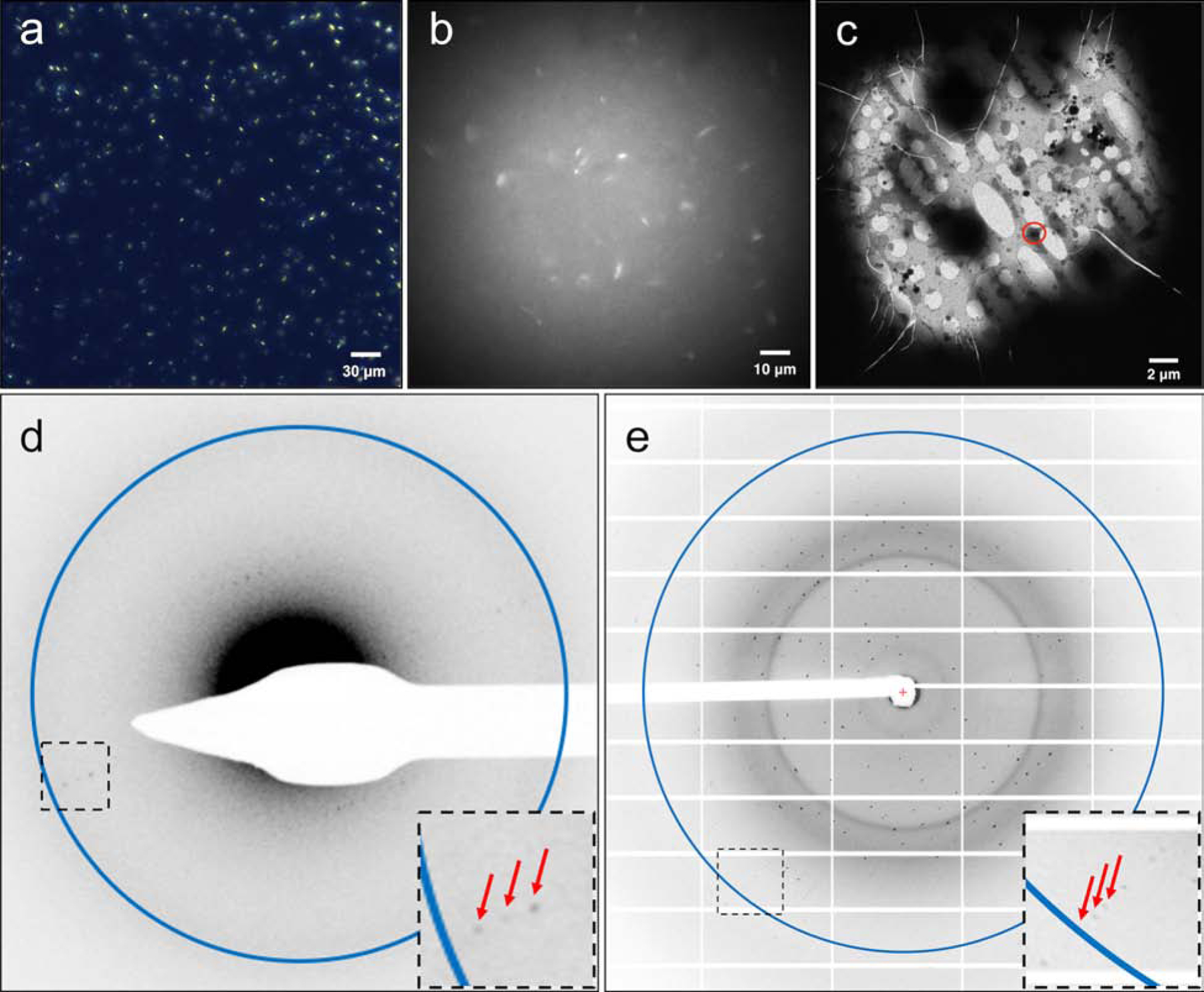
A2AAR were crystallized in LCP to a size of 5x5x2 μm3 (a, viewed with cross-polarized light), and microcrystals were survived after LCP phase conversion by the converting buffer supplemented with 7% MPD (b, viewed with UV). Microcrystals were located on the EM grids (c) and a still initial electron diffraction pattern with resolution ring of 4.5 Å (d) was recorded before the continuous-rotation MicroED data collection in cryo-TEM. Red arrows denote the diffracted spots to 4.5 Å in a closer view of the black boxed area. (e) A2AAR microcrystals, treated with the same phase conversion method, retained their diffraction power to ~2.4 Å resolution (shown as the resolution ring in the image) at a microfocus X-ray beamline (diffracted spots to the highest resolution were denoted by red arrows in a closer view of the black boxed area).
MicroED analysis of converted LCP samples
Samples prepared by the methods described above were visually analyzed in the cryo-TEM to verify the thickness and the presence of microcrystals. In all cases, regions of sample grids containing proteinase K (Figure 3a and 3e), cholesterol (Figure 4a), or A2AAR microcrystals (Figure 5c) could be visually identified and confirmed.
In the case of proteinase K, while both treatment strategies produced suitable samples, lipase-treated samples generally gave a thinner layer on the EM grid relative to the MPD-induced less viscous lipid mesophase samples. Standard MicroED diffraction screening, data collection, and data processing protocols (Hattne et al., 2015; Shi et al., 2016) were used to collect high-resolution data sets from each type of sample preparation. For the MPD converted samples, diffraction data from 4 proteinase K crystals were merged together to produce a final data set with a refined structure at 2.0 Å. In the case of the lipase treated LCP samples, data from 2 crystals were used to resolve the structure of proteinase K to also 2.0 Å. Both of these methods of LCP sample preparation ultimately produced high quality MicroED data, density maps, and models (Table 1; Figure 3). When compared to other proteinase K structures determined by MicroED (de la Cruz et al., 2017; Hattne et al., 2016; Hattne et al., 2018), both of the LCP-proteinase K structures in this work showed similar levels of data quality (e.g. resolution, R-factors), indicating that the LCP phase conversion method – whether induced by MPD or lipase – did not greatly impact the quality of vitrified crystals. Additionally, the overall RMSD is 0.59 Å between both of our models and another MicroED proteinase K structure (PDB ID: 5I9S (Hattne et al., 2016)), showing minimal differences. When we compare the MPD and lipase treated, the all atom RMSD between these two new structures is 0.47 Å.
Table 1.
Proteinase K data collection and refinement statistics
| Data Collection | ||
|---|---|---|
| Excitation voltage | 200kV | |
| Electron Source | Field Emission Gun | |
| Wavelength (Å) | 0.025079 | |
| Total dose per crystal | ~4 e−/Å2 | |
| Frame rate | 4 s/frame | |
| Rotation rate | 0.09°/s | |
| Data Processing | ||
| MPD-treated | Lipase-treated | |
| Number of crystals | 4 | 2 |
| Space group | P43212 | P43212 |
| Unit cell dimensions | ||
| a, b, c (Å) | 67.4, 67.4, 106.5 | 67.6, 67.6, 106.8 |
| α=β=γ (°) | 90 | 90° |
| Resolution (Å) | 17.4 – 2.0 | 16.6 – 2.0 |
| Total reflections | 111,081 | 85,421 |
| Total Unique Reflections | 14,491 | 16,351 |
| Rmerge (%) | 32.4 (53.2)a | 40.4 (70.5)a |
| CC1/2 | 0.937 (0.368)a | 0.900 (0.275)a |
| Multiplicity | 7.7 (6.3)a | 5.2 (5.4)a |
| Completeness (%) | 84.6 (63.3)a | 94.6 (94.8)a |
| Mean (I/σ(I)) | 5.5 (3.4)a | 4.0 (2.6)a |
| Data Refinement | ||
| Rwork/Rfree (%) | 21.7/26.7 | 24.4/28.2 |
| RMSD Bonds (Å) | 0.003 | 0.003 |
| RMSD Angles (°) | 0.571 | 0.478 |
| Ramachandran (%)b (Favored, allowed, outlier) | 96.8; 2.8; 0.4 | 97.1; 2.9; 0 |
Values for highest resolution shell of 2.05 Å – 2.0 Å
Statistics given by MolProbity(Chen et al., 2010)
Cholesterol microcrystals grown in LCP were converted to a less viscous phase using the same protocol involving MPD. The vitrified sample on EM grid (Figure 4a) was diffracted in the cryo-TEM and subsequent diffraction patterns showed well resolved spots to high resolution (slightly beyond 1Å) (Figure 4b). We were able to index the cholesterol data sets with a resulting P1 space group with unit cell dimensions of 12.257 Å, 12.343 Å, and 34.262 Å with angles of 89.551°, 83.497°, and 78.907° (Table 2). These values are consistent with previously published results on cholesterol structure (Craven, 1976; Varsano et al., 2018). Also, the X-ray structure of this cholesterol form (Craven, 1976) was used to calculate model amplitudes and they compared very well with the corresponding amplitudes obtained by MicroED (CC = 84.2%). Unfortunately, because of the low symmetry of the cholesterol crystals (P1) and the preferred orientation on the grid, the completeness of the merged data set was very low (31.1%), which prevented the use of direct methods for phasing. Due to low data completeness a reliable structure of cholesterol could not be determined, however a projection map could be calculated. By using the X-ray model of cholesterol and molecular replacement, a projection map was generated that shows good maps when viewed along the direction containing high data completeness for the cholesterol and water molecules in the crystals (Figure 4c). This is an important step as it demonstrated that the method used for the proteinase K samples previously could also be extended to the collection of MicroED data from crystals that had been grown in LCP.
Table 2.
Cholesterol data collection statistics
| Data Collection | |
|---|---|
| Excitation voltage | 300kV |
| Electron Source | Field Emission Gun |
| Wavelength (Å) | 0.019687 |
| Total dose per crystal | ~2 e−/Å2 |
| Frame rate | 2 s/frame |
| Rotation rate | 0.91 °/s |
| Data Processing | |
| Number of crystals | 2 |
| Space group | P1 |
| Unit cell dimensions | |
| a, b, c (Å) | 12.257, 12.343, 34.262 |
| α=β=γ (°) | 89.551, 83.497, 78.907 |
| Resolution (Å) | 1.00 |
| Total reflections | 8,582 |
| Total Unique Reflections | 3,293 |
| Rmerge (%) | 0.206 (0.835)a |
| CC1/2 | 0.978 (0.393)a |
| Multiplicity | 2.6 (2.7)a |
| Completeness (%) | 31.1 (30.9)a |
Values for highest resolution shell of 1.03 Å – 1.00 Å
We next applied the same MicroED data collection process to study A2AAR microcrystals and observed the diffraction to about 4.5 Å (Figure 5d). Though much fewer spots were found in the diffraction patterns within a narrow tilt angle series, we were able to collect a small tilt series that showed the unit cell parameters are consistent with previously published A2AAR models, including one from LCP-SFX experiment (Batyuk et al., 2016).
DISCUSSION
These results demonstrate, for the first time, that microcrystals embedded in LCP can yield high-resolution data and structures using MicroED. This proof-of-concept study and methodology presented here paves the way for future LCP-MicroED applications for challenging membrane protein targets that only form micro- or nanocrystals. To expand the application of LCP-MicroED to these difficult targets, further development and optimization based on these initial methods need to be explored. Because every LCP-microcrystal sample is unique, the expansion of the methods reported here into a suite of techniques will be important for its broad applicability. For the use of additives for phase conversion, a broader spectrum of chemicals should be investigated for LCP-MicroED. Certain polar solvents or other additives such as: propylene glycol (PG) and pentaerythritol propoxylate (PPO), are commonly used in membrane protein crystallization precipitant solutions, and can also be used as phase conversion additives. The identification of a suite of additives compatible with LCP-MicroED would allow users to choose chemicals already present in the crystallization precipitant solution (or components with similar chemical properties). When screening new additives, two critical factors – the viscosity of the chemical and the overall percentage needed to convert the phase – should be kept in mind. In this study, we found that MPD behaved much better than PEGs in microcrystal blotting on EM grids, which was attributed to its lower viscosity and lower concentration (6–18%) required for phase conversion to less viscous. The intrinsic viscosity is not a parameter which can be tuned easily; however, these additives typically have a wide range of concentrations that can drive conversion to a less viscous lipid mesophase (Wadsten et al., 2006). While screening additives, it is important to note that some chemicals may affect microcrystal quality at the concentrations required to trigger the desired phase conversion. Therefore, for novel additives, their effects on phase conversion and crystal quality should be carefully examined.
After phase conversion by the addition of additives, the diffraction quality of converted microcrystal samples could be evaluation in a microfocus X-ray beamline either with cryogenic frozen sample or with the equipped LCP sample injector at room temperature in a serial mode. Once the crystal diffraction quality is confirmed, vitrified EM grids can be loaded into a scanning electron microscope (SEM) to assess the distribution of blotted crystals with the excess LCP residua prior to data collection in cryo-TEM. Under a certain circumstance that the conversion by additives might disrupt the crystal diffraction quality, a lower concentration of additives in a safe range to preserve the crystal integrity could be used to partially convert the phase to a relatively lower viscosity, though not sufficient to produce a pure clean MicroED grid. The thickness of the vitrified grids could be further reduced to remove the excess LCP, generating desirable-sized lamellae, by cryo-focused ion beam (cryo-FIB) milling prior to MicroED data collection (Duyvesteyn et al., 2018) (Martynowycz et al., 2019a; Martynowycz et al., 2019b). This strategy has been applied to several structure determinations by MicroED without damage to the underlying crystal lattice. Following the proteinase K crystal diffraction data and structure determination, we crystallized, transformed the phase of, and vitrified cholesterol microcrystals grown in LCP for further MicroED data collection. The purpose of collecting cholesterol crystal diffraction is two-fold: 1) to establish that in the A2AAR-LCP crystallization condition (containing 10% (w/w) cholesterol supplemented in the host lipid monoolein) the concentration of cholesterol could not crystallize, even with MPD-induced phase conversion; and 2) that very high-resolution can be obtained following LCP conversion. The cholesterol crystals diffracted to high resolution and were able to be indexed showing small unit cell dimensions, indicating that the crystals indeed were a small molecule compound. Further, cholesterol crystallization in LCP occurred at a higher concentration (30% w/w for our samples) than that used for A2AAR-LCP crystallization, satisfying any concern that cholesterol may have crystallized and contaminated the final A2AAR crystal sample.
In this study, we present LCP-MicroED diffraction image and processed data using A2AAR microcrystals. A2AAR is a model G protein-coupled receptor that had been used extensively in diffraction method development and validation, particularly in cases of using LCP as a carrier medium, such as LCP-SFX (Batyuk et al., 2016) and -SMX (Martin-Garcia et al., 2017). A2AAR was crystallized in LCP in a high (28%) concentration of PEG400 condition that resulted in average crystal sizes of 5 x 5 x 2 mm3 as previously described for LCP-SFX data collection (Batyuk et al., 2016). Due to the high concentration of PEG400, the phase of the matrix was intermediate to that of cubic and sponge, therefore we empirically determined that 7% MPD was sufficient to reduce the viscosity of the matrix further for blotting and vitrification onto EM grids. We were able to visually confirm the presence of the A2AAR crystals on the EM grids that retained the appearance and dimensionality as we had observed prior to the MPD-mediated conversion. Further, MicroED experiments recorded diffraction spots that were visible to approximately 4.5 Å (Figure 5d). The indexed unit cell dimensions were similar to previously published results, suggesting that these are A2AAR diffraction patterns since no other protein crystals were blotted onto the grids at the time of experiment (with the exception of PK), and no other components of the LCP matrix would have possibly diffracted with such unit cell parameters.
The microfocus X-ray diffraction data show that the A2AAR crystals diffract well even after undergoing the treatment described here, whereas when analyzed by MicroED the diffraction only extended to 4.5 Å. This suggests that the weaker diffraction seen by MicroED is not a result of the sample processing procedures, but rather due to other factors. The most likely reason for the reduced diffraction is because of the thickness and shape of the A2AAR crystals. MicroED requires the crystals be thin (on the order of a few hundred nanometers) for the electron beam to penetrate and produce reliable diffraction data. When the crystals are thicker than 1 μm, as the case of A2AAR crystals in this study, much of the beam absorbed by the sample. Additionally, while not a problem for Proteinase K, excess LCP surrounding the A2AAR crystals could also increase the absorption of the electron beam. In the case of A2AAR, the average crystal size seen is approximately 5x5x2 μm3, which is too thick, and the as the crystals become smaller, the ratio of length and width to thickness remains relatively consistent. For crystals on the grid which are thin enough for MicroED, the length and width are also greatly reduced, and therefore the total number of unit cells in the electron beam is low. As described above, the use of cryo-FIB milling offers a solution to this problem, where the larger and thicker crystals could be milled leaving thin lamella that still had a reasonable area for diffraction. It is important to note that cryo-FIB milling alone is not sufficient for preparing samples from LCP as the LCP matrix would still too thick to process by cryo-FIB. Therefore protocols such as these presented in this work would still be required as upstream sample preparation prior to further sample processing by cryo-FIB milling. We expect that in future work, the A2AAR microcrystals will be the model system to further optimize MicroED data collection for LCP-embedded membrane proteins in streamlining the mesophase conversion and vitrification process, higher throughput diffraction experiments using the TEM, as well as in data collection and processing.
Because lipase hydrolyzes the lipids that make up the LCP matrix, lipase treatment generally provided thinner vitrified samples relative to MPD treatment, thereby increasing the area which was visible in the electron microscope. It has been previously shown that the lipase treatment of bacteriorhodopsin crystals could be performed without degrading the crystals (Belrhali et al., 1999). In the case of some membrane protein crystallization, lipid molecules may interact with membrane proteins and mediate protein crystal packing (Hanson et al., 2008; Liu et al., 2012), the lipase hydrolysis treatment method may cause deleterious effects to the target membrane protein crystals. Therefore, lipase treatment should be evaluated extensively for different membrane protein targets prior to LCP-MicroED studies.
The combination of the extraordinary properties of LCP and MicroED promises to facilitate the determination of high-resolution structures of challenging protein targets using just a few sub-micron thick crystals. These structural studies with MicroED could open the door to the identification of new structural information by improving resolution of poorly ordered samples (de la Cruz et al., 2017), determining structures with minimal radiation damage (Hattne et al., 2018), and facilitating the modeling of charge within structures (Yonekura et al., 2015; Yonekura and Maki-Yonekura, 2016). LCP-MicroED has the potential to be a robust method, expanding the scope of MicroED to the challenging membrane proteins, such as GPCRs, that do not exclusively grow in solutions, but instead with the great success of LCP crystallization, to solve high-resolution structures from microcrystals grown in LCP. Another intriguing potential is to apply MicroED towards the structural elucidation of small molecules, potentially complementing other EM techniques in materials studies. The successful development and use of LCP-MicroED will add another important tool to the field of structural biology.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brent L. Nannenga (Brent.Nannenga@asu.edu)
Materials Availability
Further information and requests for resources and reagents may be directed to and will be fulfilled by the Lead Contact, Brent L. Nannenga. All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate new software. Details of deposited coordinates and density are provided in the Key Resources Table. Coordinates and structure factors were deposited in the Protein Data Bank (PDB) under the accession code 6PQ0 and 6PQ4.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG M2-FITC antibody | Sigma | Cat# F4049 |
| Bacterial and Virus Strains | ||
| One Shot™ TOP10 Competent E.coli | Invitrogen | Cat# C404010 |
| MAX Efficiency™ DH10Bac Competent Cells | Invitrogen | Cat# 10361012 |
| Biological Samples | ||
| Proteinase K from Tritirachium album | Sigma | Cat# P2308 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DDM (N-Dodecyl-B-D-Maltoside) | Anatrace | Cat# D310 |
| EDTA-free complete protease inhibitor cocktail tablets | Roche | Cat# 5056489001 |
| ZM241385 | Tocris | Cat# 1036 |
| Bovine Serum Albumin | Sigma | Cat# A7030 |
| Cholesterol | Sigma | Cat# C8667 |
| CHS (Cholesteryl hemisuccinate) | Sigma | Cat# C6512 |
| Iodoacetamide | Sigma | Cat# I1149 |
| Lipase from Candida rugosa | Sigma | Cat# L1754 |
| Monoolein (1-Oleoyl-rac-glycerol) | Sigma | Cat# M7765 |
| Additive Screen | Hampton Research | Cat# HR2-428 |
| Ethylene glycol | Hampton Research | Cat# HR2-621 |
| Jeffamine M-600 | Hampton Research | Cat# HR2-501 |
| Polyethylene glycol 200 | Hampton Research | Cat# HR2-601 |
| Polyethylene glycol 400 | Hampton Research | Cat# HR2-603 |
| (+/−)-2-Methyl-2,4-pentanediol | Hampton Research | Cat# HR2-627 |
| Critical Commercial Assays | ||
| TALON Metal Affinity Resin | Clotech | Cat# 635507 |
| Deposited Data | ||
| PK-MPD | PDB, This Study | 6PQ0 |
| PK-Lipase | PDB, This Study | 6PQ4 |
| PK-Molecular replacement model | PDB, (Wang et al., 2006) | 2ID8 |
| CCDC, (Craven, 1976) | 1124488 | |
| Experimental Models: Cell Lines | ||
| Spodoptera frugiperda (Sf9) insect cells | Invitrogen | Cat# 11496-015 |
| Recombinant DNA | ||
| Human A2AAR cloned into the expression vector pFastBac1 | GenScript | N/A |
| Software and Algorithms | ||
| iMOSFLM | (Battye et al., 2011) | https://www.mrc-lmb.cam.ac.uk/harry/imosflm/ver730/introduction.html |
| AIMLESS | (Evans and Murshudov, 2013a) | http://www.ccp4.ac.uk/html/aimless.html |
| Phaser | (McCoy et al., 2007) | https://www.phenix-online.org/ |
| COOT | (Emsley and Cowtan, 2004) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PHENIX | (Afonine et al., 2012) | https://www.phenix-online.org/ |
| TVIPS | (Burnley et al., 2017) | https://www.ccpem.ac.uk/downloads/tvips-tools/0.0.3/README.html |
| Other | ||
| QUANTIFOIL Holey Carbon R 2/4, 300 Mesh, Copper | Electron Microscopy Sciences | Cat# Q310CR-4 |
| QUANTIFOIL Holey Carbon Multi A, 300 Mesh, Copper | Electron Microscopy Sciences | Cat# Q310CMA |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
A2AAR proteins were expressed and purified from baculovirus infected Spodoptera frugiperda (Sf9) insect cells, grown in ESF-921™ serum-free, protein-free insect cell medium (Expression Systems) at 27°C for 2 days.
METHOD DETAILS
Microcrystal sample preparation
Proteinase K (catalog no. P2308, Sigma) was crystallized using batch crystallization method by mixing equal volumes of proteinase K solution at 40 mg/mL in 0.02 M MES pH 6.5 and a precipitant solution composed of 0.1 M MES pH 6.5, 0.5 M sodium nitrate, 0.1 M calcium chloride. Proteinase K microcrystals appeared after 20 min incubation at 20 °C (Martin-Garcia et al., 2017). Microcrystals were pelleted by centrifugation at 500 g for 5 min, resuspended in the crystallization buffer, and then reconstituted into LCP by mixing with molten monoolein host lipid (catalog no. M7765, Sigma) in a lipid:solution ratio of 3:2 (v:v) using a dual-syringe mixer until a homogeneous and transparent LCP was formed (Caffrey and Cherezov, 2009).
The mixture of cholesterol (catalog no. CH200, Anatrace) and monoolein in a ratio of 3:7 (w/w) was co-dissolved in chloroform, and solvent was removed by evaporation under a stream of an inert gas followed by high vacuum drying at RT for 24 hours. Cholesterol was reconstituted into LCP by mixing of the premixture with water in a ratio of 3:2 (v/v) and injected into a syringe containing the precipitant solution same as A2AAR crystallization. Cholesterol crystals formed in LCP stream after 18 hours incubation at RT.
A2AAR was expressed, purified, and crystallized as described before (Batyuk et al., 2016; Liu et al., 2012; Liu et al., 2014). In brief, the A2AAR construct containing the BRIL fusion protein in intracellular loop 3 (A2AAR-BRIL) was subcloned into the pFastBac1 vector and transformed into One Shot™ TOP10 competent E.coli cells. Recombinant A2AAR bacmid DNA was prepared by transforming MAX Efficiency™ DH10Bac competent cells, followed by transfection of Sf9 cells to generate A2AAR baculoviruses. Sf9 cells were grown to 2 x 106 cells per mL before A2AAR baculovirus was added for infection. A2AAR was expressed in Sf9 cells for 48 hours at 27 °C after infection, and cells were harvested by centrifugation and stored at −80 °C until purification. Once the protein was purified and concentrated to approximately 25 mg/mL, A2AAR was reconstituted into LCP by mixing with molten monoolein, containing 40% (w/w) protein solution, 54% (w/w) monoolein and 6% (w/w) cholesterol. The protein-laden LCP were then injected into Hamilton gas-tight syringes containing precipitant solution composed of 28% (v/v) PEG400, 40 mM sodium thiocyanate, and 100 mM sodium citrate pH 5.0. A2AAR microcrystals formed in LCP stream after 24 hours incubation at 20 °C. Upon further inspection, the A2AAR microcrystals appear comparable to previously crystallized samples (Batyuk et al., 2016; Martin-Garcia et al., 2017) in different microscopy imaging modes (Figure 5a).
LCP microcrystal sample conversion set-up
Proteinase K microcrystals embedded in LCP were then either converted by mixing with additives to achieve a less viscous lipid mesophase or subjected to lipase treatment to separate into two immiscible liquid phases.
LCP-proteinase K crystal sample conversion was tested with seven additives, MPD, PEG200, PEG400, jeffamine M600, t-butanol, ethylene glycol, and 1,4-butanediol. Sample conversion was performed by syringe mixing 15–20 times of an LCP embedded proteinase K sample in one syringe and the conversion buffer in the other syringe. The conversion buffer was made from the initial crystallization buffer supplemented with each of different additives. Each conversion buffer was optimized with a gradient additive concentration series of 10% increments. Finer additive concentration optimization followed when an initial point was identified that was capable of converting the LCP phase to a less viscous lipid mesophase. Once the concentration range of supplemented additive was identified, EM grid blotting experiments were conducted to investigate the capability of the additives for producing quality MicroED samples. Among those seven additives, only MPD-induced less viscous lipid mesophase sample exhibited reproducibly good quality EM grids. MPD was then focused for further optimization to ensure crystal survival during phase conversion. Once the LCP phase was converted, LCP-microcrystal samples were transferred into a microcentrifuge tube. Crystals were centrifuged and harvested from the bottom of the tube by pipetting. These samples were applied to a glass slide to monitor crystal survival by light microscopy with cross-polarized and UV light, or deposited on EM grid for analysis in the cryo-TEM.
In the case of lipase treatment, LCP-proteinase K microcrystal samples were expelled from the LCP mixing syringe into a 0.2 mL microcentrifuge tube, followed by the addition of freshly prepared lipase solution at a volume ratio of 1:1:2 (lipase solution:LCP:crystallization buffer) directly into the same tube without mixing. The lipase used is from Candida rugosa (catalog no. L1754, Sigma) and is prepared at a concentration of 50 mg/mL in saturated K phosphate buffer. Incubation for at least 18 hours converts the lipidic cubic phase into a two-phase system consisting of two immiscible liquids: water/glycerol and oleic acid. Proteinase K crystals partitioned into the glycerol/water phase.
Once A2AAR microcrystals formed in LCP stream, excess precipitant solution was removed from syringe and the LCP- A2AAR crystal sample was directly used for MicroED sample preparation. LCP-A2AAR crystal sample conversion was tested with four additives, MPD, jeffamine M600, PEG200, and PEG400, by syringe mixing 3–5 times very gently with conversion buffer in the other syringe. Same as the conversion test of proteinase K, only MPD-treatment produced good quality EM grids with clear and thin window visualized in TEM. The conversion buffer was made from the initial crystallization buffer supplemented with 7% MPD. After syringe mixing of LCP- A2AAR crystals with conversion buffer, sample was incubated at 20 °C for 10 min allowing the MPD better diffusing in the LCP matrix. Once the LCP phase was converted, no further dilution with conversion buffer was required for A2AAR sample. X-ray crystallographic data on converted A2AAR samples were collected on the 23ID-D beamline (GM/CA) of the Advanced Photon Source at the Argonne National Laboratory using a 5 um minibeam collimator following previous protocols (Liu et al., 2012).
LCP-cholesterol crystal samples were converted with MPD treatment, following the same setup as stated above for A2AAR.
MicroED grid preparation and data collection
After MPD or LCP treatment, proteinase K microcrystals were collected by centrifugation at 500 g for 5 min. Crystal solution was then harvested by pipetting from the microfuge tube bottom into a fresh microfuge tube. For MPD samples, the crystal solution was further diluted by adding a 1:1 ratio of fresh precipitant solution supplemented with 12.5% MPD. Cholesterol and A2AAR microcrystals were directly applied to a glass slide to monitor crystal survival by light microscopy with cross-polarized and UV light, or deposited on EM grid for analysis in the cryo-TEM. Cryo-TEM samples were prepared by standard MicroED sample preparation procedures (Shi et al., 2016). Briefly, 2 μL of crystal solution was deposited on each side of a glow-discharged holey carbon EM grid (Quantifoil 2/4 for proteinase K crystals and cholesterol crystals, Quantifoil Multi A for A2AAR crystals), and the grid was processed with a Vitrobot Mark IV (Thermo Fisher) by blotting for 12–16 s followed by vitrification by plunging into liquid ethane. Sample preparation was optimized and screened in high-throughput fashion using a Titan Krios with CETA CMOS detector (Thermo Fisher). MicroED data collection of proteinase K was performed by standard methods (Shi et al., 2016) using a FEI TF20 cryo-TEM equipped with a F416 CMOS detector (TVIPS), operated at 200 kV. Diffraction data sets were collected as the stage was continuously rotated at a rate of ~0.09 °/s and the detector collected frames every 4 s at a dosage of approximately 0.01 e−/Å2 per second. Data sets covered approximately a 45°, for a total dose of no more than ~5 e−/Å2 per data set. A2AAR and cholesterol data collection were performed using a Titan Krios with CETA CMOS detector, operated at 300 kV, with continuously rotation at a rate of ~0.09 °/s and 0.91 °/s and the detector collected frames every 8 s and 2 s for A2AAR and cholesterol crystals, respectively. Data sets from A2AAR were collected to cover ~20° wedge, and those from cholesterol were collected to cover ~45°. The selected area aperture of the TEM was used to limit the area for single crystal from which data were collected.
Data processing and structure determination
MicroED data collected from MPD and lipase treated LCP-proteinase K microcrystals were indexed and integrated in iMOSFLM (Battye et al., 2011). For each treatment, data from multiple crystals (4 for MPD-treated and 2 for lipase-treated) were merged and scaled in AIMLESS (Evans and Murshudov, 2013b) to create merged data sets with high completeness. Phaser (McCoy et al., 2007) was used to perform molecular replacement using a Proteinase K model (PDB ID: 2ID8, (Wang et al., 2006)). The molecular replacement solution was refined using phenix.refine (Afonine et al., 2012) using electron scattering factors, followed by manual inspection of the model in Coot (Emsley and Cowtan, 2004; Emsley et al., 2010). This process is then repeated iteratively with attention paid to avoid phase bias from the initial Proteinase K molecular replacement template (2ID8).
A2AAR diffraction data were recorded as movie files of continuous tilt series. The images were extracted from the movie files first using the script mrc2tif from the IMOD software package (Kremer et al., 1996; Mastronarde, 2008) followed by the script tiff2smv from the TVIPS tools software suite (Hattne et al., 2015) to generate images in formats that can be processed by standard X-ray diffraction processing software. Cholesterol data sets were converted using similar conversion tools (Hattne et al., 2019). XDSGUI (Diederichs, 2018) was used for subsequent spot picking, indexing, integration, and scaling of the diffraction data. Cholesterol data from two crystals were merged using AIMLESS, and a previous model of cholesterol (CSD Entry: CHOLES20) was used to phase the diffraction data with Phaser.
QUANTIFICATION AND STATISTICAL ANALYSIS
Reported crystallographic statistics were calculated using the crystallographic data processing programs as described in the methods, and these values are reported in the crystallographic tables (Table 1 and Table 2).
Highlights.
LCP phase conversion enables the study of LCP embedded crystals by MicroED.
Lipase hydrolysis can liberate crystals from LCP for MicroED studies.
LCP-MicroED opens a path for structure study of membrane protein microcrystals.
ACKNOWLEDGMENTS
We would like to acknowledge the use of the Titan Krios at the Erying Materials Center at Arizona State University and the funding of this instrument by NSF MRI 1531991. This work was supported by the Center for Applied Structural Discovery (CASD) at the Biodesign Institute at Arizona State University (L.Z., G.B., L.J., M.-Y.L, W.L. and B.L.N.), a Mayo Clinic ASU Collaborative Seed Grant Award (W.L.), the Flinn Foundation Seed Grant (W.L.), the STC Program of the National Science Foundation through BioXFEL (No. 1231306; L.Z., L.J. and W.L.), the National Institutes of Health grants R21DA042298 (W.L.) and R01GM124152 (W.L. and B.L.N.). The Gonen lab is supported by funds from the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aherne M, Lyons JA, and Caffrey M (2012). A fast, simple and robust protocol for growing crystals in the lipidic cubic phase. Journal of Applied Crystallography 45, 1330–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sahouri Z, Lee M-Y, Li D, Liu W, and Caffrey M (2018). The Lipid Cubic Phase as a Medium for the Growth of Membrane Protein Microcrystals. In X-ray Free Electron Lasers, pp. 87–107.
- Battye TG, Kontogiannis L, Johnson O, Powell HR, and Leslie AG (2011). iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batyuk A, Galli L, Ishchenko A, Han GW, Gati C, Popov PA, Lee MY, Stauch B, White TA, Barty A, et al. (2016). Native phasing of x-ray free-electron laser data for a G protein-coupled receptor. Sci Adv 2, e1600292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belrhali H, Nollert P, Royant A, Menzel C, Rosenbusch JP, Landau EM, and Pebay-Peyroula E (1999). Protein, lipid and water organization in bacteriorhodopsin crystals: a molecular view of the purple membrane at 1.9 A resolution. Structure 7, 909–917. [DOI] [PubMed] [Google Scholar]
- Burnley T, Palmer CM, and Winn M (2017). Recent developments in the CCP-EM software suite. Acta Crystallogr D Struct Biol 73, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M (2008). On the Mechanism of Membrane Protein Crystallization in Lipidic Mesophases†. Crystal Growth & Design 8, 4244–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M (2015). A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Crystallogr F Struct Biol Commun 71, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M, and Cherezov V (2009). Crystallizing membrane proteins using lipidic mesophases. Nat Protoc 4, 706–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M, Li D, and Dukkipati A (2012). Membrane Protein Structure Determination Using Crystallography and Lipidic Mesophases: Recent Advances and Successes. Biochemistry 51, 6266–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M, Li D, Howe N, and Shah ST (2014). ‘Hit and run’ serial femtosecond crystallography of a membrane kinase in the lipid cubic phase. Philos Trans R Soc Lond B Biol Sci 369, 20130621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Clogston J, Papiz MZ, and Caffrey M (2006). Room to move: crystallizing membrane proteins in swollen lipidic mesophases. J Mol Biol 357, 1605–1618. [DOI] [PubMed] [Google Scholar]
- Craven BM (1976). Crystal structure of cholesterol monohydrate. Nature 260, 727–729. [DOI] [PubMed] [Google Scholar]
- de la Cruz MJ, Hattne J, Shi D, Seidler P, Rodriguez J, Reyes FE, Sawaya MR, Cascio D, Weiss SC, Kim SK, et al. (2017). Atomic-resolution structures from fragmented protein crystals with the cryoEM method MicroED. Nat Methods 14, 399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs K, Brehm W (2018). XDSGUI
- Duyvesteyn HME, Kotecha A, Ginn HM, Hecksel CW, Beale EV, de Haas F, Evans G, Zhang P, Chiu W, and Stuart DI (2018). Machining protein microcrystals for structure determination by electron diffraction. Proceedings of the National Academy of Sciences 115, 9569–9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ghachi M, Howe N, Huang CY, Olieric V, Warshamanage R, Touze T, Weichert D, Stansfeld PJ, Wang M, Kerff F, et al. (2018). Crystal structure of undecaprenyl-pyrophosphate phosphatase and its role in peptidoglycan biosynthesis. Nat Commun 9, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR, and Murshudov GN (2013a). How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR, and Murshudov GN (2013b). How good are my data and what is the resolution? Acta Crystallographica Section D Biological Crystallography 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmi M, Mugnaioli E, Gorelik TE, Kolb U, Palatinus L, Boullay P, Hovmöller S, and Abrahams JP (2019). 3D Electron Diffraction: The Nanocrystallography Revolution. ACS Central Science 5, 1315–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruene T, Wennmacher JTC, Zaubitzer C, Holstein JJ, Heidler J, Fecteau-Lefebvre A, De Carlo S, Muller E, Goldie KN, Regeni I, et al. (2018). Rapid Structure Determination of Microcrystalline Molecular Compounds Using Electron Diffraction. Angew Chem Int Ed Engl 57, 16313–16317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, and Stevens RC (2008). A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 16, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattne J, Martynowycz MW, Penczek PA, and Gonen T (2019). MicroED with the Falcon III direct electron detector. IUCrJ 6, 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattne J, Reyes FE, Nannenga BL, Shi D, de la Cruz MJ, Leslie AG, and Gonen T (2015). MicroED data collection and processing. Acta Crystallogr A Found Adv 71, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattne J, Shi D, de la Cruz MJ, Reyes FE, and Gonen T (2016). Modeling truncated pixel values of faint reflections in MicroED images. J Appl Crystallogr 49, 1029–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattne J, Shi D, Glynn C, Zee CT, Gallagher-Jones M, Martynowycz MW, Rodriguez JA, and Gonen T (2018). Analysis of Global and Site-Specific Radiation Damage in Cryo-EM. Structure 26, 759–766 e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C-Y, Olieric V, Howe N, Warshamanage R, Weinert T, Panepucci E, Vogeley L, Basu S, Diederichs K, Caffrey M, et al. (2018). In situ serial crystallography for rapid de novo membrane protein structure determination. Communications Biology 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Olieric V, Ma P, Panepucci E, Diederichs K, Wang M, and Caffrey M (2015). In meso in situ serial X-ray crystallography of soluble and membrane proteins. Acta Crystallogr D Biol Crystallogr 71, 1238–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishchenko A, Abola EE, and Cherezov V (2017a). Crystallization of Membrane Proteins: An Overview. Methods Mol Biol 1607, 117–141. [DOI] [PubMed] [Google Scholar]
- Ishchenko A, Peng L, Zinovev E, Vlasov A, Lee SC, Kuklin A, Mishin A, Borshchevskiy V, Zhang Q, and Cherezov V (2017b). Chemically Stable Lipids for Membrane Protein Crystallization. Cryst Growth Des 17, 3502–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger K, Bruenle S, Weinert T, Guba W, Muehle J, Miyazaki T, Weber M, Furrer A, Haenggi N, Tetaz T, et al. (2019). Structural Basis for Allosteric Ligand Recognition in the Human CC Chemokine Receptor 7. Cell 178, 1222–1230 e1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson LC, Stauch B, Ishchenko A, and Cherezov V (2017). A Bright Future for Serial Femtosecond Crystallography with XFELs. Trends Biochem Sci 42, 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CG, Martynowycz MW, Hattne J, Fulton TJ, Stoltz BM, Rodriguez JA, Nelson HM, and Gonen T (2018). The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Central Science 4, 1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, and McIntosh JR (1996). Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116, 71–76. [DOI] [PubMed] [Google Scholar]
- Lan Z, Lee MY, Chun E, Liu B, and Liu W (2018). Overview of Biochemical Assays in Lipidic Cubic Phase. Trends Biochem Sci [DOI] [PubMed]
- Landau EM, and Rosenbusch JP (1996). Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci U S A 93, 14532–14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AM, Bu G, Biswas S, Tsai EHR, Braunschweig AB, and Nannenga BL (2020). Crystal structure and orientation of organic semiconductor thin films by microcrystal electron diffraction and grazing-incidence wide-angle X-ray scattering. Chemical Communications 56, 4204–4207. [DOI] [PubMed] [Google Scholar]
- Li D, and Caffrey M (2011). Lipid cubic phase as a membrane mimetic for integral membrane protein enzymes. Proc Natl Acad Sci U S A 108, 8639–8644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Stansfeld PJ, Sansom MS, Keogh A, Vogeley L, Howe N, Lyons JA, Aragao D, Fromme P, Fromme R, et al. (2015). Ternary structure reveals mechanism of a membrane diacylglycerol kinase. Nat Commun 6, 10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, AP IJ, et al. (2012). Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Ishchenko A, and Cherezov V (2014). Preparation of microcrystals in lipidic cubic phase for serial femtosecond crystallography. Nat Protoc 9, 2123–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Wacker D, Gati C, Han GW, James D, Wang D, Nelson G, Weierstall U, Katritch V, Barty A, et al. (2013). Serial femtosecond crystallography of G protein-coupled receptors. Science 342, 1521–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma P, Weichert D, Aleksandrov LA, Jensen TJ, Riordan JR, Liu X, Kobilka BK, and Caffrey M (2017). The cubicon method for concentrating membrane proteins in the cubic mesophase. Nature Protocols 12, 1745–1762. [DOI] [PubMed] [Google Scholar]
- Martin-Garcia JM, Conrad CE, Nelson G, Stander N, Zatsepin NA, Zook J, Zhu L, Geiger J, Chun E, Kissick D, et al. (2017). Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ 4, 439–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Garcia JM, Zhu L, Mendez D, Lee MY, Chun E, Li C, Hu H, Subramanian G, Kissick D, Ogata C, et al. (2019). High-viscosity injector-based pink-beam serial crystallography of microcrystals at a synchrotron radiation source. IUCrJ 6, 412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynowycz MW, Zhao W, Hattne J, Jensen GJ, and Gonen T (2019a). Collection of Continuous Rotation MicroED Data from Ion Beam-Milled Crystals of Any Size. Structure 27, 545–548 e542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynowycz MW, Zhao W, Hattne J, Jensen GJ, and Gonen T (2019b). Qualitative Analyses of Polishing and Precoating FIB Milled Crystals for MicroED. Structure 27, 1594–1600.e1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2008). Correction for non-perpendicularity of beam and tilt axis in tomographic reconstructions with the IMOD package. J Microsc 230, 212–217. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nannenga BL, and Gonen T (2016). MicroED opens a new era for biological structure determination. Curr Opin Struct Biol 40, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nannenga BL, and Gonen T (2018). MicroED: a versatile cryoEM method for structure determination. Emerging Topics in Life Sciences. [DOI] [PMC free article] [PubMed]
- Nannenga BL, and Gonen T (2019). The cryo-EM method microcrystal electron diffraction (MicroED). Nature Methods 16, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nannenga BL, Shi D, Leslie AGW, and Gonen T (2014). High-resolution structure determination by continuous-rotation data collection in MicroED. Nat Methods 11, 927–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogly P, Weinert T, James D, Carbajo S, Ozerov D, Furrer A, Gashi D, Borin V, Skopintsev P, Jaeger K, et al. (2018). Retinal isomerization in bacteriorhodopsin captured by a femtosecond x-ray laser. Science 361. [DOI] [PubMed] [Google Scholar]
- Nollert P, and Landau EM (1998). Enzymic release of crystals from lipidic cubic phases. Biochem Soc Trans 26, 709–713. [DOI] [PubMed] [Google Scholar]
- Nollert P, Navarro J, and Landau EM (2002). Crystallization of membrane proteins in cubo. Methods Enzymol 343, 183–199. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula E, Rummel G, Rosenbusch JP, and Landau EM (1997). X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science 277, 1676–1681. [DOI] [PubMed] [Google Scholar]
- Qiu H, and Caffrey M (2000). The phase diagram of the monoolein/water system: metastability and equilibrium aspects. Biomaterials 21, 223–234. [DOI] [PubMed] [Google Scholar]
- Shi D, Nannenga BL, de la Cruz MJ, Liu J, Sawtelle S, Calero G, Reyes FE, Hattne J, and Gonen T (2016). The collection of MicroED data for macromolecular crystallography. Nat Protoc 11, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi D, Nannenga BL, Iadanza MG, and Gonen T (2013). Three-dimensional electron crystallography of protein microcrystals. Elife 2, e01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauch B, and Cherezov V (2018). Serial Femtosecond Crystallography of G Protein-Coupled Receptors. Annu Rev Biophys 47, 377–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varsano N, Beghi F, Elad N, Pereiro E, Dadosh T, Pinkas I, Perez-Berna AJ, Jin X, Kruth HS, Leiserowitz L, et al. (2018). Two polymorphic cholesterol monohydrate crystal structures form in macrophage culture models of atherosclerosis. Proceedings of the National Academy of Sciences 115, 7662–7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogeley L, El Arnaout T, Bailey J, Stansfeld PJ, Boland C, and Caffrey M (2016). Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science 351, 876–880. [DOI] [PubMed] [Google Scholar]
- Wadsten P, Wohri AB, Snijder A, Katona G, Gardiner AT, Cogdell RJ, Neutze R, and Engstrom S (2006). Lipidic sponge phase crystallization of membrane proteins. J Mol Biol 364, 44–53. [DOI] [PubMed] [Google Scholar]
- Wang J, Dauter M, and Dauter Z (2006). What can be done with a good crystal and an accurate beamline? Acta Crystallographica Section D 62, 1475–1483. [DOI] [PubMed] [Google Scholar]
- Weierstall U, James D, Wang C, White TA, Wang D, Liu W, Spence JC, Bruce Doak R, Nelson G, Fromme P, et al. (2014). Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat Commun 5, 3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T, Olieric N, Cheng R, Brunle S, James D, Ozerov D, Gashi D, Vera L, Marsh M, Jaeger K, et al. (2017). Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat Commun 8, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T, Skopintsev P, James D, Dworkowski F, Panepucci E, Kekilli D, Furrer A, Brunle S, Mous S, Ozerov D, et al. (2019). Proton uptake mechanism in bacteriorhodopsin captured by serial synchrotron crystallography. Science 365, 61–65. [DOI] [PubMed] [Google Scholar]
- Winkler MBL, Kidmose RT, Szomek M, Thaysen K, Rawson S, Muench SP, Wustner D, and Pedersen BP (2019). Structural Insight into Eukaryotic Sterol Transport through Niemann-Pick Type C Proteins. Cell 179, 485–497 e418. [DOI] [PubMed] [Google Scholar]
- Xiang J, Chun E, Liu C, Jing L, Al-Sahouri Z, Zhu L, and Liu W (2016). Successful Strategies to Determine High-Resolution Structures of GPCRs. Trends Pharmacol Sci 37, 1055–1069. [DOI] [PubMed] [Google Scholar]
- Yonekura K, Kato K, Ogasawara M, Tomita M, and Toyoshima C (2015). Electron crystallography of ultrathin 3D protein crystals: atomic model with charges. Proc Natl Acad Sci U S A 112, 3368–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura K, and Maki-Yonekura S (2016). Refinement of cryo-EM structures using scattering factors of charged atoms. Journal of Applied Crystallography 49. [Google Scholar]
- Zabara A, Chong JTY, Martiel I, Stark L, Cromer BA, Speziale C, Drummond CJ, and Mezzenga R (2018). Design of ultra-swollen lipidic mesophases for the crystallization of membrane proteins with large extracellular domains. Nat Commun 9, 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate new software. Details of deposited coordinates and density are provided in the Key Resources Table. Coordinates and structure factors were deposited in the Protein Data Bank (PDB) under the accession code 6PQ0 and 6PQ4.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG M2-FITC antibody | Sigma | Cat# F4049 |
| Bacterial and Virus Strains | ||
| One Shot™ TOP10 Competent E.coli | Invitrogen | Cat# C404010 |
| MAX Efficiency™ DH10Bac Competent Cells | Invitrogen | Cat# 10361012 |
| Biological Samples | ||
| Proteinase K from Tritirachium album | Sigma | Cat# P2308 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DDM (N-Dodecyl-B-D-Maltoside) | Anatrace | Cat# D310 |
| EDTA-free complete protease inhibitor cocktail tablets | Roche | Cat# 5056489001 |
| ZM241385 | Tocris | Cat# 1036 |
| Bovine Serum Albumin | Sigma | Cat# A7030 |
| Cholesterol | Sigma | Cat# C8667 |
| CHS (Cholesteryl hemisuccinate) | Sigma | Cat# C6512 |
| Iodoacetamide | Sigma | Cat# I1149 |
| Lipase from Candida rugosa | Sigma | Cat# L1754 |
| Monoolein (1-Oleoyl-rac-glycerol) | Sigma | Cat# M7765 |
| Additive Screen | Hampton Research | Cat# HR2-428 |
| Ethylene glycol | Hampton Research | Cat# HR2-621 |
| Jeffamine M-600 | Hampton Research | Cat# HR2-501 |
| Polyethylene glycol 200 | Hampton Research | Cat# HR2-601 |
| Polyethylene glycol 400 | Hampton Research | Cat# HR2-603 |
| (+/−)-2-Methyl-2,4-pentanediol | Hampton Research | Cat# HR2-627 |
| Critical Commercial Assays | ||
| TALON Metal Affinity Resin | Clotech | Cat# 635507 |
| Deposited Data | ||
| PK-MPD | PDB, This Study | 6PQ0 |
| PK-Lipase | PDB, This Study | 6PQ4 |
| PK-Molecular replacement model | PDB, (Wang et al., 2006) | 2ID8 |
| CCDC, (Craven, 1976) | 1124488 | |
| Experimental Models: Cell Lines | ||
| Spodoptera frugiperda (Sf9) insect cells | Invitrogen | Cat# 11496-015 |
| Recombinant DNA | ||
| Human A2AAR cloned into the expression vector pFastBac1 | GenScript | N/A |
| Software and Algorithms | ||
| iMOSFLM | (Battye et al., 2011) | https://www.mrc-lmb.cam.ac.uk/harry/imosflm/ver730/introduction.html |
| AIMLESS | (Evans and Murshudov, 2013a) | http://www.ccp4.ac.uk/html/aimless.html |
| Phaser | (McCoy et al., 2007) | https://www.phenix-online.org/ |
| COOT | (Emsley and Cowtan, 2004) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PHENIX | (Afonine et al., 2012) | https://www.phenix-online.org/ |
| TVIPS | (Burnley et al., 2017) | https://www.ccpem.ac.uk/downloads/tvips-tools/0.0.3/README.html |
| Other | ||
| QUANTIFOIL Holey Carbon R 2/4, 300 Mesh, Copper | Electron Microscopy Sciences | Cat# Q310CR-4 |
| QUANTIFOIL Holey Carbon Multi A, 300 Mesh, Copper | Electron Microscopy Sciences | Cat# Q310CMA |